

A Cross-Sectional Study of Potential Antimicrobial Resistance and Ecology in Gastrointestinal and Oral Microbial Communities of Young Normoweight Pakistani Individuals
 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bioinformatics
2.2. Statistical Analysis
2.2.1. Prefiltering
2.2.2. Microbial Diversity Analyses
2.2.3. Contribution of Antimicrobial Resistance Genes to Microbial Diversity
2.2.4. Interaction between the Microbial Communities or Predictive Antimicrobial Resistance Genes (piARGs) with Study Participant Variables
2.2.5. Differential Taxa
2.2.6. Microbial Niche Width
2.2.7. Null-Modelling Analyses
2.2.8. Complexity Stability Analysis
3. Results
3.1. Diversity Patterns Differ across Gender and Sample Types
3.2. Intersample Variability in AMR Genes Is Attributed to Both Antibiotic Usage and Gender
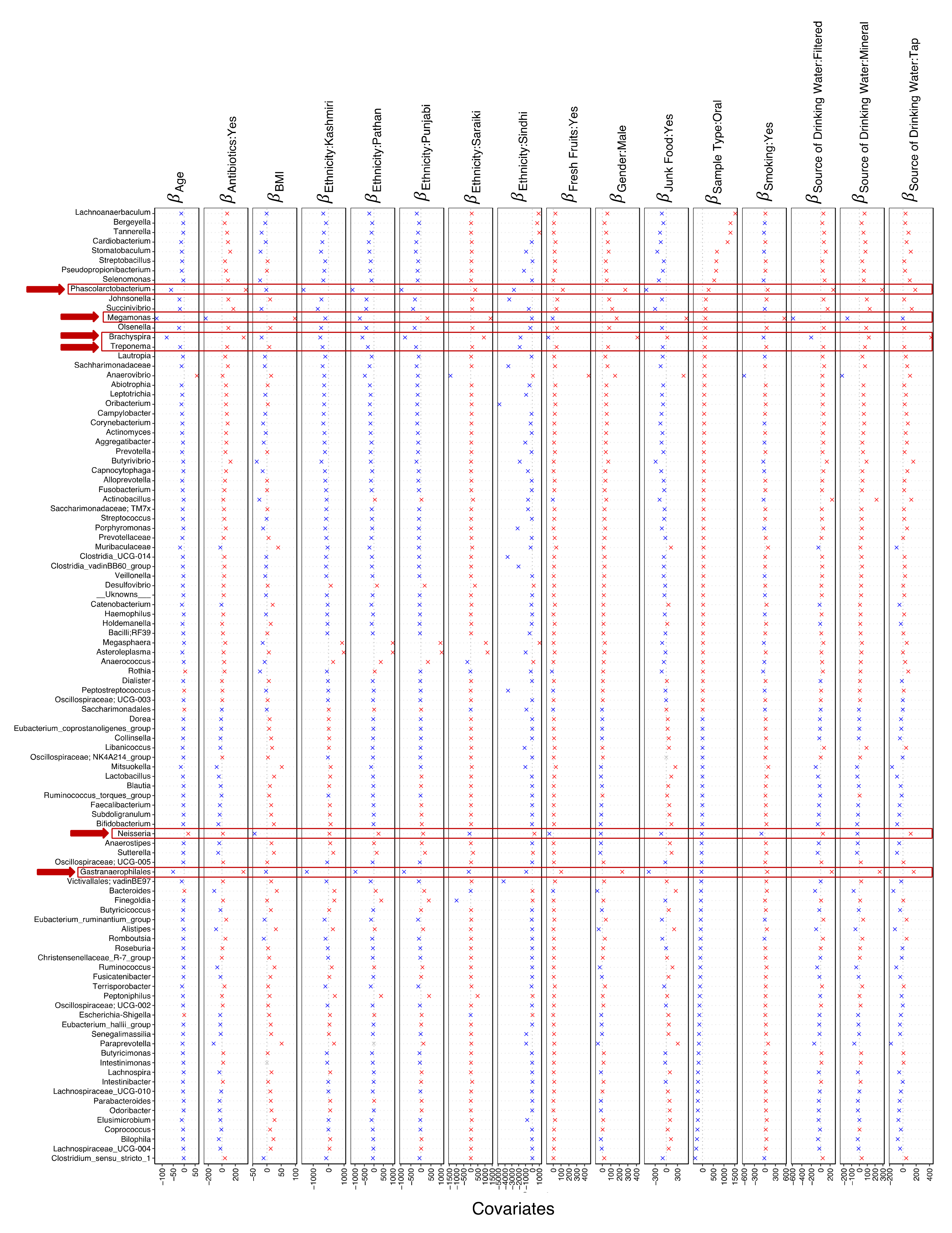
3.3. Sources of Variations Implicated in the Prevalence of Antimicrobial Resistance Genes
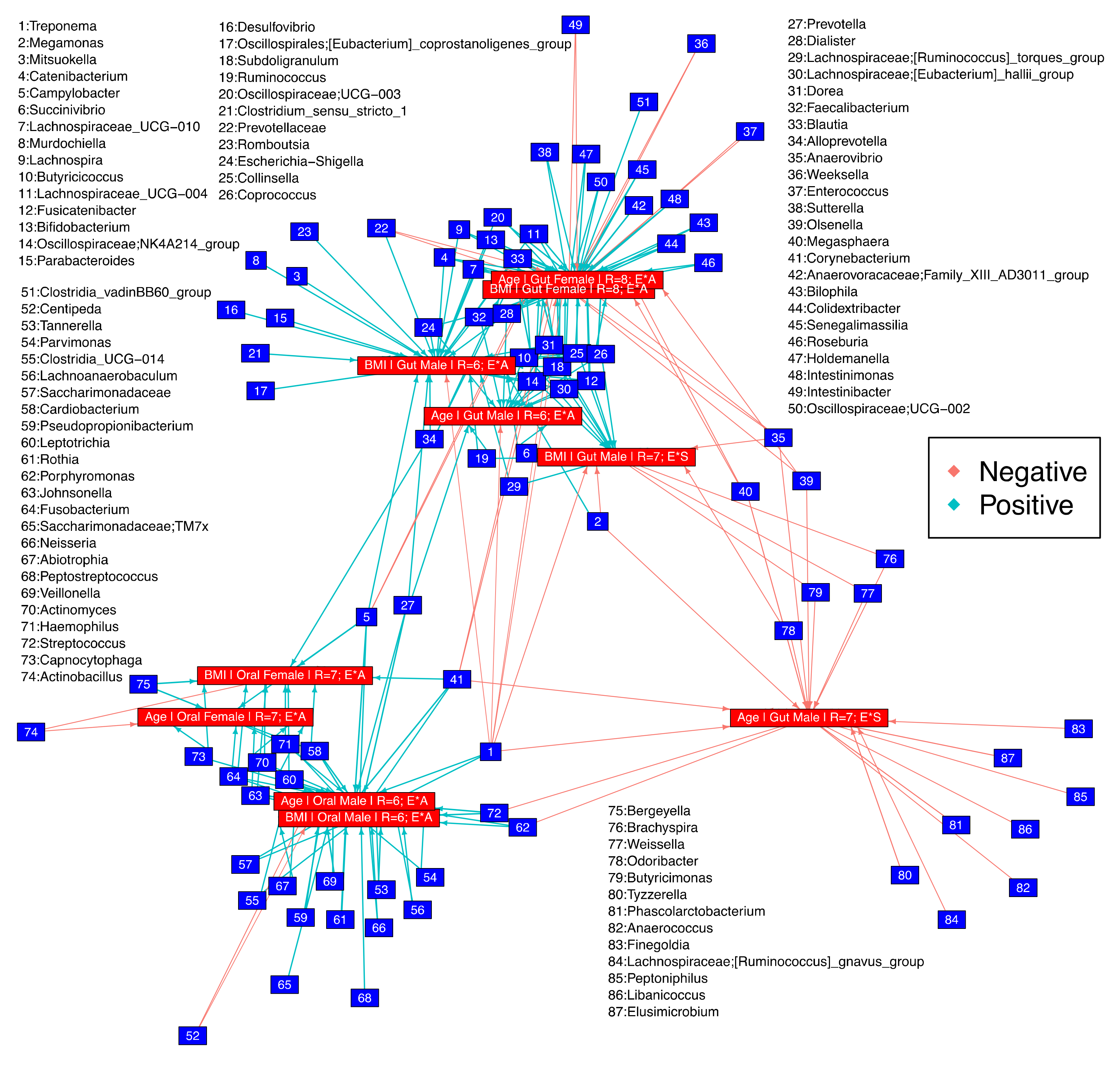
3.4. Key Microbes Associated with Sources of Variation
3.5. Microbial Niche Exploration Reveals Signature Taxa for Oral and Gut Communities Associated with Age and BMI
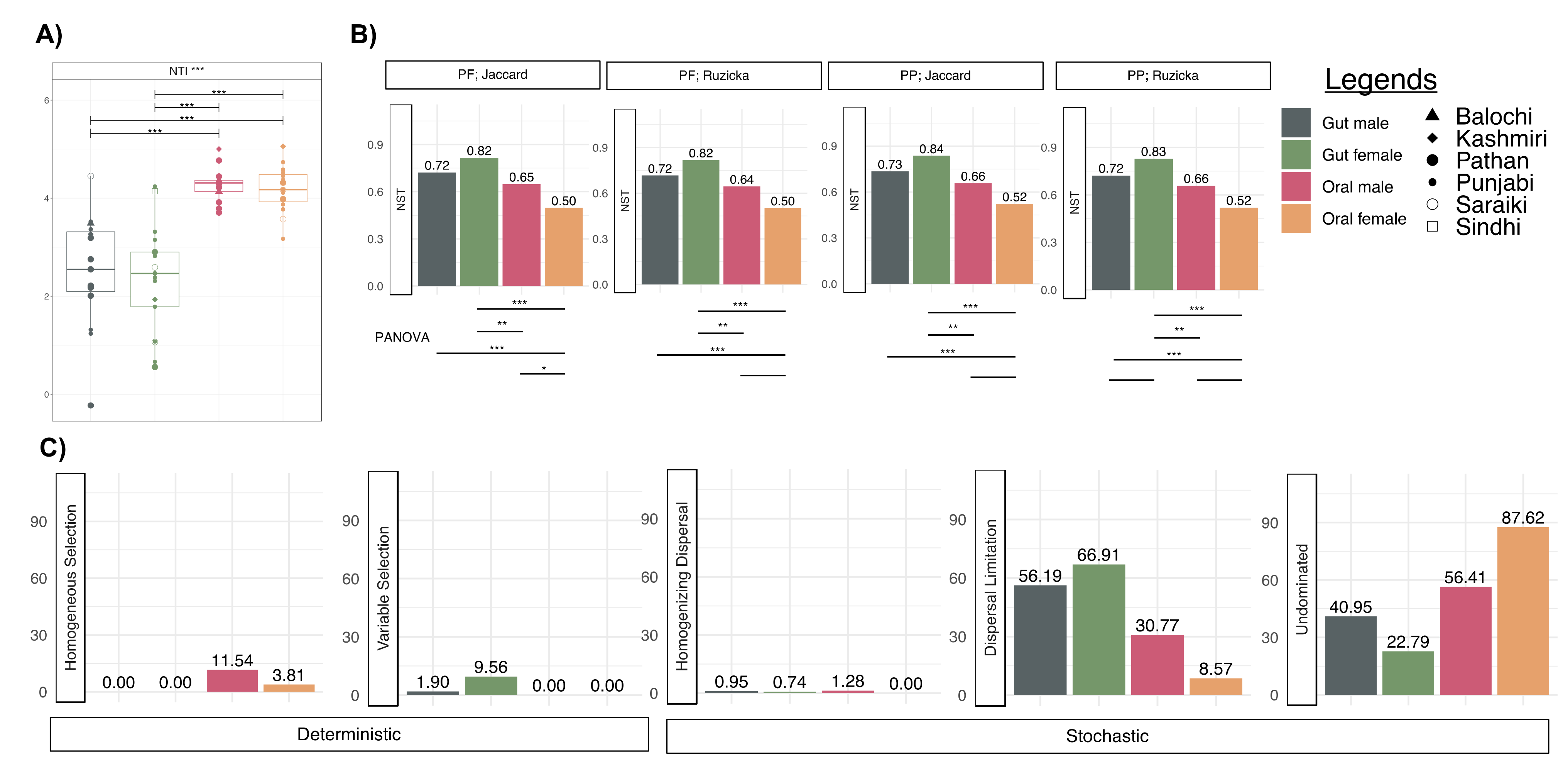
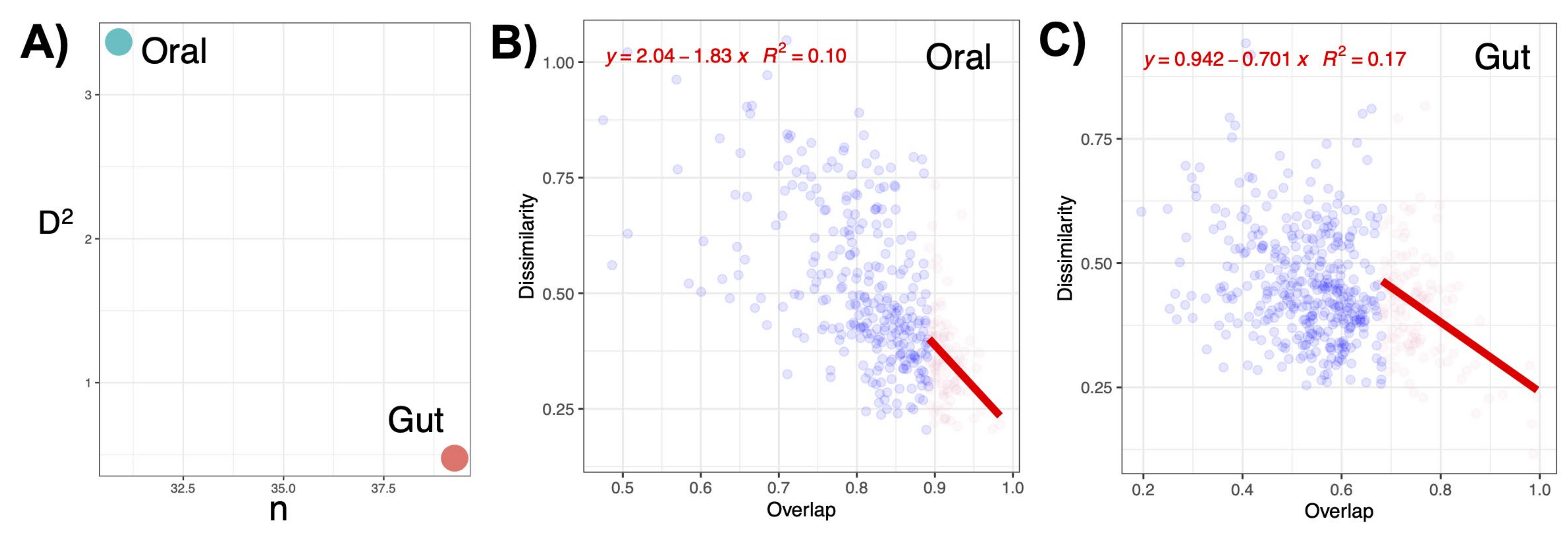
3.6. More Environmental Pressures on Oral Communities than Gut Communities
3.7. Oral Microbiota Is Less Stable as Compared to the Gut Microbiota
3.8. The Link between Oral and Gut Microbial Communities
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Adedeji, W.A. The Treasure called Antibiotics. Ann. Ib. Postgrad. Med. 2016, 14, 56–57. [Google Scholar] [PubMed]
- Sandoval-Motta, S.; Aldana, M. Adaptive Resistance to Antibiotics in Bacteria: A Systems Biology Perspective. WIREs Syst. Biol. Med. 2016, 8, 253–267. [Google Scholar] [CrossRef] [PubMed]
- Ferri, M.; Ranucci, E.; Romagnoli, P.; Giaccone, V. Antimicrobial Resistance: A Global Emerging Threat to Public Health Systems. Crit. Rev. Food Sci. Nutr. 2017, 57, 2857–2876. [Google Scholar] [CrossRef] [PubMed]
- Murray, C.J.; Ikuta, K.S.; Sharara, F.; Swetschinski, L.; Aguilar, G.R.; Gray, A.; Han, C.; Bisignano, C.; Rao, P.; Wool, E.; et al. Global Burden of Bacterial Antimicrobial Resistance in 2019: A Systematic Analysis. Lancet 2022, 399, 629–655. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, J. Tackling Drug-Resistant Infections Globally: Final Report and Recommendations. Review on Antimicrobial Resistance, chaired by Jim O’Neill; Government of the United Kingdom: London, UK, 2016. [Google Scholar]
- Dunachie, S.J.; Day, N.P.; Dolecek, C. The Challenges of Estimating the Human Global Burden of Disease of Antimicrobial Resistant Bacteria. Curr. Opin. Microbiol. 2020, 57, 95–101. [Google Scholar] [CrossRef]
- World Health Organization. Global Antimicrobial Resistance Surveillance System: Manual for Early Implementation; World Health Organization: Geneva, Switzerland, 2015; ISBN 978-92-4-154940-0. [Google Scholar]
- Bhatia, R.; Narain, J.P. The Growing Challenge of Antimicrobial Resistance in the South-East Asia Region-Are We Losing the Battle? Indian J. Med. Res. 2010, 132, 482–486. [Google Scholar] [CrossRef]
- Iskandar, K.; Molinier, L.; Hallit, S.; Sartelli, M.; Hardcastle, T.C.; Haque, M.; Lugova, H.; Dhingra, S.; Sharma, P.; Islam, S.; et al. Surveillance of Antimicrobial Resistance in Low- and Middle-Income Countries: A Scattered Picture. Antimicrob. Resist. Infect. Control 2021, 10, 63. [Google Scholar] [CrossRef]
- Ayukekbong, J.A.; Ntemgwa, M.; Atabe, A.N. The Threat of Antimicrobial Resistance in Developing Countries: Causes and Control Strategies. Antimicrob. Resist. Infect. Control 2017, 6, 47. [Google Scholar] [CrossRef]
- Bilal, H.; Khan, M.N.; Rehman, T.; Hameed, M.F.; Yang, X. Antibiotic Resistance in Pakistan: A Systematic Review of Past Decade. BMC Infect. Dis. 2021, 21, 244. [Google Scholar] [CrossRef]
- Saleem, Z.; Saeed, H.; Hassali, M.A.; Godman, B.; Asif, U.; Yousaf, M.; Ahmed, Z.; Riaz, H.; Raza, S.A. Pattern of Inappropriate Antibiotic Use among Hospitalized Patients in Pakistan: A Longitudinal Surveillance and Implications. Antimicrob. Resist. Infect. Control 2019, 8, 188. [Google Scholar] [CrossRef]
- Atif, M.; Asghar, S.; Mushtaq, I.; Malik, I.; Amin, A.; Babar, Z.-U.-D.; Scahill, S. What Drives Inappropriate Use of Antibiotics? A Mixed Methods Study from Bahawalpur, Pakistan. Infect. Drug Resist. 2019, 12, 687–699. [Google Scholar] [CrossRef] [Green Version]
- Khan, S.; Shehzad, A.; Shehzad, O.; Al-Suhaimi, E.A. Inpatient Antibiotics Pharmacology and Physiological Use in Hayatabad Medical Complex, Pakistan. Int. J. Physiol. Pathophysiol. Pharmacol. 2013, 5, 120–127. [Google Scholar]
- Saeed, D.K.; Farooqi, J.; Shakoor, S.; Hasan, R. Antimicrobial Resistance among GLASS Priority Pathogens from Pakistan: 2006–2018. BMC Infect. Dis. 2021, 21, 1231. [Google Scholar] [CrossRef]
- Karah, N.; Khalid, F.; Wai, S.N.; Uhlin, B.E.; Ahmad, I. Molecular Epidemiology and Antimicrobial Resistance Features of Acinetobacter Baumannii Clinical Isolates from Pakistan. Ann. Clin. Microbiol. Antimicrob. 2020, 19, 2. [Google Scholar] [CrossRef] [Green Version]
- Centers for Disease Control and Prevention (U.S.). Antibiotic Resistance Threats in the United States; Centers for Disease Control and Prevention (U.S.): Atlanta, GA, USA, 2019. [Google Scholar]
- Belkaid, Y.; Hand, T. Role of the Microbiota in Immunity and Inflammation. Cell 2014, 157, 121–141. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.-J.; Wu, E. The Role of Gut Microbiota in Immune Homeostasis and Autoimmunity. Gut Microbes 2012, 3, 4–14. [Google Scholar] [CrossRef] [Green Version]
- Blaser, M.J. Antibiotic Use and Its Consequences for the Normal Microbiome. Science 2016, 352, 544–545. [Google Scholar] [CrossRef] [Green Version]
- Stecher, B.; Maier, L.; Hardt, W.-D. “Blooming” in the Gut: How Dysbiosis Might Contribute to Pathogen Evolution. Nat. Rev. Microbiol. 2013, 11, 277–284. [Google Scholar] [CrossRef] [Green Version]
- Huttenhower, C.; Gevers, D.; Knight, R.; Abubucker, S.; Badger, J.H.; Chinwalla, A.T.; Creasy, H.H.; Earl, A.M.; FitzGerald, M.G.; Fulton, R.S.; et al. Structure, Function and Diversity of the Healthy Human Microbiome. Nature 2012, 486, 207–214. [Google Scholar] [CrossRef] [Green Version]
- Lange, K.; Buerger, M.; Stallmach, A.; Bruns, T. Effects of Antibiotics on Gut Microbiota. Dig. Dis. 2016, 34, 260–268. [Google Scholar] [CrossRef]
- Faith, J.J.; Guruge, J.L.; Charbonneau, M.; Subramanian, S.; Seedorf, H.; Goodman, A.L.; Clemente, J.C.; Knight, R.; Heath, A.C.; Leibel, R.L.; et al. The Long-Term Stability of the Human Gut Microbiota. Science 2013, 341, 1237439. [Google Scholar] [CrossRef] [PubMed]
- Gacesa, R.; Kurilshikov, A.; Vich Vila, A.; Sinha, T.; Klaassen, M.A.Y.; Bolte, L.A.; Andreu-Sánchez, S.; Chen, L.; Collij, V.; Hu, S.; et al. Environmental Factors Shaping the Gut Microbiome in a Dutch Population. Nature 2022, 604, 732–739. [Google Scholar] [CrossRef] [PubMed]
- Lozupone, C.A.; Stombaugh, J.I.; Gordon, J.I.; Jansson, J.K.; Knight, R. Diversity, Stability and Resilience of the Human Gut Microbiota. Nature 2012, 489, 220–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stegen, J.C.; Lin, X.; Konopka, A.E.; Fredrickson, J.K. Stochastic and Deterministic Assembly Processes in Subsurface Microbial Communities. ISME J. 2012, 6, 1653–1664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stegen, J.C.; Lin, X.; Fredrickson, J.K.; Konopka, A.E. Estimating and Mapping Ecological Processes Influencing Microbial Community Assembly. Front. Microbiol. 2015, 6, 370. [Google Scholar] [CrossRef] [Green Version]
- Stegen, J.C.; Lin, X.; Fredrickson, J.K.; Chen, X.; Kennedy, D.W.; Murray, C.J.; Rockhold, M.L.; Konopka, A. Quantifying Community Assembly Processes and Identifying Features That Impose Them. ISME J. 2013, 7, 2069–2079. [Google Scholar] [CrossRef]
- Stegen, J.C.; Bottos, E.M.; Jansson, J.K. A Unified Conceptual Framework for Prediction and Control of Microbiomes. Curr. Opin. Microbiol. 2018, 44, 20–27. [Google Scholar] [CrossRef]
- Martínez, I.; Stegen, J.C.; Maldonado-Gómez, M.X.; Eren, A.M.; Siba, P.M.; Greenhill, A.R.; Walter, J. The Gut Microbiota of Rural Papua New Guineans: Composition, Diversity Patterns, and Ecological Processes. Cell Rep. 2015, 11, 527–538. [Google Scholar] [CrossRef] [Green Version]
- Leibold, M.A. The Niche Concept Revisited: Mechanistic Models and Community Context. Ecology 1995, 76, 1371–1382. [Google Scholar] [CrossRef]
- Finn, D.R.; Yu, J.; Ilhan, Z.E.; Fernandes, V.M.C.; Penton, C.R.; Krajmalnik-Brown, R.; Garcia-Pichel, F.; Vogel, T.M. MicroNiche: An R Package for Assessing Microbial Niche Breadth and Overlap from Amplicon Sequencing Data. FEMS Microbiol. Ecol. 2020, 96, fiaa131. [Google Scholar] [CrossRef]
- Batool, M.; Ali, S.B.; Jaan, A.; Khalid, K.; Ali, S.A.; Kamal, K.; Raja, A.A.; Gul, F.; Nasir, A. Initial Sequencing and Characterization of the Gastrointestinal and Oral Microbiota in Urban Pakistani Adults. Front. Cell. Infect. Microbiol. 2020, 10, 409. [Google Scholar] [CrossRef]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PICRUSt2 for Prediction of Metagenome Functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, Interactive, Scalable and Extensible Microbiome Data Science Using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Amir, A.; McDonald, D.; Navas-Molina, J.A.; Kopylova, E.; Morton, J.T.; Zech Xu, Z.; Kightley, E.P.; Thompson, L.R.; Hyde, E.R.; Gonzalez, A.; et al. Deblur Rapidly Resolves Single-Nucleotide Community Sequence Patterns. mSystems 2017, 2, e00191-e16. [Google Scholar] [CrossRef] [Green Version]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA Ribosomal RNA Gene Database Project: Improved Data Processing and Web-Based Tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2–Approximately Maximum-Likelihood Trees for Large Alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria. Available online: https://www.R-project.org (accessed on 19 November 2022).
- Dixon, P. VEGAN, a Package of R Functions for Community Ecology. J. Veg. Sci. 2003, 14, 927–930. [Google Scholar] [CrossRef]
- McMurdie, P.J.; Holmes, S. Phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [Green Version]
- Su, Z.; Huang, B.; Mu, Q.; Wen, D. Evaluating the Potential Antibiotic Resistance Status in Environment Based on the Trait of Microbial Community. Front. Microbiol. 2020, 11, 575707. [Google Scholar] [CrossRef]
- Yang, S. OtuSummary: Summarizing OTU Table Regarding the Composition, Abundance and Beta Diversity of Abundant and Rare Biospheres. 2018. [Google Scholar]
- Niku, J.; Hui, F.K.C.; Taskinen, S.; Warton, D.I. Gllvm: Fast Analysis of Multivariate Abundance Data with Generalized Linear Latent Variable Models in r. Methods Ecol. Evol. 2019, 10, 2173–2182. [Google Scholar] [CrossRef]
- Tang, Z.-Z.; Chen, G. Robust and Powerful Differential Composition Tests for Clustered Microbiome Data. Stat. Biosci. 2021, 13, 200–216. [Google Scholar] [CrossRef]
- Csardi, G.; Nepusz, T. The Igraph Software Package for Complex Network Research. InterJournal, Complex Systems. 2005; 1695, 1–9. [Google Scholar]
- Wang, B.; Yao, M.; Lv, L.; Ling, Z.; Li, L. The Human Microbiota in Health and Disease. Engineering 2017, 3, 71–82. [Google Scholar] [CrossRef]
- Kembel, S.W.; Cowan, P.D.; Helmus, M.R.; Cornwell, W.K.; Morlon, H.; Ackerly, D.D.; Blomberg, S.P.; Webb, C.O. Picante: R Tools for Integrating Phylogenies and Ecology. Bioinforma. Oxf. Engl. 2010, 26, 1463–1464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yonatan, Y.; Amit, G.; Friedman, J.; Bashan, A. Complexity-Stability Trade-off in Empirical Microbial Ecosystems. Nat. Ecol. Evol. 2022, 6, 693–700. [Google Scholar] [CrossRef]
- May, R.M. Will a Large Complex System Be Stable? Nature 1972, 238, 413–414. [Google Scholar] [CrossRef]
- Wickham, H. Ggplot2. WIREs Comput. Stat. 2011, 3, 180–185. [Google Scholar] [CrossRef]
- Kim, S.-M.; Kim, H.C.; Lee, S.-W.S. Characterization of Antibiotic Resistance Determinants in Oral Biofilms. J. Microbiol. 2011, 49, 595. [Google Scholar] [CrossRef]
- Carr, V.R.; Witherden, E.A.; Lee, S.; Shoaie, S.; Mullany, P.; Proctor, G.B.; Gomez-Cabrero, D.; Moyes, D.L. Abundance and Diversity of Resistomes Differ between Healthy Human Oral Cavities and Gut. Nat. Commun. 2020, 11, 693. [Google Scholar] [CrossRef] [Green Version]
- Penders, J.; Stobberingh, E.; Savelkoul, P.; Wolffs, P. The Human Microbiome as a Reservoir of Antimicrobial Resistance. Front. Microbiol. 2013, 4, 87. [Google Scholar] [CrossRef] [Green Version]
- Daud, M.K.; Nafees, M.; Ali, S.; Rizwan, M.; Bajwa, R.A.; Shakoor, M.B.; Arshad, M.U.; Chatha, S.A.S.; Deeba, F.; Murad, W.; et al. Drinking Water Quality Status and Contamination in Pakistan. BioMed Res. Int. 2017, 2017, 7908183. [Google Scholar] [CrossRef]
- Atif, M.; Azeem, M.; Sarwar, M.R.; Shahid, S.; Javaid, S.; Ikram, H.; Baig, U.; Scahill, S. WHO/INRUD Prescribing Indicators and Prescribing Trends of Antibiotics in the Accident and Emergency Department of Bahawal Victoria Hospital, Pakistan. SpringerPlus 2016, 5, 1928. [Google Scholar] [CrossRef] [Green Version]
- Khan, K.; Lu, Y.; Saeed, M.A.; Bilal, H.; Sher, H.; Khan, H.; Ali, J.; Wang, P.; Uwizeyimana, H.; Baninla, Y.; et al. Prevalent Fecal Contamination in Drinking Water Resources and Potential Health Risks in Swat, Pakistan. J. Environ. Sci. China 2018, 72, 1–12. [Google Scholar] [CrossRef]
- Khan, G.A.; Berglund, B.; Khan, K.M.; Lindgren, P.-E.; Fick, J. Occurrence and Abundance of Antibiotics and Resistance Genes in Rivers, Canal and near Drug Formulation Facilities–A Study in Pakistan. PLoS ONE 2013, 8, e62712. [Google Scholar] [CrossRef]
- Taviani, E.; Pedro, O. Impact of the Aquatic Pathobiome in Low-Income and Middle-Income Countries (LMICs) Quest for Safe Water and Sanitation Practices. Curr. Opin. Biotechnol. 2022, 73, 220–224. [Google Scholar] [CrossRef]
- McInnes, R.S.; Alam, I.T.; Ho SF, S.; Moran, R.A.; Clemens, J.D.; Islam, M.S.; van Schaik, W. Microbiota Analysis of Rural and Urban Surface Waters and Sediments in Bangladesh Identifies Human Waste as Driver of Antibiotic Resistance. bioRxiv 2021. [Google Scholar] [CrossRef]
- Bennett, P.M. Plasmid Encoded Antibiotic Resistance: Acquisition and Transfer of Antibiotic Resistance Genes in Bacteria. Br. J. Pharmacol. 2008, 153, S347–S357. [Google Scholar] [CrossRef] [Green Version]
- Palmas, V.; Pisanu, S.; Madau, V.; Casula, E.; Deledda, A.; Cusano, R.; Uva, P.; Vascellari, S.; Loviselli, A.; Manzin, A.; et al. Gut Microbiota Markers Associated with Obesity and Overweight in Italian Adults. Sci. Rep. 2021, 11, 5532. [Google Scholar] [CrossRef]
- Yan, H.; Qin, Q.; Yan, S.; Chen, J.; Yang, Y.; Li, T.; Gao, X.; Ding, S. Comparison Of The Gut Microbiota In Different Age Groups In China. Front. Cell. Infect. Microbiol. 2022, 12. [Google Scholar] [CrossRef]
- Yang, H.-T.; Xiu, W.-J.; Liu, J.-K.; Yang, Y.; Zhang, Y.-J.; Zheng, Y.-Y.; Wu, T.-T.; Hou, X.-G.; Wu, C.-X.; Ma, Y.-T.; et al. Characteristics of the Intestinal Microorganisms in Middle-Aged and Elderly Patients: Effects of Smoking. ACS Omega 2022, 7, 1628–1638. [Google Scholar] [CrossRef] [PubMed]
- Määttä, A.-M.; Salminen, A.; Pietiäinen, M.; Leskelä, J.; Palviainen, T.; Sattler, W.; Sinisalo, J.; Salomaa, V.; Kaprio, J.; Pussinen, P.J. Endotoxemia Is Associated with an Adverse Metabolic Profile. Innate Immun. 2021, 27, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Sakon, H.; Nagai, F.; Morotomi, M.; Tanaka, R. Sutterella Parvirubra Sp. Nov. and Megamonas Funiformis Sp. Nov., Isolated from Human Faeces. Int. J. Syst. Evol. Microbiol. 2008, 58, 970–975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, Y.P.; Bernardi, A.; Frozza, R.L. The Role of Short-Chain Fatty Acids From Gut Microbiota in Gut-Brain Communication. Front. Endocrinol. 2020, 11, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, X.; Jiang, Z.; Yang, F.; Wang, Y.; Gao, X.; Wang, Y.; Chai, X.; Pan, G.; Zhu, Y. Sensitive and Simplified Detection of Antibiotic Influence on the Dynamic and Versatile Changes of Fecal Short-Chain Fatty Acids. PLoS ONE 2016, 11, e0167032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsinganou, E.; Gebbers, J.-O. Human Intestinal Spirochetosis--A Review. Ger. Med. Sci. GMS E-J. 2010, 8, Doc01. [Google Scholar] [CrossRef]
- Kraaz, W.; Pettersson, B.; Thunberg, U.; Engstrand, L.; Fellström, C. Brachyspira Aalborgi Infection Diagnosed by Culture and 16S Ribosomal DNA Sequencing Using Human Colonic Biopsy Specimens. J. Clin. Microbiol. 2000, 38, 3555–3560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Margawani, K.R.; Robertson, I.D.; Brooke, C.J.; Hampson, D.J.Y. Prevalence, Risk Factors and Molecular Epidemiology of Brachyspira Pilosicoli in Humans on the Island of Bali, Indonesia. J. Med. Microbiol. 2004. 53, 325–332. [CrossRef]
- Orazi, G.; Collins, A.J.; Whitaker, R.J. Prediction of Prophages and Their Host Ranges in Pathogenic and Commensal Neisseria Species. mSystems 2022, 7, e00083-22. [Google Scholar] [CrossRef]
- Zhang, L. Oral Campylobacter Species: Initiators of a Subgroup of Inflammatory Bowel Disease? World J. Gastroenterol. WJG 2015, 21, 9239–9244. [Google Scholar] [CrossRef]
- Arimatsu, K.; Yamada, H.; Miyazawa, H.; Minagawa, T.; Nakajima, M.; Ryder, M.I.; Gotoh, K.; Motooka, D.; Nakamura, S.; Iida, T.; et al. Oral Pathobiont Induces Systemic Inflammation and Metabolic Changes Associated with Alteration of Gut Microbiota. Sci. Rep. 2014, 4, 4828. [Google Scholar] [CrossRef] [Green Version]
- Bartlett, A.; Gullickson, R.G.; Singh, R.; Ro, S.; Omaye, S.T. The Link between Oral and Gut Microbiota in Inflammatory Bowel Disease and a Synopsis of Potential Salivary Biomarkers. Appl. Sci. 2020, 10, 6421. [Google Scholar] [CrossRef]
- Gao, L.; Xu, T.; Huang, G.; Jiang, S.; Gu, Y.; Chen, F. Oral Microbiomes: More and More Importance in Oral Cavity and Whole Body. Protein Cell 2018, 9, 488–500. [Google Scholar] [CrossRef] [Green Version]
- Takagi, T.; Naito, Y.; Inoue, R.; Kashiwagi, S.; Uchiyama, K.; Mizushima, K.; Tsuchiya, S.; Dohi, O.; Yoshida, N.; Kamada, K.; et al. Differences in Gut Microbiota Associated with Age, Sex, and Stool Consistency in Healthy Japanese Subjects. J. Gastroenterol. 2019, 54, 53–63. [Google Scholar] [CrossRef]
- Gao, X.; Zhang, M.; Xue, J.; Huang, J.; Zhuang, R.; Zhou, X.; Zhang, H.; Fu, Q.; Hao, Y. Body Mass Index Differences in the Gut Microbiota Are Gender Specific. Front. Microbiol. 2018, 9, 1250. [Google Scholar] [CrossRef]
- Spielman, L.J.; Gibson, D.L.; Klegeris, A. Unhealthy Gut, Unhealthy Brain: The Role of the Intestinal Microbiota in Neurodegenerative Diseases. Neurochem. Int. 2018, 120, 149–163. [Google Scholar] [CrossRef]
- Feng, T.; Hilal, M.G.; Wang, Y.; Zhou, R.; Yu, Q.; Qu, J.; Li, H. Differences in Gut Microbiome Composition and Antibiotic Resistance Gene Distribution between Chinese and Pakistani University Students from a Common Peer Group. Microorganisms 2021, 9, 1152. [Google Scholar] [CrossRef]
- Jeraldo, P.; Sipos, M.; Chia, N.; Brulc, J.M.; Dhillon, A.S.; Konkel, M.E.; Larson, C.L.; Nelson, K.E.; Qu, A.; Schook, L.B.; et al. Quantification of the Relative Roles of Niche and Neutral Processes in Structuring Gastrointestinal Microbiomes. Proc. Natl. Acad. Sci. USA 2012, 109, 9692–9698. [Google Scholar] [CrossRef] [Green Version]
- Shafquat, A.; Joice, R.; Simmons, S.L.; Huttenhower, C. Functional and Phylogenetic Assembly of Microbial Communities in the Human Microbiome. Trends Microbiol. 2014, 22, 261–266. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Batool, M.; Keating, C.; Javed, S.; Nasir, A.; Muddassar, M.; Ijaz, U.Z. A Cross-Sectional Study of Potential Antimicrobial Resistance and Ecology in Gastrointestinal and Oral Microbial Communities of Young Normoweight Pakistani Individuals. Microorganisms 2023, 11, 279. https://doi.org/10.3390/microorganisms11020279
Batool M, Keating C, Javed S, Nasir A, Muddassar M, Ijaz UZ. A Cross-Sectional Study of Potential Antimicrobial Resistance and Ecology in Gastrointestinal and Oral Microbial Communities of Young Normoweight Pakistani Individuals. Microorganisms. 2023; 11(2):279. https://doi.org/10.3390/microorganisms11020279
Chicago/Turabian StyleBatool, Maria, Ciara Keating, Sundus Javed, Arshan Nasir, Muhammad Muddassar, and Umer Zeeshan Ijaz. 2023. "A Cross-Sectional Study of Potential Antimicrobial Resistance and Ecology in Gastrointestinal and Oral Microbial Communities of Young Normoweight Pakistani Individuals" Microorganisms 11, no. 2: 279. https://doi.org/10.3390/microorganisms11020279