
Genomic Analysis and In Vitro Investigation of the Hop Resistance Phenotype of Two Novel Loigolactobacillus backii Strains, Isolated from Spoiled Beer
 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Strains Isolation and Growth Conditions
2.2. DNA Extraction and Molecular Identification
2.3. Whole Genome Sequencing Analysis and De Novo Assembly
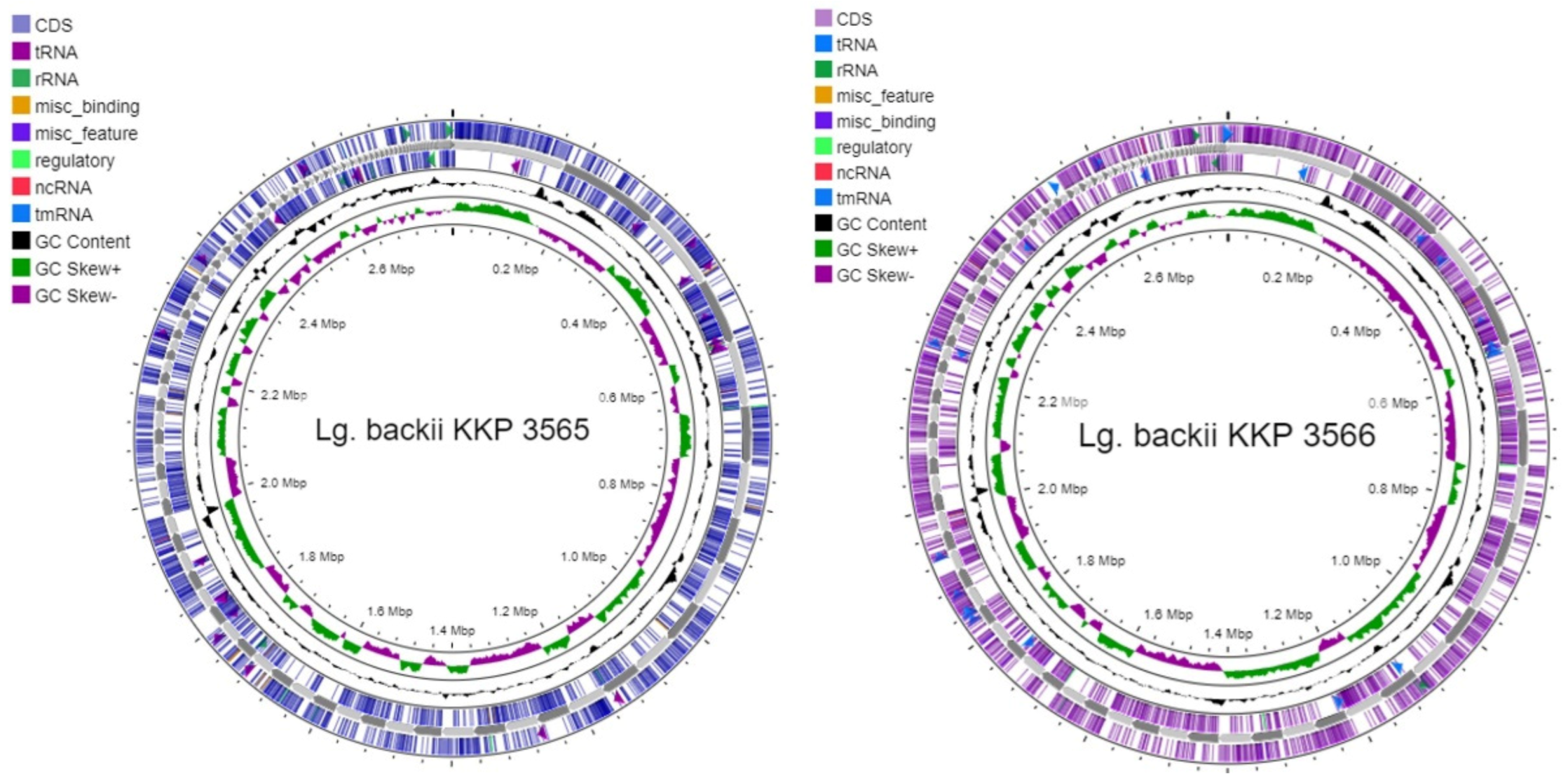
2.3.1. Genome Annotation
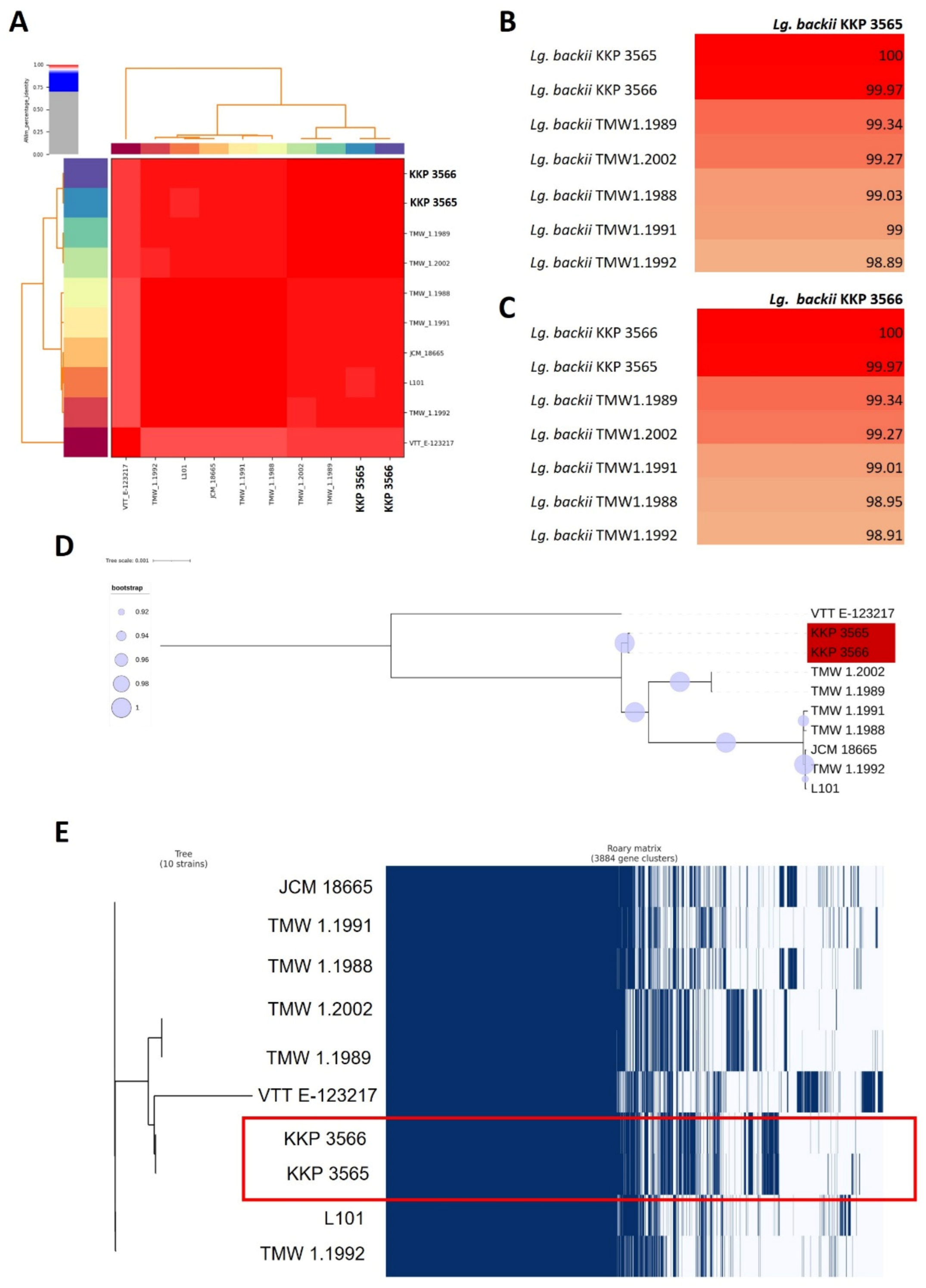
2.3.2. Comparative Genomics
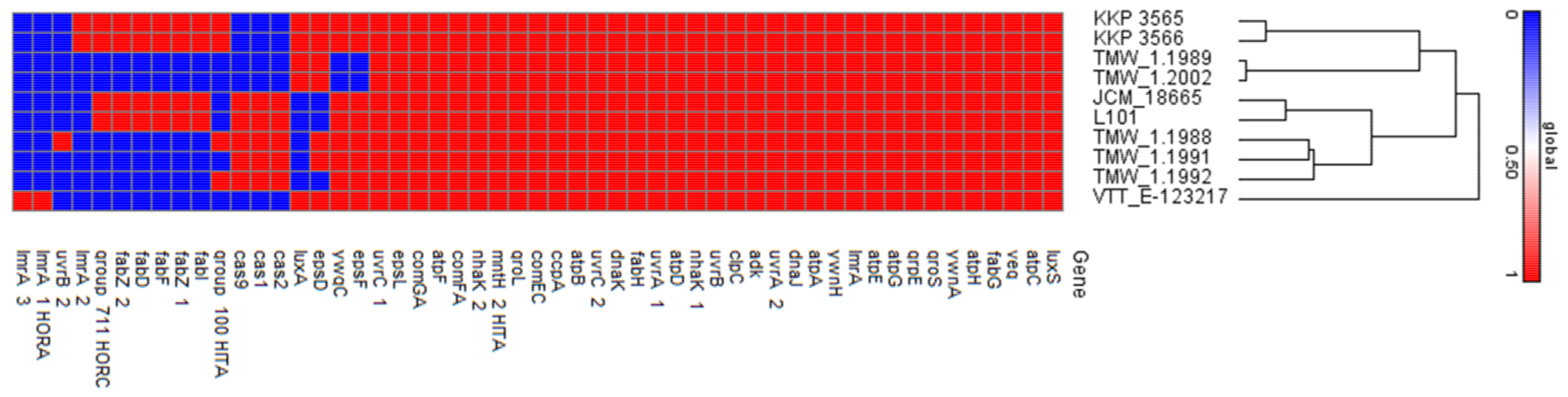
2.3.3. In Silico Investigation of Properties Related to Beer-Spoiling Capacity
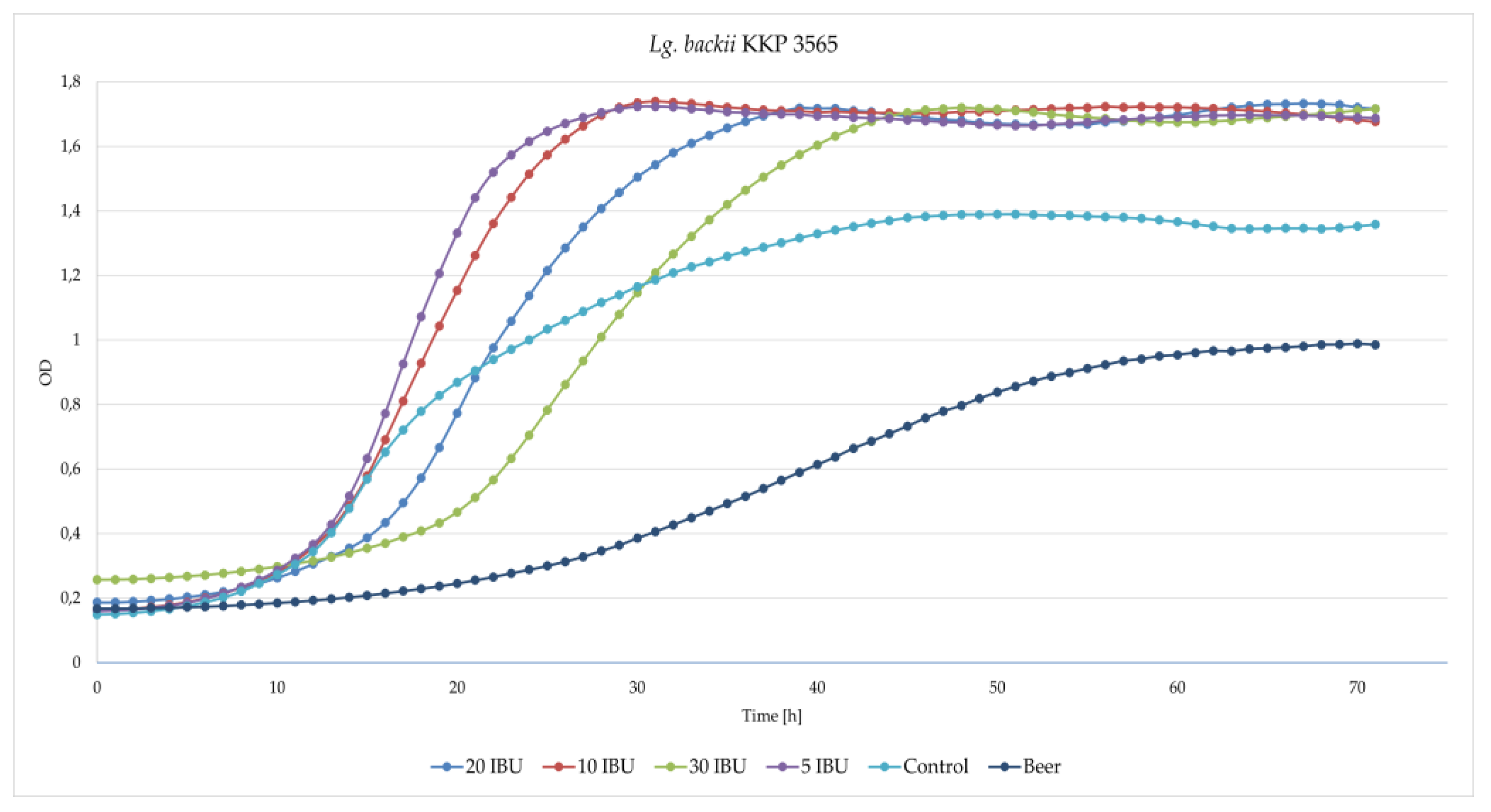
2.4. Determination of Lg. backii Strains Growth Inhibition in 5, 10, 20 and 30 IBU Hop Concentrations
2.5. Statistical Analysis
3. Results and Discussion
3.1. Whole Genome Annotation and Gene Clustering
3.2. Phylogenomic and Pangenome Analysis
3.3. Comparative Genomic Analysis of Genes Related to Beer Spoilage and Adaptation to the Brewery Microenvironment
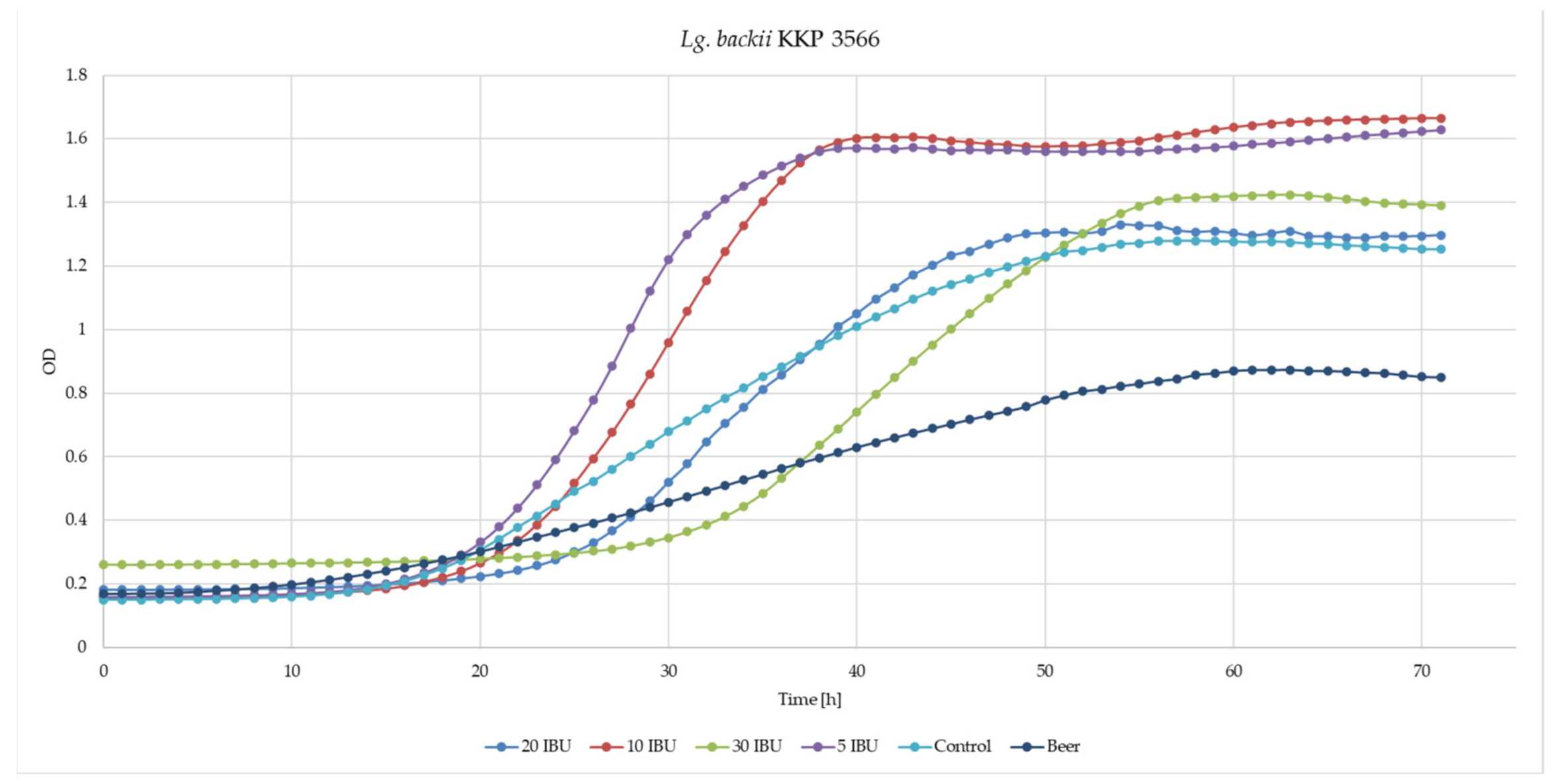
3.4. Beer-Spoilage Ability of Lg. backii KKP3565 and KKP3566 Strains
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schönberger, C.; Kostelecky, T. 125th Anniversary Review: The Role of Hops in Brewing. J. Inst. Brew. 2011, 117, 259–267. [Google Scholar] [CrossRef]
- Sakamoto, K.; Konings, W.N. Beer spoilage bacteria and hop resistance. Int. J. Food Microbiol. 2003, 89, 105–124. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Iijima, K.; Sakamoto, K.; Sami, M.; Yamashita, H. A Review of Hop Resistance in Beer Spoilage Lactic Acid Bacteria. J. Inst. Brew. 2006, 112, 173–191. [Google Scholar] [CrossRef]
- Wieme, A.D.; Spitaels, F.; Aerts, M.; De Bruyne, K.; Van Landschoot, A.; Vandamme, P. Identification of beer-spoilage bacteria using matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Int. J. Food Microbiol. 2014, 185, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K. Emergence of New Spoilage Microorganisms in the Brewing Industry and Development of Microbiological Quality Control Methods to Cope with This Phenomenon: A Review. J. Am. Soc. Brew. Chem. 2020, 78, 245–259. [Google Scholar] [CrossRef]
- Umegatani, M.; Takesue, N.; Asano, S.; Tadami, H.; Uemura, K. Study of Beer Spoilage Lactobacillus nagelii Harboring Hop Resistance Gene horA. J. Am. Soc. Brew. Chem. 2021, 80, 92–98. [Google Scholar] [CrossRef]
- Suzuki, K.; Koyanagi, M.; Yamashita, H. Genetic characterization of non-spoilage variant isolated from beer-spoilage Lactobacillus brevis ABBC45C. J. Appl. Microbiol. 2004, 96, 946–953. [Google Scholar] [CrossRef]
- Suzuki, K.; Shinohara, Y.; Kurniawan, Y.N. Role of Plasmids in Beer Spoilage Lactic Acid Bacteria: A Review. J. Am. Soc. Brew. Chem. 2020, 79, 1–16. [Google Scholar] [CrossRef]
- Bucka-Kolendo, J.; Juszczuk-Kubiak, E.; Sokołowska, B. Effect of High Hydrostatic Pressure on Stress-Related dnaK, hrcA, and ctsR Expression Patterns in Selected Lactobacilli Strains. Genes 2021, 12, 12. [Google Scholar] [CrossRef]
- Bucka-Kolendo, J.; Sokołowska, B. Lactic acid bacteria stress response to preservation processes in the beverage and juice industry. Acta Biochim. Pol. 2017, 64, 459–464. [Google Scholar] [CrossRef]
- Bucka-Kolendo, J.; Sokołowska, B.; Winiarczyk, S. Influence of High Hydrostatic Pressure on the Identification of Lactobacillus by MALDI-TOF MS- Preliminary Study. Microorganisms 2020, 8, 813. [Google Scholar] [CrossRef] [PubMed]
- Babraham Bioinformatics—FastQC A Quality Control Tool for High Throughput Sequence Data. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 19 March 2022).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. Original Articles SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boetzer, M.; Henkel, C.V.; Jansen, H.J.; Butler, D.; Pirovano, W. Scaffolding pre-assembled contigs using SSPACE. Bioinform. Appl. Note 2011, 27, 578–579. [Google Scholar] [CrossRef] [Green Version]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef] [Green Version]
- Seemann, T. Genome Analysis Prokka: Rapid Prokaryotic Genome Annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [Green Version]
- Tatusova, T.; DiCuccio, M.; Badretdin, A.; Chetvernin, V.; Nawrocki, E.P.; Zaslavsky, L.; Lomsadze, A.; Pruitt, K.D.; Borodovsky, M.; Ostell, J. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 2016, 44, 6614–6624. [Google Scholar] [CrossRef]
- Carattoli, A.; Zankari, E.; Garcìa-Fernandez, A.; Larsen, M.; Lund, O.; Voldby Villa, L.; Møller Aarestrup, F.; Hasman, H. In Silico Detection and Typing of Plasmids. Antimicrob using PlasmidFinder and plasmid multilocus sequence typing. Antimicrob. Agents Chemother. 2014, 58, 3895–3903. [Google Scholar] [CrossRef] [Green Version]
- Johansson, M.H.K.; Bortolaia, V.; Tansirichaiya, S.; Aarestrup, F.M.; Roberts, A.P.; Petersen, T.N. Detection of mobile genetic elements associated with antibiotic resistance in Salmonella enterica using a newly developed web tool: MobileElementFinder. J. Antimicrob. Chemother. 2020, 76, 101–109. [Google Scholar] [CrossRef]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef]
- Siguier, P.; Perochon, J.; Lestrade, L.; Mahillon, J.; Chandler, M. ISfinder: The reference centre for bacterial insertion sequences. Nucleic Acids Res. 2006, 34, D32–D36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biswas, A.; Staals, R.H.; Morales, S.E.; Fineran, P.C.; Brown, C.M. CRISPRDetect: A flexible algorithm to define CRISPR arrays. BMC Genom. 2016, 17, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.C. PILER-CR: Fast and accurate identification of CRISPR repeats. BMC Bioinform. 2007, 8, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Zankari, E.; Hasman, H.; Cosentino, S.; Vestergaard, M.; Rasmussen, S.; Lund, O.; Aarestrup, F.M.; Larsen, M.V. Identification of acquired antimicrobial resistance genes. J. Antimicrob. Chemother. 2012, 67, 2640–2644. [Google Scholar] [CrossRef] [PubMed]
- Bortolaia, V.; Kaas, R.S.; Ruppe, E.; Roberts, M.C.; Schwarz, S.; Cattoir, V.; Philippon, A.; Allesoe, R.L.; Rebelo, A.R.; Florensa, A.F.; et al. ResFinder 4.0 for predictions of phenotypes from genotypes. J. Antimicrob. Chemother. 2020, 75, 3491–3500. [Google Scholar] [CrossRef] [PubMed]
- Cosentino, S.; Voldby Larsen, M.; Møller Aarestrup, F.; Lund, O. PathogenFinder—Distinguishing Friend from Foe Using Bacterial Whole Genome Sequence Data. PLoS ONE 2013, 8, e77302. [Google Scholar] [CrossRef]
- Huerta-Cepas, J.; Szklarczyk, D.; Heller, D.; Hernández-Plaza, A.; Forslund, S.K.; Cook, H.V.; Mende, D.R.; Letunic, I.; Rattei, T.; Jensen, L.J.; et al. eggNOG 5.0: A hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 2019, 47, D309–D314. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016, 44, D457–D462. [Google Scholar] [CrossRef] [Green Version]
- Grant, J.R.; Stothard, P. The CGView Server: A comparative genomics tool for circular genomes. Nucleic Acids Res. 2008, 36, W181–W184. [Google Scholar] [CrossRef]
- Pritchard, L.; Glover, R.H.; Humphris, S.; Elphinstone, J.G.; Toth, I.K. Genomics and taxonomy in diagnostics for food security: Soft-rotting enterobacterial plant pathogens. Anal. Methods 2015, 8, 12–24. [Google Scholar] [CrossRef]
- Page, A.J.; Cummins, C.A.; Hunt, M.; Wong, V.K.; Reuter, S.; Holden, M.T.G.; Fookes, M.; Falush, D.; Keane, J.A.; Parkhill, J. Roary: Rapid large-scale prokaryote pan genome analysis. Bioinformatics 2015, 31, 3691–3693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately Maximum-Likelihood Trees for Large Alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef] [PubMed]
- Darling, A.E.; Mau, B.; Perna, N.T. progressiveMauve: Multiple Genome Alignment with Gene Gain, Loss and Rearrangement. PLoS ONE 2010, 5, e11147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Letunic, I.; Bork, P. Interactive tree of life (iTOL) v3: An online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 2016, 44, W242–W245. [Google Scholar] [CrossRef] [PubMed]
- Van Heel, A.J.; De Jong, A.; Song, C.; Viel, J.H.; Kok, J.; Kuipers, O.P. BAGEL4: A user-friendly web server to thoroughly mine RiPPs and bacteriocins. Nucleic Acids Res. 2018, 46, W278–W281. [Google Scholar] [CrossRef]
- Kajala, I.; Bergsveinson, J.; Friesen, V.; Redekop, A.; Juvonen, R.; Storgårds, E.; Ziola, B. Lactobacillus backii and Pediococcus damnosus isolated from 170-year-old beer recovered from a shipwreck lack the metabolic activities required to grow in modern lager beer. FEMS Microbiol. Ecol. 2018, 94, fix152. [Google Scholar] [CrossRef]
- Xu, Z.; Luo, Y.; Mao, Y.; Peng, R.; Chen, J.; Soteyome, T.; Bai, C.; Chen, L.; Liang, Y.; Su, J.; et al. Spoilage Lactic Acid Bacteria in the Brewing Industry. J. Microbiol. Biotechnol. 2020, 30, 955–961. [Google Scholar] [CrossRef]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef] [Green Version]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.A.; Clamp, M.; Barton, G.J. Jalview Version 2—A multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef] [Green Version]
- Gientka, I.; Bugajewska, A.; Chlebowska-Śmigiel, A.; Kieliszek, M.; Misiura, S. Ocena Zdolności Przeciwdrobnoustrojowych i Bakteriocynogennych Lactobacillus rhamnosus ATCC 7469. Zesz. Probl. Postępów Nauk Rol. 2016, 585, 65–73. [Google Scholar]
- Duar, R.M.; Lin, X.B.; Zheng, J.; Martino, M.E.; Grenier, T.; Pérez-Muñoz, M.E.; Leulier, F.; Gänzle, M.; Walter, J. Lifestyles in transition: Evolution and natural history of the genus Lactobacillus. FEMS Microbiol. Rev. 2017, 41, S27–S48. [Google Scholar] [CrossRef] [PubMed]
- Kant, R.; Blom, J.; Palva, A.; Siezen, R.J.; de Vos, W.M. Comparative genomics of Lactobacillus. Microb. Biotechnol. 2011, 4, 323–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stefanovic, E.; Fitzgerald, G.; McAuliffe, O. Advances in the genomics and metabolomics of dairy lactobacilli: A review. Food Microbiol. 2017, 61, 33–49. [Google Scholar] [CrossRef] [PubMed]
- Hampton, H.G.; Watson, B.N.J.; Fineran, P.C. The arms race between bacteria and their phage foes. Nature 2020, 577, 327–336. [Google Scholar] [CrossRef]
- Ciufo, S.; Kannan, S.; Sharma, S.; Badretdin, A.; Clark, K.; Turner, S.; Brover, S.; Schoch, C.L.; Kimchi, A.; DiCuccio, M. Using average nucleotide identity to improve taxonomic assignments in prokaryotic genomes at the NCBI. Int. J. Syst. Evol. Microbiol. 2018, 68, 2386–2392. [Google Scholar] [CrossRef]
- Vandecraen, J.; Chandler, M.; Aertsen, A.; Van Houdt, R. The impact of insertion sequences on bacterial genome plasticity and adaptability. Crit. Rev. Microbiol. 2017, 43, 709–730. [Google Scholar] [CrossRef] [Green Version]
- Kaleta, P.; O’Callaghan, J.; Fitzgerald, G.F.; Beresford, T.P.; Ross, R.P. Crucial Role for Insertion Sequence Elements in Lactobacillus helveticus Evolution as Revealed by Interstrain Genomic Comparison. Appl. Environ. Microbiol. 2010, 76, 212–220. [Google Scholar] [CrossRef] [Green Version]
- Barbieri, F.; Montanari, C.; Gardini, F.; Tabanelli, G. Biogenic Amine Production by Lactic Acid Bacteria: A Review. Foods 2019, 8, 17. [Google Scholar] [CrossRef] [Green Version]
- Dimopoulou, M.; Dols-Lafargue, M. Exopolysaccharides Producing Lactic Acid Bacteria in Wine and Other Fermented Beverages: For Better or for Worse? Foods 2021, 10, 2204. [Google Scholar] [CrossRef]
- Wirtanen, G.; Storgårds, E.; Saarela, M.; Salo, S. Detection of Biofilms in the Food and Beverage Industry MIRRI-Microbial Resource Research Infrastructure (Preparatory Phase) View Project BIODAM View Project; John Wiley & Sons Ltd: Hoboken, NJ, USA, 2000. [Google Scholar]
- Kiousi, D.E.; Efstathiou, C.; Tegopoulos, K.; Mantzourani, I.; Alexopoulos, A.; Plessas, S.; Kolovos, P.; Koffa, M.; Galanis, A. Genomic Insight Into Lacticaseibacillus paracasei SP5, Reveals Genes and Gene Clusters of Probiotic Interest and Biotechnological Potential. Front. Microbiol. 2022, 13, 922689. [Google Scholar] [CrossRef]
- Pöntinen, A.; Aalto-Araneda, M.; Lindström, M.; Korkeala, H. Heat Resistance Mediated by pLM58 Plasmid-Borne ClpL in Listeria monocytogenes. mSphere 2017, 2, e00364-17. [Google Scholar] [CrossRef] [PubMed]
- Tran, T.D.-H.; Kwon, H.-Y.; Kim, E.-H.; Kim, K.-W.; Briles, D.E.; Pyo, S.; Rhee, D.-K. Decrease in Penicillin Susceptibility Due to Heat Shock Protein ClpL in Streptococcus pneumoniae. Antimicrob. Agents Chemother. 2011, 55, 2714–2728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Behr, J.; Geissler, A.J.; Schmid, J.; Zehe, A.; Vogel, R.F. The Identification of Novel Diagnostic Marker Genes for the Detection of Beer Spoiling Pediococcus damnosus Strains Using the BlAst Diagnostic Gene findEr. PLoS ONE 2016, 11, e0152747. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Asano, S.; Iijima, K.; Kitamoto, K. Sake and Beer Spoilage Lactic Acid Bacteria—A Review. J. Inst. Brew. 2008, 114, 209–223. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 5 IBU | 10 IBU | 20 IBU | 30 IBU | Beer 43.6 IBU | Control | |
|---|---|---|---|---|---|---|
| MRS broth concentrate (2×) | 50% | 50% | 50% | - | - | 50% |
| MRS broth concentrate (4×) | - | - | - | 25% | - | - |
| Water | 37,5% | 25% | - | - | 50% | |
| Beer (40 IBU) | 12,5% | 25% | 50% | 75% | - | - |
| Beer (43.6 IBU) | - | - | - | - | 100% | - |
| Element | Lg. backii KKP 3565 | Lg. backii KKP 3566 |
|---|---|---|
| Length | 2,796,657 bp | 2,798,617 bp |
| GC content (%) | 40.68 | 40.68 |
| Genes (total) | 2640 | 2633 |
| CDSs | 2593 | 2582 |
| tRNAs | 39 | 42 |
| ncRNAs | 4 | 4 |
| Pseudogenes | 47 | 50 |
| Cas arrays | 0 | 0 |
| Insertion Elements | 124 | 175 |
| Mobile Elements | 19 | 20 |
| Prophages | ||
| Intact | 2 | 2 |
| Incomplete | 1 | 0 |
| Questionable | 0 | 0 |
| Bacteriocin Production | No | No |
| Bacteriocin Immunity | Yes | Yes |
| Pathogenicity (%) | 0.106 | 0.099 |
| Strain | Plasmid AN | PlasmidFinder Annotation | Origin | Identity |
|---|---|---|---|---|
| Lg. backii KKP 3565 | NZ_CP014896.1 | rep28_2_LBPp6g007 (LBPp6) | Lg. backi TMW 1.1992 | 0.99928 |
| NZ_CP031183.1 | rep38_1_rep (pLBUC03) | Lv. brevis UCCLB95 | 0.99447 | |
| NZ_CP014889.1 | rep38_1_rep (pLBUC03) | Lg. backii TMW 1.1991 | 0.99145 | |
| Lg. backii KKP 3566 | NZ_CP014896.1 | rep28_2_LBPp6g007 (LBPp6) | Lg. backii TMW 1.1992 | 0.99918 |
| NZ_CP031183.1 | rep38_1_rep (pLBUC03), | Lv. brevis UCCLB95 | 0.99345 | |
| NZ_CP014889.1 | rep38_1_rep (pLBUC03) | Lg. backii TMW 1.1991 | 0.99048 |
| COG | Lg. backii KKP 3565 | Lg. backii KKP 3566 |
|---|---|---|
| C—Energy production and conversion | 95 (4.3%) | 95 (4.26%) |
| D—Cell cycle control and mitosis | 46 (2.07%) | 47 (2.11%) |
| E—Amino acid metabolism and transport | 203 (9.13%) | 204 (9.14%) |
| F—Nucleotide metabolism and transport | 102 (4.58%) | 107 (4.8%) |
| G—Carbohydrate metabolism and transport | 128 (5.75%) | 130 (5.82%) |
| H—Coenzyme metabolism | 89 (4%) | 90 (4.03%) |
| I—Lipid metabolism | 64 (2.88%) | 64 (2.87%) |
| J—Translation | 173 (7.78%) | 173 (7.75%) |
| K—Transcription | 176 (7.92%) | 177 (7.93%) |
| L—Replication and repair | 195 (8.77%) | 195 (8.74%) |
| M—Cell wall/membrane/envelop biogenesis | 136 (6.12%) | 133 (5.96%) |
| N—Cell motility | 9 (0.4%) | 9 (0.4%) |
| O—Post-translational modification, protein turnover, chaperone functions | 49 (2.2%) | 49 (2.2%) |
| P—Inorganic ion transport and metabolism | 129 (5.8%) | 131 (5.87%) |
| Q—Secondary structure | 24 (1.08%) | 24 (1.08%) |
| T—Signal transduction | 47 (2.11%) | 47 (2.11%) |
| U—Intracellular trafficking and secretion | 52 (2.34%) | 52 (2.34%) |
| V—Defense mechanisms | 34 (1.53%) | 34 (1.52%) |
| S—Function unknown | 472 (21.23%) | 471 (21.1%) |
| Total | 2223 (100%) | 2232 (100%) |
| Gene | Function | Locus Tag (Lg. backii) | Reference (AN) | Identity (%) | E-Value |
|---|---|---|---|---|---|
| hitA | Nramp family divalent metal transporter | KKP3565_001038 | J7LK56_LEVBR | 83 | 0.0 |
| hitA | Nramp family divalent metal transporter | KKP3566_001304 | J7LK56_LEVBR | 83 | 0.0 |
| horA | ABC transporter ATP binding protein/permease | KKP3565_002144 | O32748_LEVBR | 97 | 0.0 |
| horA | ABC transporter ATP binding protein/permease | KKP3566_002171 | O32748_LEVBR | 97 | 0.0 |
| horC | ABC transporter permease | KKP3565_001952 | Q6I7K2_LEVBR | 95 | 0.0 |
| horC | ABC transporter permease | KKP3566_001675 | Q6I7K2_LEVBR | 95 | 0.0 |
| µ | Control | 5 IBU | 10 IBU | 20 IBU | 30 IBU | Beer 43.6 IBU |
|---|---|---|---|---|---|---|
| KKP 3565 | 0.05013 ± 0.0002 cB | 0.0829 ± 0.0003 eB | 0.0825 ± 0.0005 eB | 0.0607 ± 0.0002 dB | 0.0437 ± 0.0003 bB | 0.0304 ± 0.0005 aB |
| KKP 3566 | 0.0475 ± 0.0018 dA | 0.0629 ± 0.0008 eA | 0.0429 ± 0.0010 cA | 0.0420 ± 0.0024 cA | 0.0318 ± 0.0006 bA | 0.0280 ± 0.0005 aA |
| OD max | Control | 5 IBU | 10 IBU | 20 IBU | 30 IBU | Beer 43.6 IBU |
|---|---|---|---|---|---|---|
| KKP 3565 | 1.3833 ± 0.0223 bB | 1.7240 ± 0.0210 cB | 1.7398 ± 0.0021 cB | 1.7190 ± 0.0055 cB | 1.7125 ± 0.0839 cB | 0.9855 ± 0.0217 aB |
| KKP 3566 | 1.2438 ± 0.0945 bA | 1.5725 ± 0.0414 cdA | 1.6420 ± 0.0810 dA | 1.3298 ± 0.1482 bA | 1.4190 ± 0.0508 bcA | 0.8495 ± 0.0133 aA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kiousi, D.E.; Bucka-Kolendo, J.; Wojtczak, A.; Sokołowska, B.; Doulgeraki, A.I.; Galanis, A. Genomic Analysis and In Vitro Investigation of the Hop Resistance Phenotype of Two Novel Loigolactobacillus backii Strains, Isolated from Spoiled Beer. Microorganisms 2023, 11, 280. https://doi.org/10.3390/microorganisms11020280
Kiousi DE, Bucka-Kolendo J, Wojtczak A, Sokołowska B, Doulgeraki AI, Galanis A. Genomic Analysis and In Vitro Investigation of the Hop Resistance Phenotype of Two Novel Loigolactobacillus backii Strains, Isolated from Spoiled Beer. Microorganisms. 2023; 11(2):280. https://doi.org/10.3390/microorganisms11020280
Chicago/Turabian StyleKiousi, Despoina Eugenia, Joanna Bucka-Kolendo, Adrian Wojtczak, Barbara Sokołowska, Agapi I. Doulgeraki, and Alex Galanis. 2023. "Genomic Analysis and In Vitro Investigation of the Hop Resistance Phenotype of Two Novel Loigolactobacillus backii Strains, Isolated from Spoiled Beer" Microorganisms 11, no. 2: 280. https://doi.org/10.3390/microorganisms11020280