

Comparative Genomic Analyses of Virulence and Antimicrobial Resistance in Citrobacter werkmanii, an Emerging Opportunistic Pathogen
 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Strain Isolation and Growth Conditions
2.2. Genome Sequencing and Annotation
2.3. Phylogenetic, Comparative Genomics, and Pangenome Analyses
2.4. Statistical Analyses
3. Results
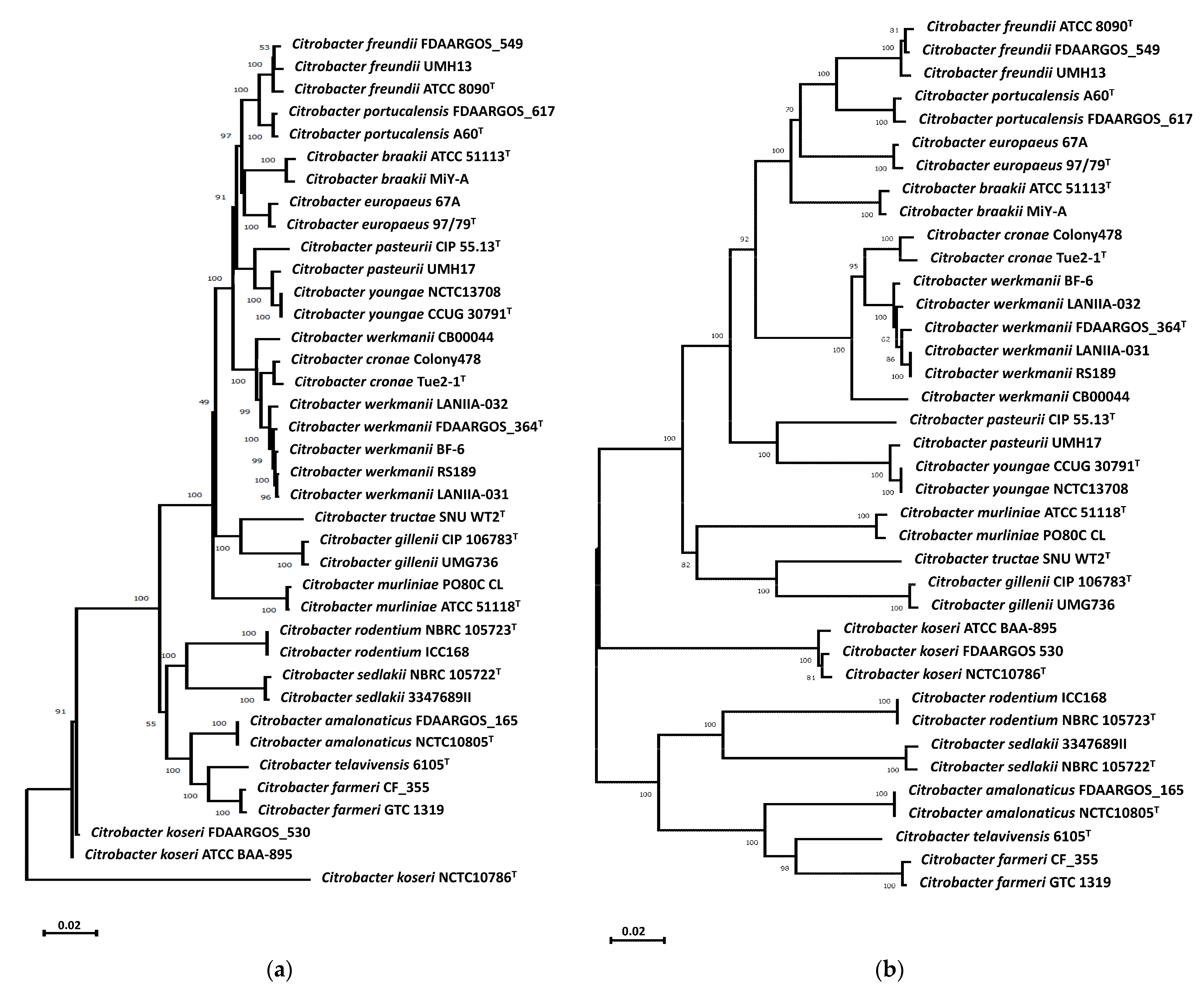
3.1. Characterization of Citrobacter Strains from River Water
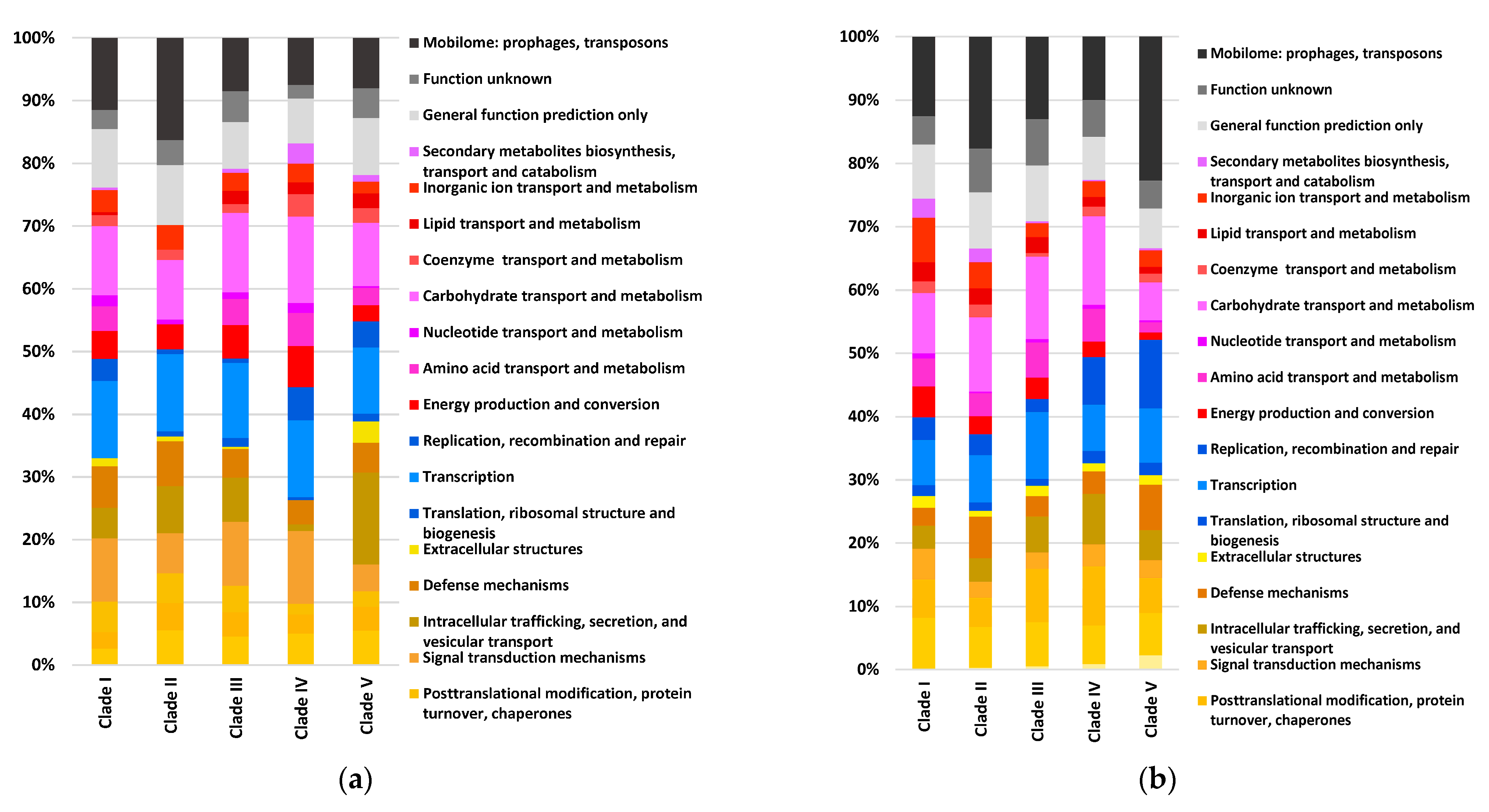
3.2. Pangenome Analyses of C. werkmanii Strains from Diverse Sources and Locations
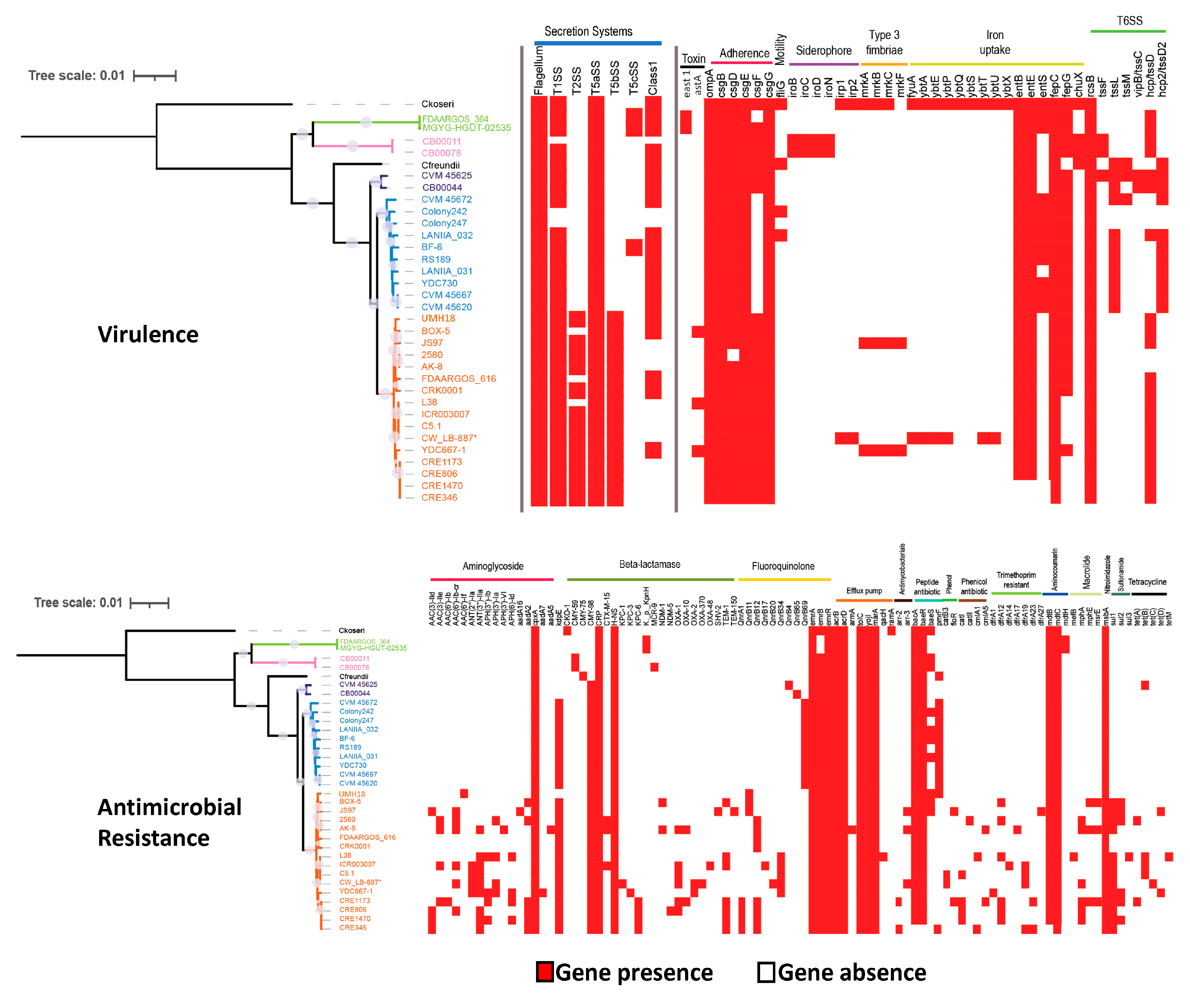
3.3. Virulence and Antimicrobial Resistance Gene Identification
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rogers, L.; Power, K.; Gaora, P.Ó.; Fanning, S. Escherichia coli and other Enterobacteriaceae: Occurrence and detection. In Encyclopedia of Food and Health; Caballero, B., Finglas, P.M., Toldrá, F., Eds.; Academic Press: Oxford, UK, 2016; pp. 545–551. [Google Scholar]
- Kus, J.V. Infections due to Citrobacter and Enterobacter. In Reference Module in Biomedical Sciences; Elsevier: Amsterdam, The Netherlands, 2014. [Google Scholar]
- Janda, J.M.; Abbott Sharon, L. The changing face of the family Enterobacteriaceae (Order: “Enterobacterales”): New members, taxonomic issues, geographic expansion, and new diseases and disease syndromes. Clin. Microbiol. Rev. 2021, 34, e00174-20. [Google Scholar] [CrossRef] [PubMed]
- Jung, W.J.; Kim, H.J.; Giri, S.S.; Kim, S.G.; Kim, S.W.; Kang, J.W.; Kwon, J.; Lee, S.B.; Oh, W.T.; Jun, J.W.; et al. Citrobacter tructae sp. nov. isolated from kidney of diseased rainbow trout (Oncorhynchus mykiss). Microorganisms 2021, 9, 275. [Google Scholar] [CrossRef] [PubMed]
- Oberhettinger, P.; Schüle, L.; Marschal, M.; Bezdan, D.; Ossowski, S.; Dörfel, D.; Vogel, W.; Rossen, J.W.; Willmann, M.; Peter, S. Description of Citrobacter cronae sp. nov., isolated from human rectal swabs and stool samples. Int. J. Syst. Evol. Microbiol. 2020, 70, 2998–3003. [Google Scholar] [CrossRef]
- Ribeiro, T.G.; Izdebski, R.; Urbanowicz, P.; Carmeli, Y.; Gniadkowski, M.; Peixe, L. Citrobacter telavivum sp. nov. with chromosomal mcr-9 from hospitalized patients. Eur. J. Clin. Microbiol. Infect Dis. 2021, 40, 123–131. [Google Scholar] [CrossRef]
- Ong, C.C.H.; Farhanah, S.; Linn, K.Z.; Tang, Y.W.; Poon, C.Y.; Lim, A.Y.; Tan, H.R.; Binte Hamed, N.H.; Huan, X.; Puah, S.H.; et al. Nosocomial infections among COVID-19 patients: An analysis of intensive care unit surveillance data. Antimicrob. Resist. Infect Control 2021, 10, 119. [Google Scholar] [CrossRef]
- Pletz, M.W.; Wollny, A.; Dobermann, U.-H.; Rödel, J.; Neubauer, S.; Stein, C.; Brandt, C.; Hartung, A.; Mellmann, A.; Trommer, S.; et al. A nosocomial foodborne outbreak of a VIM carbapenemase-expressing Citrobacter freundii. Clin. Infect Dis. 2018, 67, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Ranjan, K.P.; Ranjan, N. Citrobacter: An emerging health care associated urinary pathogen. Urol. Ann. 2013, 5, 313. [Google Scholar] [CrossRef]
- Tchidjou, H.K.; Romeo, B. Infant case of co-infection with SARS-CoV-2 and Citrobacter koseri urinary infection. J. Trop. Pediatr. 2021, 67, fmaa032. [Google Scholar] [CrossRef]
- Anderson, M.T.; Mitchell, L.A.; Zhao, L.; Mobley, H.L.T. Citrobacter freundii fitness during bloodstream infection. Sci. Rep. 2018, 8, 11792. [Google Scholar] [CrossRef] [PubMed]
- Ariza-Prota, M.A.; Pando-Sandoval, A.; García-Clemente, M.; Fernández, R.; Casan, P. Community-acquired pneumonia and empyema caused by Citrobacter koseri in an immunocompetent patient. Case Rep. Pulmonol. 2015, 2015, 670373. [Google Scholar] [CrossRef]
- Pepperell, C.; Kus, J.V.; Gardam, M.A.; Humar, A.; Burrows, L.L. Low-virulence Citrobacter species encode resistance to multiple antimicrobials. Antimicrob. Agents Chemother. 2002, 46, 3555–3560. [Google Scholar] [CrossRef] [PubMed]
- Gardam, M.A.; Burrows, L.L.; Kus, J.V.; Brunton, J.; Low, D.E.; Conly, J.M.; Humar, A. Is surveillance for multidrug-resistant Enterobacteriaceae an effective infection control strategy in the absence of an outbreak? J. Infect. Dis. 2002, 186, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Hammerum, A.M.; Hansen, F.; Nielsen, H.L.; Jakobsen, L.; Stegger, M.; Andersen, P.S.; Jensen, P.; Nielsen, T.K.; Hansen, L.H.; Hasman, H.; et al. Use of WGS data for investigation of a long-term NDM-1-producing Citrobacter freundii outbreak and secondary in vivo spread of blaNDM-1 to Escherichia coli, Klebsiella pneumoniae and Klebsiella oxytoca. J. Antimicrob. Chemother. 2016, 71, 3117–3124. [Google Scholar] [CrossRef]
- Jones, M.E.; Karlowsky, J.A.; Draghi, D.C.; Thornsberry, C.; Sahm, D.F.; Nathwani, D. Epidemiology and antibiotic susceptibility of bacteria causing skin and soft tissue infections in the USA and Europe: A guide to appropriate antimicrobial therapy. Int. J. Antimicrob. Agents 2003, 22, 406–419. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.; Choi, S.M.; Jo, S.J.; Lee, J.; Cho, S.Y.; Kim, S.H.; Lee, D.G.; Jeong, H.S. Clinical characteristics and antimicrobial susceptibility trends in Citrobacter bacteremia: An 11-year single-center experience. Infect. Chemother. 2019, 51, 1–9. [Google Scholar] [CrossRef]
- Mohanty, S.; Singhal, R.; Sood, S.; Dhawan, B.; Kapil, A.; Das, B.K. Citrobacter infections in a tertiary care hospital in Northern India. J. Infect. 2007, 54, 58–64. [Google Scholar] [CrossRef]
- Nayar, R.; Shukla, I.; Sultan, A. Epidemiology, prevalence and identification of Citrobacter species in clinical specimens in a tertiary care hospital in India. Int. J. Sci. Res. Publ. 2014, 4, 1–6. [Google Scholar]
- Parvez, S.; Khan, A.U.; Kaur, G.; Barakat, M.; Ortet, P.; Mayilraj, S. An insight into the genome of extensively drug-resistant and uropathogenic Citrobacter werkmanii. J. Glob. Antimicrob. Resist. 2020, 22, 785–791. [Google Scholar] [CrossRef]
- González-López, I.; Medrano-Félix, J.A.; Castro-Del Campo, N.; López-Cuevas, O.; González-Gómez, J.P.; Valdez-Torres, J.B.; Aguirre-Sánchez, J.R.; Martínez-Urtaza, J.; Gómez-Gil, B.; Lee, B.G.; et al. Prevalence and genomic diversity of Salmonella enterica recovered from river water in a major sgricultural region in Northwestern Mexico. Microorganisms 2022, 10, 1214. [Google Scholar] [CrossRef] [PubMed]
- Aguirre-Sánchez, J.R.; Valdez-Torres, J.B.; del Campo, N.C.; Martínez-Urtaza, J.; del Campo, N.C.; Lee, B.G.; Quiñones, B.; Chaidez-Quiroz, C. Phylogenetic group and virulence profile classification in Escherichia coli from distinct isolation sources in Mexico. Infect. Genet. Evol. 2022, 106, 105380. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 1 May 2021).
- Krueger, F.; Trim Galore. A Wrapper Tool around Cutadapt FastQC to Consistently Apply QUAL Adapt Trimming to FastQ Files. Available online: https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/ (accessed on 1 May 2021).
- Bushnell, B. BBTools Software Package. Available online: Sourceforge.net/projects/bbmap/ (accessed on 1 May 2021).
- Coil, D.; Jospin, G.; Darling, A.E. A5-miseq: An updated pipeline to assemble microbial genomes from Illumina MiSeq data. Bioinformatics 2015, 31, 587–589. [Google Scholar] [CrossRef]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Brettin, T.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Olsen, G.J.; Olson, R.; Overbeek, R.; Parrello, B.; Pusch, G.D.; et al. RASTtk: A modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes. Sci. Rep. 2015, 5, 8365. [Google Scholar] [CrossRef] [PubMed]
- Jia, B.; Raphenya, A.R.; Alcock, B.; Waglechner, N.; Guo, P.; Tsang, K.K.; Lago, B.A.; Dave, B.M.; Pereira, S.; Sharma, A.N.; et al. CARD 2017: Expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Res. 2017, 45, D566–D573. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Zheng, D.; Jin, Q.; Chen, L.; Yang, J. VFDB 2019: A comparative pathogenomic platform with an interactive web interface. Nucleic Acids Res. 2019, 47, D687–D692. [Google Scholar] [CrossRef]
- Seemann, T. ABRicate. Available online: https://github.com/tseemann/abricate (accessed on 1 May 2021).
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019, 47, W256–W259. [Google Scholar] [CrossRef]
- Abby, S.S.; Néron, B.; Ménager, H.; Touchon, M.; Rocha, E.P.C. MacSyFinder: A program to mine genomes for molecular dystems with an spplication to CRISPR-Cas systems. PLoS ONE 2014, 9, e110726. [Google Scholar] [CrossRef]
- Abby, S.S.; Rocha, E.P.C. Identification of protein secretion systems in bacterial genomes using MacSyFinder. In Bacterial Protein Secretion Systems: Methods and Protocols; Journet, L., Cascales, E., Eds.; Springer New York: New York, NY, USA, 2017; pp. 1–21. [Google Scholar]
- Clermont, D.; Motreff, L.; Passet, V.; Fernandez, J.-C.; Bizet, C.; Brisse, S. Multilocus sequence analysis of the genus Citrobacter and description of Citrobacter pasteurii sp. nov. Int. J. Syst. Evol. Microbiol. 2015, 65, 1486–1490. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Dash, H.R.; Mangwani, N.; Chakraborty, J.; Kumari, S. Understanding molecular identification and polyphasic taxonomic approaches for genetic relatedness and phylogenetic relationships of microorganisms. J. Microbiol. Methods 2014, 103, 80–100. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Nei, M.; Kumar, S. Prospects for inferring very large phylogenies by using the neighbor-joining method. Proc. Natl. Acad. Sci. USA 2004, 101, 11030–11035. [Google Scholar] [CrossRef]
- Chun, J.; Oren, A.; Ventosa, A.; Christensen, H.; Arahal, D.R.; da Costa, M.S.; Rooney, A.P.; Yi, H.; Xu, X.W.; De Meyer, S.; et al. Proposed minimal standards for the use of genome data for the taxonomy of prokaryotes. Int. J. Syst. Evol. Microbiol. 2018, 68, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Meier-Kolthoff, J.P.; Carbasse, J.S.; Peinado-Olarte, R.L.; Göker, M. TYGS and LPSN: A database tandem for fast and reliable genome-based classification and nomenclature of prokaryotes. Nucleic Acids Res. 2022, 50, D801–D807. [Google Scholar] [CrossRef] [PubMed]
- Richter, M.; Rossello-Mora, R.; Oliver Glockner, F.; Peplies, J. JSpeciesWS: A web server for prokaryotic species circumscription based on pairwise genome comparison. Bioinformatics 2016, 32, 929–931. [Google Scholar] [CrossRef] [PubMed]
- Bertelli, C.; Laird, M.R.; Williams, K.P.; Simon Fraser University Research Computing Group; Lau, B.Y.; Hoad, G.; Winsor, G.L.; Brinkman, F.S.L. IslandViewer 4: Expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res. 2017, 45, W30–W35. [Google Scholar] [CrossRef]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef]
- Darling, A.E.; Mau, B.; Perna, N.T. progressiveMauve: Multiple genome alignment with gene gain, loss and rearrangement. PLoS ONE 2010, 5, e11147. [Google Scholar] [CrossRef]
- Page, A.J.; Cummins, C.A.; Hunt, M.; Wong, V.K.; Reuter, S.; Holden, M.T.G.; Fookes, M.; Falush, D.; Keane, J.A.; Parkhill, J. Roary: Rapid large-scale prokaryote pan genome analysis. Bioinformatics 2015, 31, 3691–3693. [Google Scholar] [CrossRef]
- Eren, A.M.; Esen, Ö.C.; Quince, C.; Vineis, J.H.; Morrison, H.G.; Sogin, M.L.; Delmont, T.O. Anvi’o: An advanced analysis and visualization platform for ‘omics data. PeerJ 2015, 3, e1319. [Google Scholar] [CrossRef]
- Treangen, T.J.; Ondov, B.D.; Koren, S.; Phillippy, A.M. The Harvest suite for rapid core-genome alignment and visualization of thousands of intraspecific microbial genomes. Genome Biol. 2014, 15, 524. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Hadfield, J.; Croucher, N.J.; Goater, R.J.; Abudahab, K.; Aanensen, D.M.; Harris, S.R. Phandango: An interactive viewer for bacterial population genomics. Bioinformatics 2018, 34, 292–293. [Google Scholar] [CrossRef] [PubMed]
- Tettelin, H.; Masignani, V.; Cieslewicz, M.J.; Donati, C.; Medini, D.; Ward, N.L.; Angiuoli, S.V.; Crabtree, J.; Jones, A.L.; Durkin, A.S.; et al. Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: Implications for the microbial “pan-genome”. Proc. Natl. Acad. Sci. USA 2005, 102, 13950–13955. [Google Scholar] [CrossRef] [PubMed]
- Mehta, C.R.; Patel, N.R. Algorithm 643. FEXACT: A FORTRAN subroutine for Fisher’s exact test on unordered r × c contingency tables. ACM Trans. Math Softw. 1986, 12, 154–161. [Google Scholar] [CrossRef]
- Goris, J.; Konstantinidis, K.T.; Klappenbach, J.A.; Coenye, T.; Vandamme, P.; Tiedje, J.M. DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int. J. Syst. Evol. Microbiol. 2007, 57, 81–91. [Google Scholar] [CrossRef]
- Smets, B.F.; Barkay, T. Horizontal gene transfer: Perspectives at a crossroads of scientific disciplines. Nat. Rev. Microbiol. 2005, 3, 675–678. [Google Scholar] [CrossRef] [PubMed]
- Pilar, A.V.C.; Petronella, N.; Dussault, F.M.; Verster, A.J.; Bekal, S.; Levesque, R.C.; Goodridge, L.; Tamber, S. Similar yet different: Phylogenomic analysis to delineate Salmonella and Citrobacter species boundaries. BMC Genom. 2020, 21, 377. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Peng, H.; Wang, Y.-s.; Huang, X.-m.; Xie, X.-b.; Shi, Q.-s. Complete genome sequence of Citrobacter werkmanii strain BF-6 isolated from industrial putrefaction. BMC Genom. 2017, 18, 765. [Google Scholar] [CrossRef]
- Tantoso, E.; Eisenhaber, B.; Kirsch, M.; Shitov, V.; Zhao, Z.; Eisenhaber, F. To kill or to be killed: Pangenome analysis of Escherichia coli strains reveals a tailocin specific for pandemic ST131. BMC Biol. 2022, 20, 146. [Google Scholar] [CrossRef]
- Laing, C.R.; Whiteside, M.D.; Gannon, V.P.J. Pan-genome analyses of the species Salmonella enterica, and identification of genomic markers predictive for species, subspecies, and serovar. Front Microbiol. 2017, 8, 1345. [Google Scholar] [CrossRef] [PubMed]
- Vernikos, G.; Medini, D.; Riley, D.R.; Tettelin, H. Ten years of pan-genome analyses. Curr. Opin. Microbiol. 2015, 23, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Yuan, C.; Yin, Z.; Wang, J.; Qian, C.; Wei, Y.; Zhang, S.; Jiang, L.; Liu, B. Comparative genomic analysis of Citrobacter and key genes essential for the pathogenicity of Citrobacter koseri. Front Microbiol. 2019, 10, 2774. [Google Scholar] [CrossRef]
- Thoden, J.B.; Kim, J.; Raushel, F.M.; Holden, H.M. The catalytic mechanism of galactose mutarotase. Protein Sci. 2003, 12, 1051–1059. [Google Scholar] [CrossRef]
- Kocharova, N.A.; Knirel, Y.A.; Stanislavsky, E.S.; Kholodkova, E.V.; Lugowski, C.; Jachymek, W.; Romanowska, E. Structural and serological studies of lipopolysaccharides of Citrobacter O35 and O38 antigenically related to Salmonella. FEMS Microbiol. Immunol. 1996, 13, 1–8. [Google Scholar] [CrossRef]
- Raff, R.A.; Wheat, R.W. Carbohydrate composition of the phenol-soluble lipopolysaccharides of Citrobacter freundii. J. Bacteriol. 1968, 95, 2035–2043. [Google Scholar] [CrossRef] [PubMed]
- Aguirre-Sánchez, J.R.; Ibarra-Rodriguez, J.R.; Vega-López, I.F.; Martínez-Urtaza, J.; Chaidez-Quiroz, C. Genomic signatures of adaptation to natural settings in non-typhoidal Salmonella enterica serovars Saintpaul, Thompson and Weltevreden. Infect. Genet. Evol. 2021, 90, 104771. [Google Scholar] [CrossRef]
- Subedi, D.; Kohli, G.S.; Vijay, A.K.; Willcox, M.; Rice, S.A. Accessory genome of the multi-drug resistant ocular isolate of Pseudomonas aeruginosa PA34. PLoS ONE 2019, 14, e0215038. [Google Scholar] [CrossRef]
- Colavecchio, A.; Cadieux, B.; Lo, A.; Goodridge, L.D. Bacteriophages contribute to the spread of antibiotic resistance genes among foodborne pathogens of the Enterobacteriaceae family—A review. Front Microbiol. 2017, 8, 1108. [Google Scholar] [CrossRef]
- Miller, D.; Stern, A.; Burstein, D. Deciphering microbial gene function using natural language processing. Nat. Commun. 2022, 13, 5731. [Google Scholar] [CrossRef]
- Vanni, C.; Schechter, M.S.; Acinas, S.G.; Barberán, A.; Buttigieg, P.L.; Casamayor, E.O.; Delmont, T.O.; Duarte, C.M.; Eren, A.M.; Finn, R.D.; et al. Unifying the known and unknown microbial coding sequence space. eLife 2022, 11, e67667. [Google Scholar] [CrossRef] [PubMed]
- Korotkov, K.V.; Sandkvist, M.; Hol, W.G.J. The type II secretion system: Biogenesis, molecular architecture and mechanism. Nat. Rev. Microbiol. 2012, 10, 336–351. [Google Scholar] [CrossRef] [PubMed]
- Meuskens, I.; Saragliadis, A.; Leo, J.C.; Linke, D. Type V secretion systems: An overview of passenger domain functions. Front Microbiol. 2019, 10, 1163. [Google Scholar] [CrossRef] [PubMed]
- Chernyatina, A.A.; Low, H.H. Core architecture of a bacterial type II secretion system. Nat. Commun. 2019, 10, 5437. [Google Scholar] [CrossRef]
- Liu, L.; Hao, S.; Lan, R.; Wang, G.; Xiao, D.; Sun, H.; Xu, J. The type VI secretion system modulates flagellar gene expression and secretion in Citrobacter freundii and contributes to adhesion and cytotoxicity to host cells. Infect. Immun. 2015, 83, 2596–2604. [Google Scholar] [CrossRef]
- Chen, C.; Yang, X.; Shen, X. Confirmed and potential roles of bacterial T6SSs in the intestinal ecosystem. Front. Microbiol. 2019, 10, 1484. [Google Scholar] [CrossRef]
- Barnhart, M.M.; Chapman, M.R. Curli biogenesis and function. Annu. Rev. Microbiol. 2006, 60, 131–147. [Google Scholar] [CrossRef]
- Zhou, G.; Wang, Y.S.; Peng, H.; Li, S.J.; Sun, T.L.; Shen, P.F.; Xie, X.B.; Shi, Q.S. Roles of ompA of Citrobacter werkmanii in bacterial growth, biocide resistance, biofilm formation and swimming motility. Appl. Microbiol. Biotechnol. 2021, 105, 2841–2854. [Google Scholar] [CrossRef]
- Kortman, G.A.; Boleij, A.; Swinkels, D.W.; Tjalsma, H. Iron availability increases the pathogenic potential of Salmonella typhimurium and other enteric pathogens at the intestinal epithelial interface. PLoS ONE 2012, 7, e29968. [Google Scholar] [CrossRef]
- Qin, J.; Zhao, Y.; Wang, A.; Chi, X.; Wen, P.; Li, S.; Wu, L.; Bi, S.; Xu, H. Comparative genomic characterization of multidrug-resistant Citrobacter spp. strains in Fennec fox imported to China. Gut Pathog. 2021, 13, 59. [Google Scholar] [CrossRef]
- Liu, L.; Lan, R.; Liu, L.; Wang, Y.; Zhang, Y.; Wang, Y.; Xu, J. Antimicrobial resistance and cytotoxicity of Citrobacter spp. in Maanshan Anhui Province, China. Front Microbiol. 2017, 8, 1357. [Google Scholar] [CrossRef]
- Aoyama, H.; Fujimaki, K.; Sato, K.; Fujii, T.; Inoue, M.; Hirai, K.; Mitsuhashi, S. Clinical isolate of Citrobacter freundii highly resistant to new quinolones. Antimicrob. Agents Chemother. 1988, 32, 922–924. [Google Scholar] [CrossRef] [PubMed]
- Metri, B.C.; Jyothi, P.; Peerapur, B.V. Antibiotic resistance in Citrobacter spp. isolated from urinary tract infection. Urol. Ann. 2013, 5, 312. [Google Scholar] [PubMed]
- Butler Mark, S.; Gigante, V.; Sati, H.; Paulin, S.; Al-Sulaiman, L.; Rex John, H.; Fernandes, P.; Arias Cesar, A.; Paul, M.; Thwaites Guy, E.; et al. Analysis of the clinical pipeline of treatments for drug-resistant bacterial infections: Despite progress, more action is needed. Antimicrob. Agents Chemother. 2022, 66, e01991-21. [Google Scholar] [CrossRef]
- Velázquez-Meza, M.E.; Galarde-López, M.; Carrillo-Quiróz, B.; Alpuche-Aranda, C.M. Antimicrobial resistance: One Health approach. Vet World 2022, 15, 743–749. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Citrobacter Species 1 | C. werkmanii LANIIA-032 | C. werkmanii FDAARGOS_364 T | C. freundii ATCC 8090 T | C. youngae CCUG30791 T | C. pasteurii CIP55.33T | C. braakii ATCC 51,113 T | C. europaeus 97/79T | C. portucalensis A60 T | C. tructae SNU WT2 T | C.cronae Tue2-1T |
|---|---|---|---|---|---|---|---|---|---|---|
| C. tructae SNU WT2 T | 32.6 | |||||||||
| C. portucalensis A60 T | 33.0 | 42.4 | ||||||||
| C. europaeus 97/79 T | 50.3 | 32.9 | 42.9 | |||||||
| C. braakii ATCC 51113 T | 52.9 | 48.5 | 33.1 | 42.7 | ||||||
| C. pasteurii CIP 55.13 T | 38.8 | 38.5 | 38.4 | 32.9 | 36.2 | |||||
| C. youngae CCUG 30791 T | 59.6 | 39.0 | 39.0 | 39.4 | 32.9 | 36.5 | ||||
| C. freundii ATCC 8090T | 39.8 | 35.2 | 45.0 | 45.2 | 52.4 | 29.8 | 37.6 | |||
| C. werkmanii FDAARGOS_364 T | 37.8 | 36.4 | 36.1 | 42.7 | 43.4 | 42.4 | 32.4 | 70.0 | ||
| C. werkmanii LANIIA-032 | 92.2 | 37.7 | 36.5 | 36.1 | 42.6 | 43.3 | 42.5 | 32.4 | 70.0 | |
| C. werkmanii LANIIA-031 | 92.3 | 92.2 | 37.9 | 36.5 | 36.2 | 42.7 | 43.3 | 42.5 | 32.4 | 70.0 |
| Clade 1 | Strain 2 | GenBank Accession Number | Isolation Source Description | Sample Source Type | Date 3 | Country |
|---|---|---|---|---|---|---|
| Clade I | CB00011 | GCA_016505505.1 | Sputum | Human | 2017 | United States |
| CB00078 | GCA_016507625.1 | Wound | Human | 2018 | United States | |
| Clade II | FDAARGOS_364 T | GCA_002386385.1 | Stool | Human | 2014 | United States |
| MGYG-HGUT-02535 | GCA_902388105.1 | Gut | Human | 2019 | United States | |
| Clade III | CB00044 | GCA_016505055.1 | Not collected | Human | 2017 | United States |
| CVM 45625 | GCA_015942525.1 | Unknown | Environmental | 2019 | United States | |
| Clade IV | BF-6 | GCA_002025225.1 | Industrial water | Environmental | 2012 | China |
| Colony242 | GCA_016893825.1 | Food | Food | 2019 | Thailand | |
| Colony247 | GCA_016893645.1 | Food | Food | 2019 | Thailand | |
| CVM 45620 | GCA_015943365.1 | Unknown | Environmental | 2019 | United States | |
| CVM 45667 | GCA_015943485.1 | Unknown | Environmental | 2019 | United States | |
| CVM 45672 | GCA_015943405.1 | Unknown | Environmental | 2020 | United States | |
| LANIIA-031 | JAUJUK000000000 | River water | Environmental | 2018 | Mexico | |
| LANIIA-032 | JAUJUL000000000 | River water | Environmental | 2018 | Mexico | |
| RS189 | GCA_015958985.1 | Not collected | Human | 2017 | United States | |
| YDC730 | GCA_015958865.1 | Pelvic abscess | Human | 2015 | United States | |
| Clade V | 2580 | GCA_009907085.1 | Urine | Human | 2015 | Nigeria |
| AK-8 | GCA_002114305.1 | Human urine | Human | 2014 | India | |
| BOX-5 | GCA_009856875.1 | Hospital sink | Environmental | 2016 | France | |
| C5.1 | GCA_008364715.1 | Sprouts | Food | 2015 | Germany | |
| CRE1173 | GCA_018106225.1 | Pus | Human | 2015 | Malaysia | |
| CRE1470 | GCA_018106165.1 | Peritoneal fluid | Human | 2016 | Malaysia | |
| CRE346 | GCA_018106145.1 | Foot ulcer | Human | 2015 | Malaysia | |
| CRE806 | GCA_018106185.1 | Foot ulcer | Human | 2014 | Malaysia | |
| CRK0001 | GCA_002185305.2 | Blood | Human | 2014 | United States | |
| CW_LB-887 | GCA_013303045.1 | Coastal water | Environmental | 2014 | Brazil | |
| FDAARGOS_616 | GCA_008693645.1 | Clinical isolate | Human | NA | United States | |
| ICR003007 | GCA_004146135.1 | Hospital patient | Human | 2017 | France | |
| JS97 | GCA_009821535.1 | Chicken | Animal | NA | Unknown | |
| L38 | GCA_013618825.1 | Chicken liver | Animal | 2019 | Nigeria | |
| UMH18 | GCA_003665555.1 | Bacteremia | Human | 2013 | United States | |
| YDC667-1 | GCA_013336965. | Lung tissue | Human | 2014 | United States |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aguirre-Sánchez, J.R.; Quiñones, B.; Ortiz-Muñoz, J.A.; Prieto-Alvarado, R.; Vega-López, I.F.; Martínez-Urtaza, J.; Lee, B.G.; Chaidez, C. Comparative Genomic Analyses of Virulence and Antimicrobial Resistance in Citrobacter werkmanii, an Emerging Opportunistic Pathogen. Microorganisms 2023, 11, 2114. https://doi.org/10.3390/microorganisms11082114
Aguirre-Sánchez JR, Quiñones B, Ortiz-Muñoz JA, Prieto-Alvarado R, Vega-López IF, Martínez-Urtaza J, Lee BG, Chaidez C. Comparative Genomic Analyses of Virulence and Antimicrobial Resistance in Citrobacter werkmanii, an Emerging Opportunistic Pathogen. Microorganisms. 2023; 11(8):2114. https://doi.org/10.3390/microorganisms11082114
Chicago/Turabian StyleAguirre-Sánchez, José R., Beatriz Quiñones, José A. Ortiz-Muñoz, Rogelio Prieto-Alvarado, Inés F. Vega-López, Jaime Martínez-Urtaza, Bertram G. Lee, and Cristóbal Chaidez. 2023. "Comparative Genomic Analyses of Virulence and Antimicrobial Resistance in Citrobacter werkmanii, an Emerging Opportunistic Pathogen" Microorganisms 11, no. 8: 2114. https://doi.org/10.3390/microorganisms11082114