
Targeted Integration of siRNA against Porcine Cytomegalovirus (PCMV) Enhances the Resistance of Porcine Cells to PCMV

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
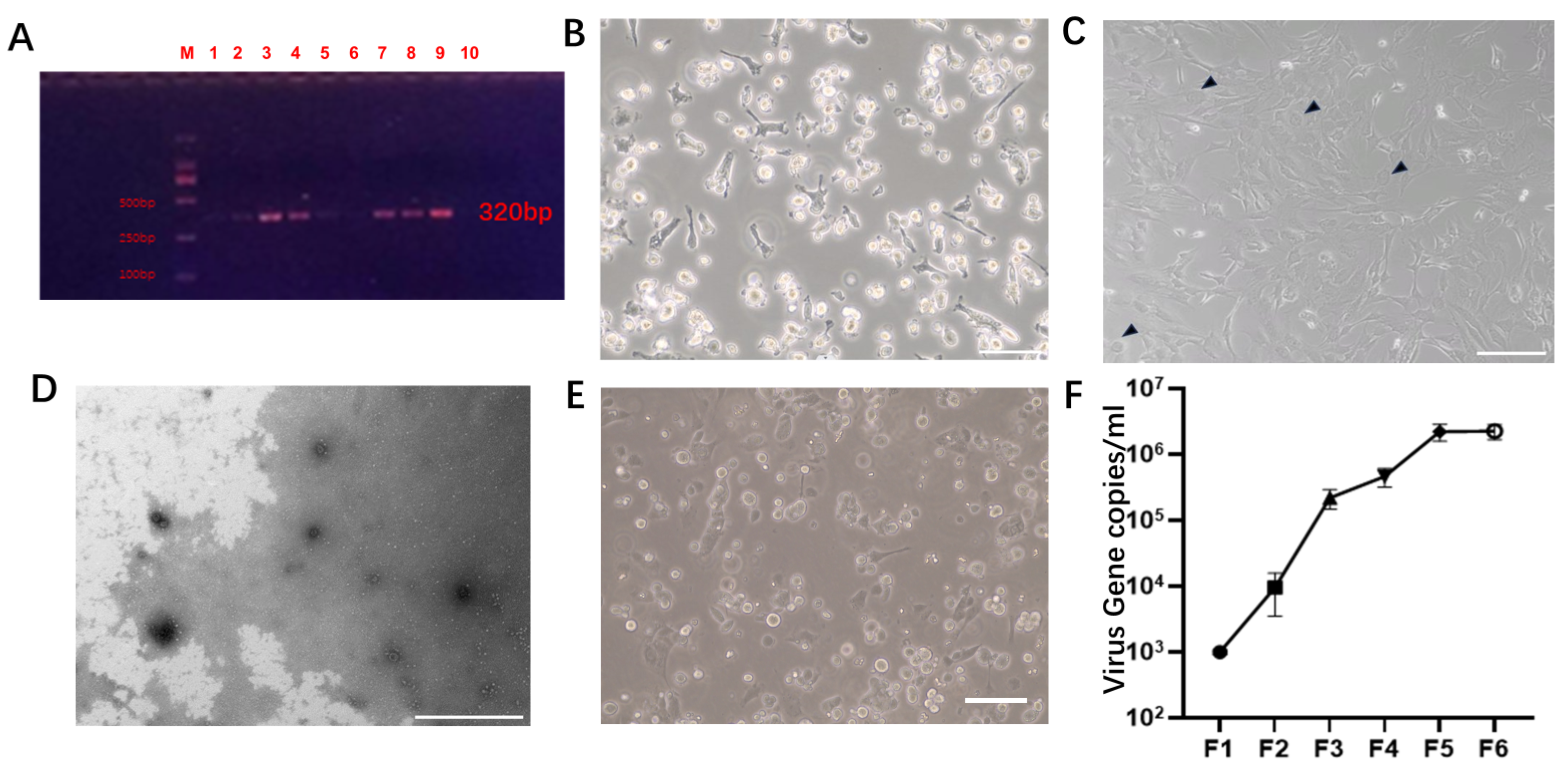
2.1. Establishment of the Virus Infection Model
2.2. Validation and Selection of siRNAs
2.3. Selection of the Gene Editing Site
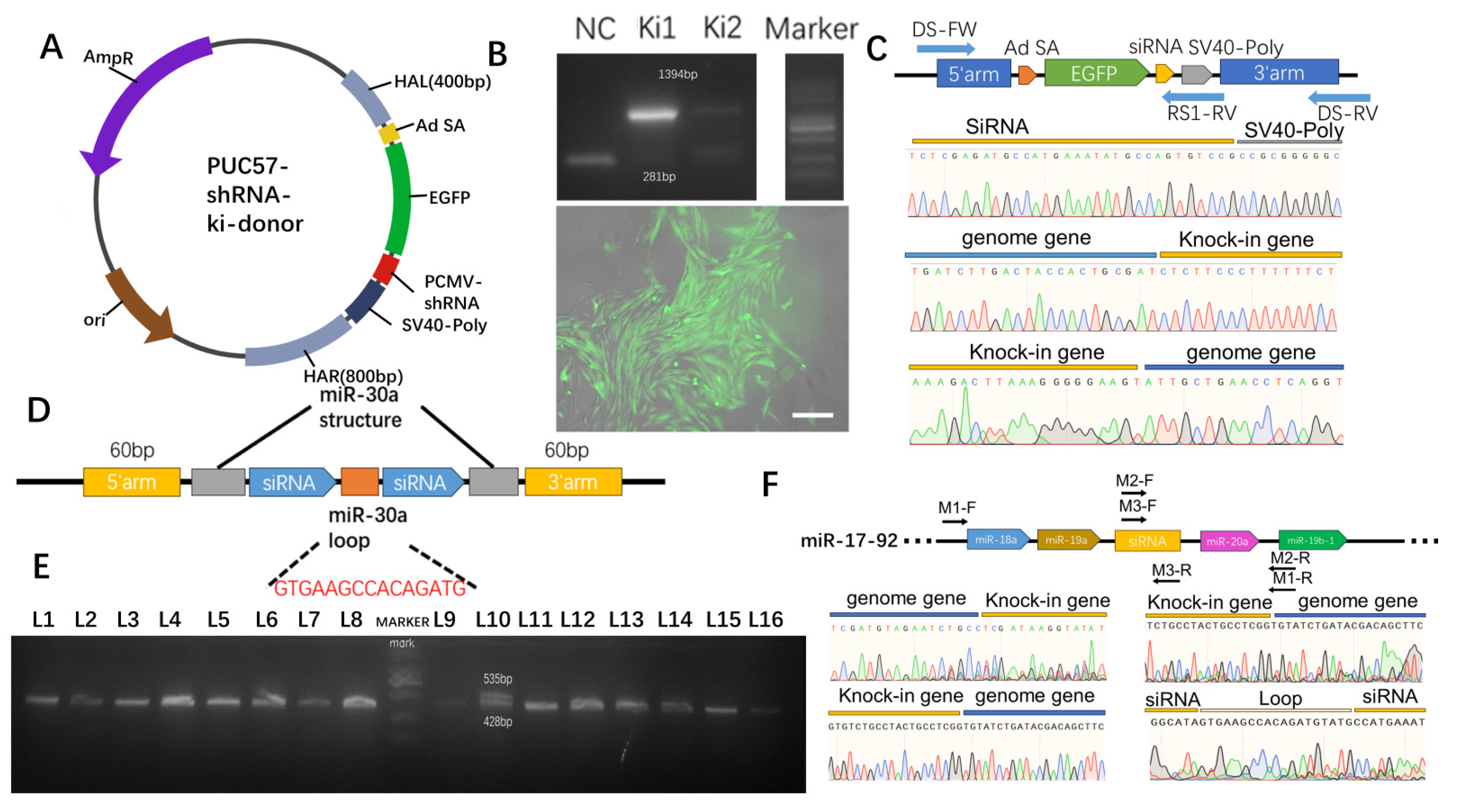
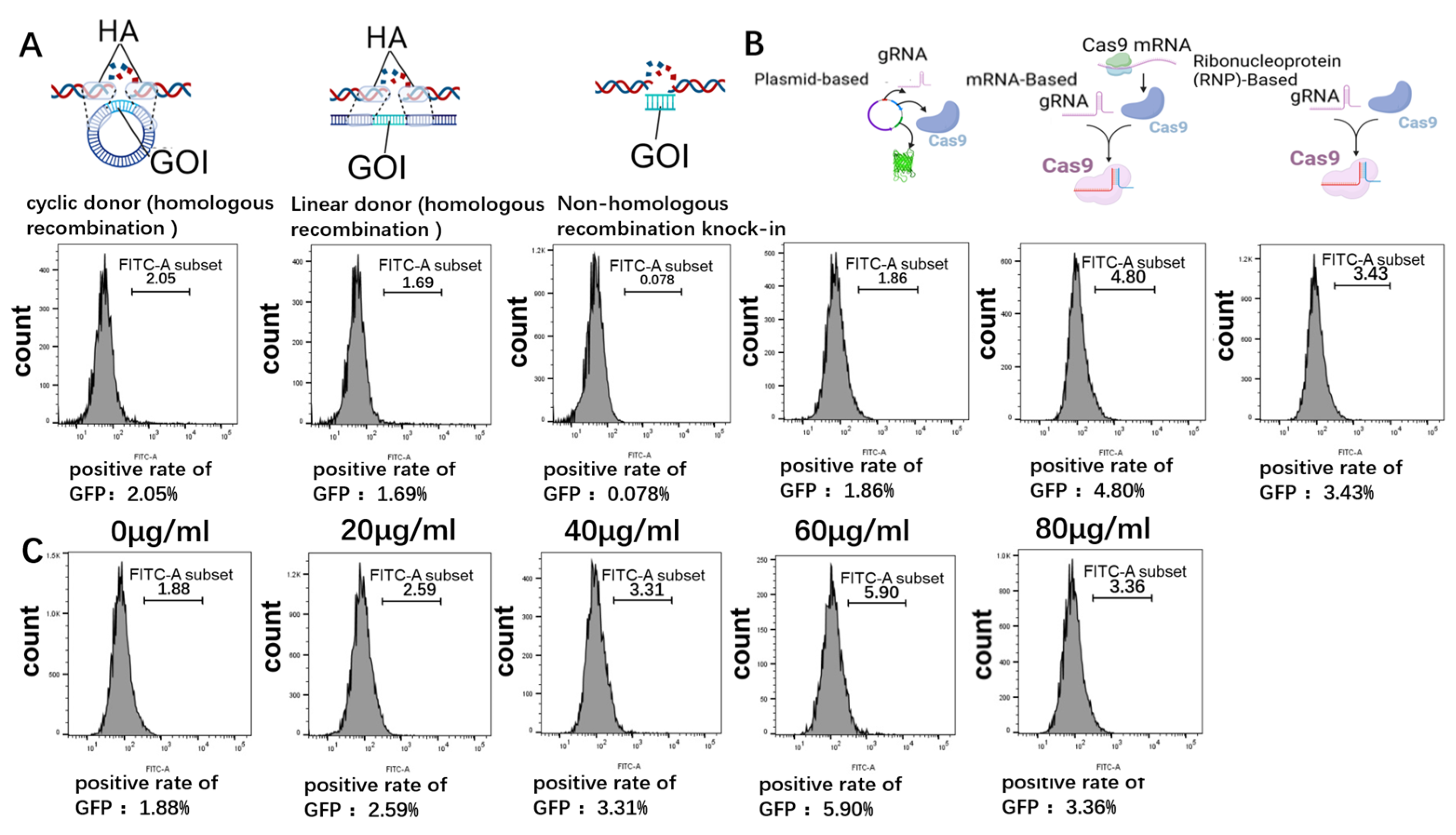
2.4. Optimization of Gene-Targeted Integration Efficiency
2.5. Targeted Integration of shRNA Genes
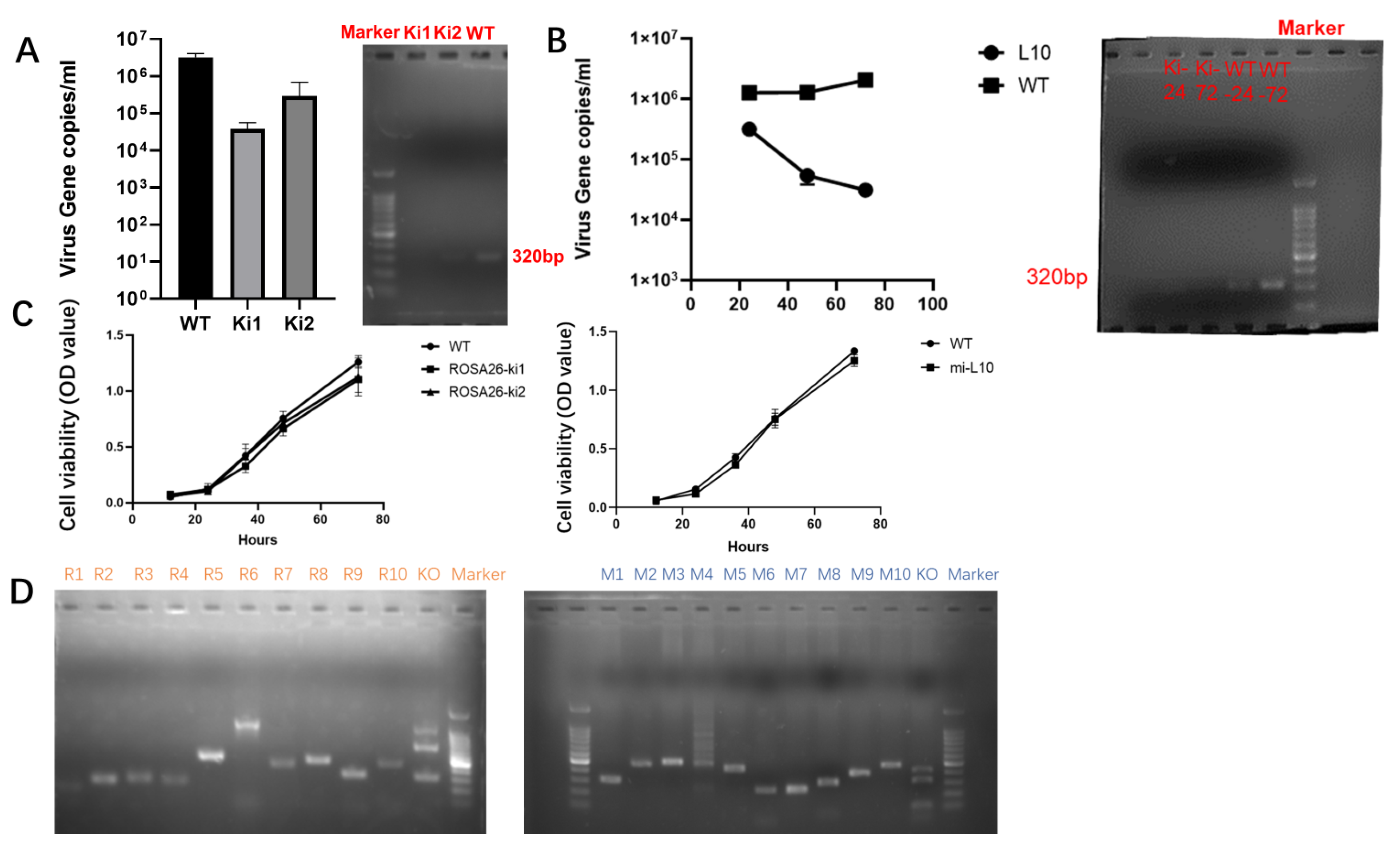
2.6. Assessment of Antiviral Capability after shRNA Knock-In
2.7. Safety Evaluation
2.8. Statistical Analysis
3. Results
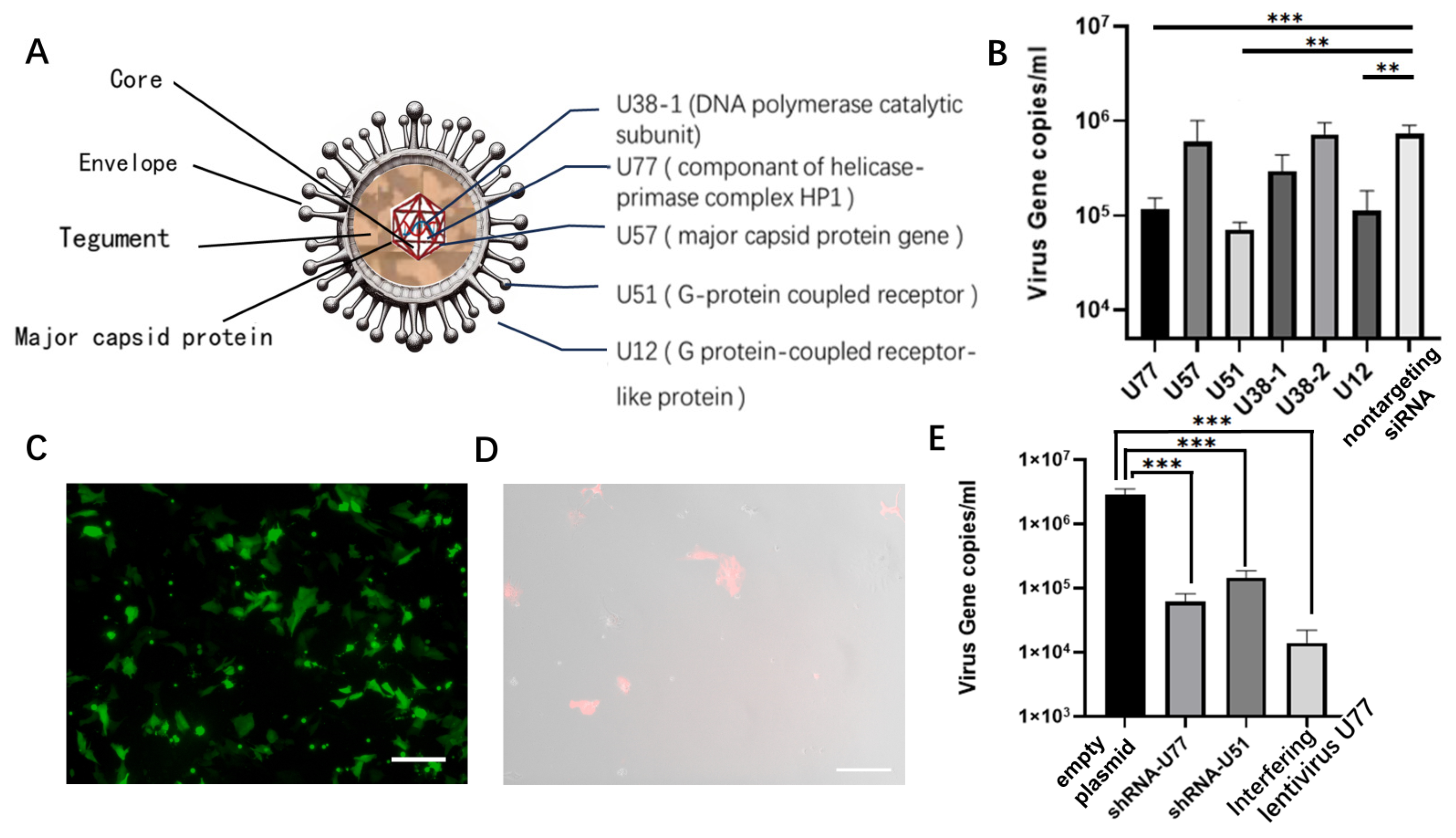
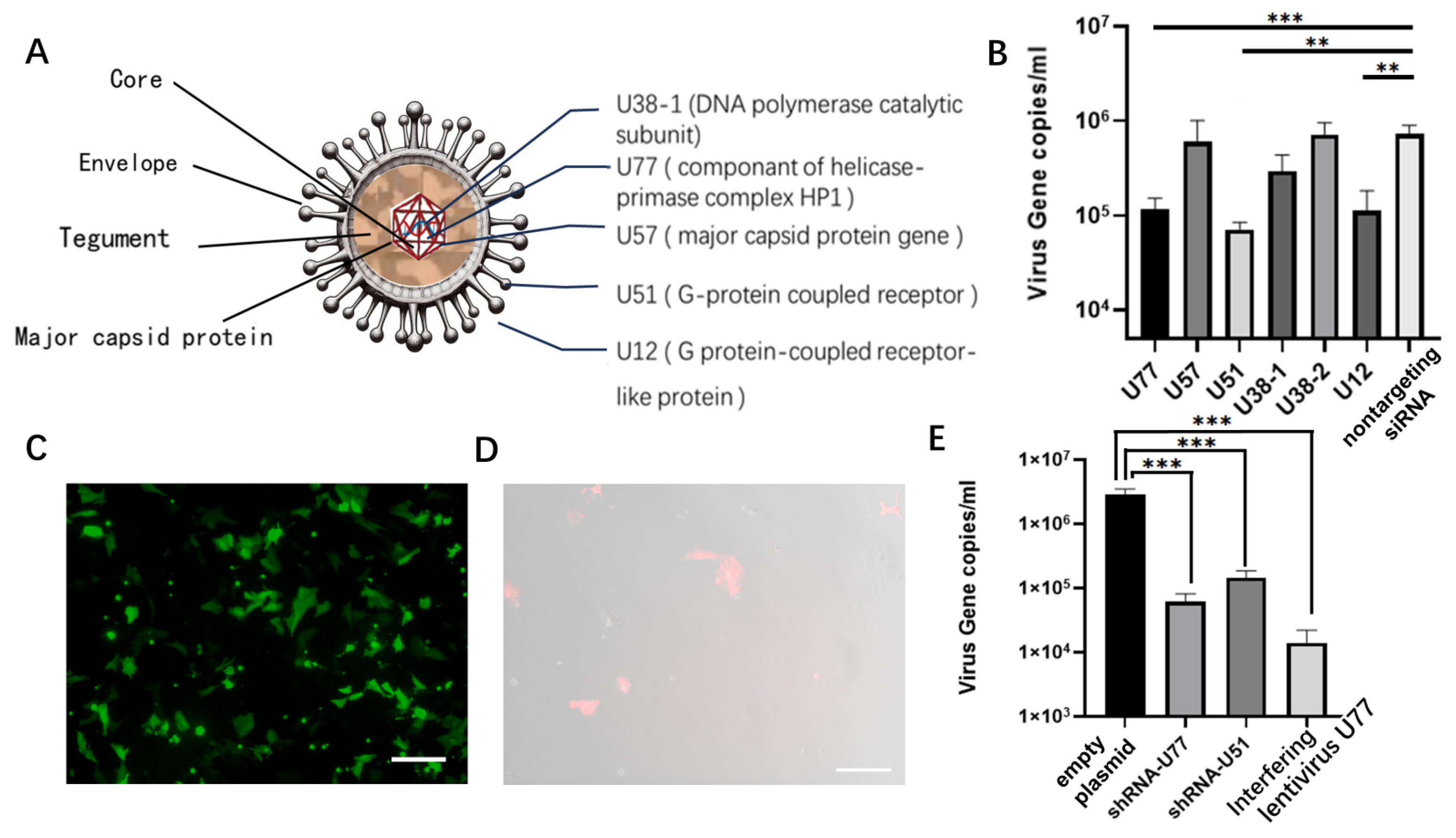
3.1. Establishment of the Virus Infection Model
3.2. Selection and Validation of Antiviral siRNAs
3.3. Interference with RNA Site-Specific Integration
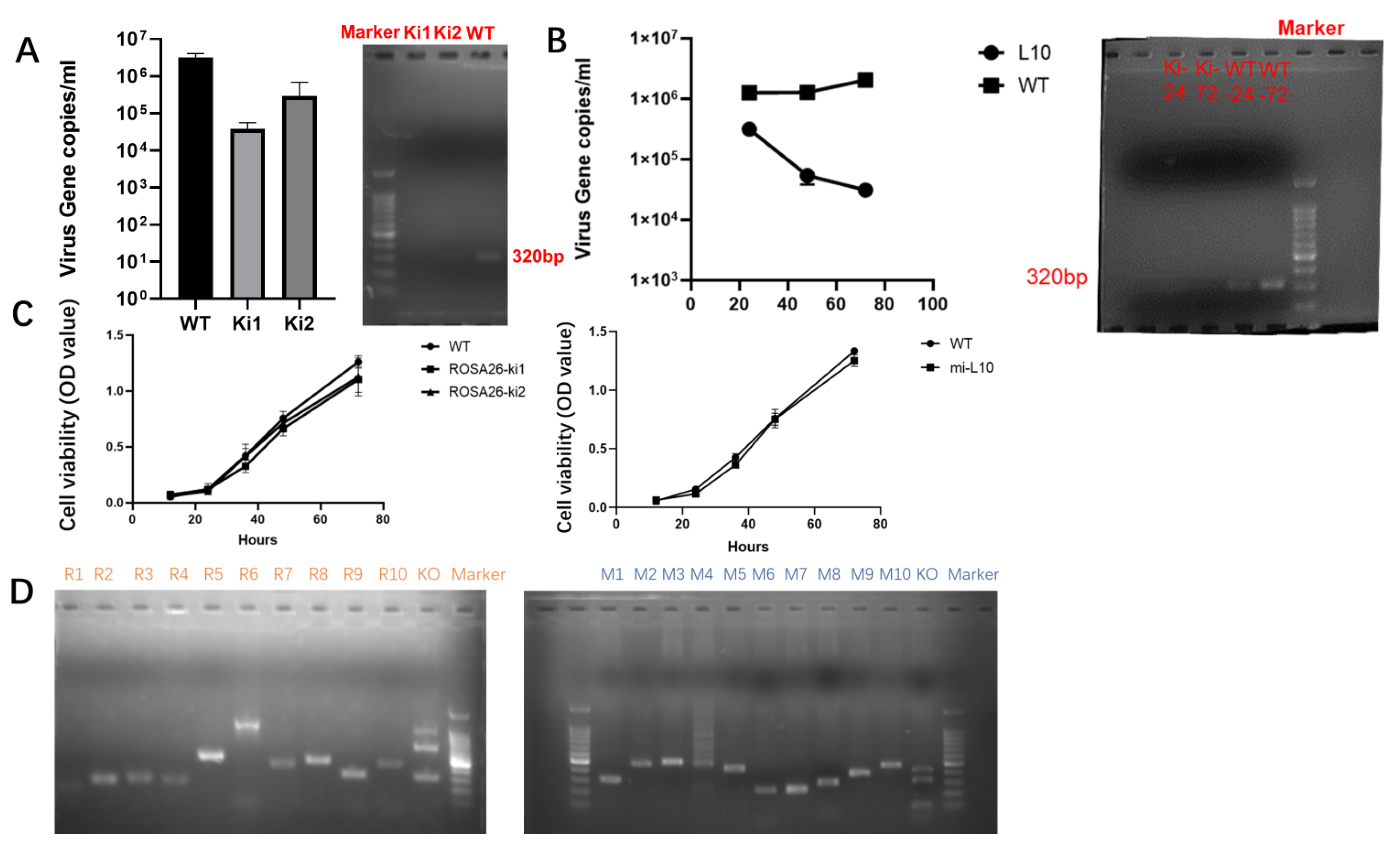
3.4. Validation of Antiviral Activity and Safety Assessment after Interfering with Gene Site-Specific Integration
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Griffith, B.P.; Goerlich, C.E.; Singh, A.K.; Rothblatt, M.; Lau, C.L.; Shah, A.; Lorber, M.; Grazioli, A.; Saharia, K.K.; Hong, S.N.; et al. Genetically Modified Porcine-to-Human Cardiac Xenotransplantation. N. Engl. J. Med. 2022, 387, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Liao, S.; Zhu, L.; Xu, Z.; Zhou, Y. Molecular epidemiology of porcine Cytomegalovirus (PCMV) in Sichuan Province, China: 2010–2012. PLoS ONE 2013, 8, e64648. [Google Scholar] [CrossRef] [PubMed]
- Hansen, S.; Fischer, K.; Krabben, L.; Rinke Carrapeiro, A.; Klinger, B.; Schnieke, A.; Kaufer, B.; Denner, J. Detection of porcine cytomegalovirus, a roseolovirus, in pig ovaries and follicular fluid: Implications for somatic cells nuclear transfer, cloning and xenotransplantation. Virol. J. 2023, 20, 15. [Google Scholar] [CrossRef] [PubMed]
- Mueller, N.J.; Denner, J. Porcine cytomegalovirus/porcine roseolovirus (PCMV/PRV): A threat for xenotransplantation? Xenotransplantation 2022, 29, e12775. [Google Scholar] [CrossRef] [PubMed]
- Ryu, J.; Prather, R.S.; Lee, K. Use of gene-editing technology to introduce targeted modifications in pigs. J. Anim. Sci. Biotechnol. 2018, 9, 5. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Pang, D.; Yuan, H.; Jiao, H.; Lu, C.; Wang, K.; Yang, Q.; Li, M.; Chen, X.; Yu, T.; et al. Genetically modified pigs are protected from classical swine fever virus. PLoS Pathog. 2018, 14, e1007193. [Google Scholar] [CrossRef] [PubMed]
- Knott, G.J.; Doudna, J.A. CRISPR-Cas guides the future of genetic engineering. Science 2018, 361, 866–869. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.D.; Lander, E.S.; Zhang, F.J.C. Development and applications of CRISPR-Cas9 for genome engineering. Cell 2014, 157, 1262–1278. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.W.; Gao, C.; Zheng, Y.M.; Yi, L.; Lu, J.C.; Huang, X.Y.; Cai, J.B.; Zhang, P.F.; Cui, Y.H.; Ke, A.W. Current applications and future perspective of CRISPR/Cas9 gene editing in cancer. Mol. Cancer 2022, 21, 57. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Zhang, J.; Wang, R.; Yang, G.; Bao, J. Pig-to-Human xenotransplantation: Moving toward organ customization. Precis. Clin. Med. 2023, 6, pbad013. [Google Scholar] [CrossRef]
- Zhang, J.; Zhu, Z.; Yue, W.; Li, J.; Chen, Q.; Yan, Y.; Lei, A.; Hua, J. Establishment of CRISPR/Cas9-Mediated Knock-in System for Porcine Cells with High Efficiency. Appl. Biochem. Biotechnol. 2019, 189, 26–36. [Google Scholar] [CrossRef] [PubMed]
- Cerchia, C.; Nasso, R.; Mori, M.; Villa, S.; Gelain, A.; Capasso, A.; Aliotta, F.; Simonetti, M.; Rullo, R.; Masullo, M.; et al. Discovery of Novel Naphthylphenylketone and Naphthylphenylamine Derivatives as Cell Division Cycle 25B (CDC25B) Phosphatase Inhibitors: Design, Synthesis, Inhibition Mechanism, and in Vitro Efficacy against Melanoma Cell Lines. J. Med. Chem. 2019, 62, 7089–7110. [Google Scholar] [CrossRef] [PubMed]
- Ryczek, N.; Hryhorowicz, M.; Zeyland, J.; Lipiński, D.; Słomski, R. CRISPR/Cas Technology in Pig-to-Human Xenotransplantation Research. Int. J. Mol. Sci. 2021, 22, 3196. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Pang, D.; Li, M.; Yuan, H.; Yu, T.; Huang, P.; Li, J.; Chen, X.; Jiao, H.; Xie, Z.; et al. CRISPR/Cas9-Mediated Hitchhike Expression of Functional shRNAs at the Porcine miR-17-92 Cluster. Cells 2019, 8, 113. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.; Marran, K.; Valentine, A.; Hannon, G.J. Creating an miR30-based shRNA vector. Cold Spring Harb. Protoc. 2013, 2013, 631–635. [Google Scholar] [CrossRef] [PubMed]
- Ramsoondar, J.; Vaught, T.; Ball, S.; Mendicino, M.; Monahan, J.; Jobst, P.; Vance, A.; Duncan, J.; Wells, K.; Ayares, D.J.X. Production of transgenic pigs that express porcine endogenous retrovirus small interfering RNAs. Xenotransplantation 2009, 16, 164–180. [Google Scholar] [CrossRef] [PubMed]
- Fiebig, U.; Holzer, A.; Ivanusic, D.; Plotzki, E.; Hengel, H.; Neipel, F.; Denner, J. Antibody Cross-Reactivity between Porcine Cytomegalovirus (PCMV) and Human Herpesvirus-6 (HHV-6). Viruses 2017, 9, 317. [Google Scholar] [CrossRef]
- Wen, D.; Ding, L.S.; Zhang, Y.; Li, X.; Zhang, X.; Yuan, F.; Zhao, T.; Zheng, A. Suppression of flavivirus transmission from animal hosts to mosquitoes with a mosquito-delivered vaccine. Nat. Commun. 2022, 13, 7780. [Google Scholar] [CrossRef]
- Wang, X.; Xu, L.; Chen, Y.; Liu, A.; Wang, L.; Xu, P.; Liu, Y.; Li, L.; Meng, F. Integrating nested PCR with high-throughput sequencing to characterize mutations of HBV genome in low viral load samples. Medicine 2017, 96, e7588. [Google Scholar] [CrossRef]
- Motohashi, K. Development of highly sensitive and low-cost DNA agarose gel electrophoresis detection systems, and evaluation of non-mutagenic and loading dye-type DNA-staining reagents. PLoS ONE 2019, 14, e0222209. [Google Scholar] [CrossRef]
- Liu, H.; Liu, J.; Huang, J.; Bai, X.; Wang, Q. Heterogeneity and plasticity of porcine alveolar macrophage and pulmonary interstitial macrophage isolated from healthy pigs in vitro. Biol. Open 2019, 8, bio046342. [Google Scholar] [CrossRef]
- Kavanová, L.; Moutelíková, R.; Prodělalová, J.; Faldyna, M.; Toman, M.; Salát, J. Monocyte derived macrophages as an appropriate model for porcine cytomegalovirus immunobiology studies. Vet. Immunol. Immunopathol. 2018, 197, 58–62. [Google Scholar] [CrossRef]
- Chen, R.-J.; Zhou, L.-J.; Wu, X.-M.; Che, Y.-L.; Wang, C.-Y.; Wang, L.-B.; Huang, X.-F.; Yan, S.; Liu, Y.-T.; Wei, H. Cloning and Sequencing of gB Gene from Porcine Cytomegalovirus Isolated in Fujian. Fujian J. Agric. Sci. 2017, 32, 7–11. [Google Scholar] [CrossRef]
- Gao, M.; Zhu, X.; Peng, W.; He, Y.; Li, Y.; Wu, Q.; Zhou, Y.; Liao, G.; Yang, G.; Bao, J.; et al. Kidney ECM Pregel Nanoarchitectonics for Microarrays to Accelerate Harvesting Gene-Edited Porcine Primary Monoclonal Spheres. ACS Omega 2022, 7, 23156–23169. [Google Scholar] [CrossRef]
- Jin, L.; Wang, Q.; Chen, J.; Wang, Z.; Xin, H.; Zhang, D.J.P. Efficient delivery of therapeutic siRNA by Fe3O4 magnetic nanoparticles into oral cancer cells. Pharmaceutics 2019, 11, 615. [Google Scholar] [CrossRef]
- Shi, B.; Xue, M.; Wang, Y.; Wang, Y.; Li, D.; Zhao, X.; Li, X. An improved method for increasing the efficiency of gene transfection and transduction. Int. J. Physiol. Pathophysiol. Pharmacol. 2018, 10, 95–104. [Google Scholar]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef]
- Lu, C. Research on Generating Gene-Modified Pigs Resistant to CSFV/PEDV Based on the Pig miR-17-92 Cluster. Ph.D. thesis. University of Jilin, Jilin, China, 2019.
- Xie, Z.; Pang, D.; Wang, K.; Li, M.; Guo, N.; Yuan, H.; Li, J.; Zou, X.; Jiao, H.; Ouyang, H.; et al. Optimization of a CRISPR/Cas9-mediated Knock-in Strategy at the Porcine Rosa26 Locus in Porcine Foetal Fibroblasts. Sci. Rep. 2017, 7, 3036. [Google Scholar] [CrossRef]
- Tyumentseva, M.A.; Tyumentsev, A.I.; Akimkin, V.G. Protocol for assessment of the efficiency of CRISPR/Cas RNP delivery to different types of target cells. PLoS ONE 2021, 16, e0259812. [Google Scholar] [CrossRef]
- Liang, Z.; Chen, K.; Zhang, Y.; Liu, J.; Yin, K.; Qiu, J.-L.; Gao, C. Genome editing of bread wheat using biolistic delivery of CRISPR/Cas9 in vitro transcripts or ribonucleoproteins. Nat. Protoc. 2018, 13, 413–430. [Google Scholar] [CrossRef]
- Barrows, N.J.; Campos, R.K.; Powell, S.T.; Prasanth, K.R.; Schott-Lerner, G.; Soto-Acosta, R.; Galarza-Muñoz, G.; McGrath, E.L.; Urrabaz-Garza, R.; Gao, J.; et al. A screen of FDA-approved drugs for inhibitors of Zika virus infection. Cell Host Microbe 2016, 20, 259–270. [Google Scholar] [CrossRef]
- Matson, A.W.; Hosny, N.; Swanson, Z.A.; Hering, B.J.; Burlak, C. Optimizing sgRNA length to improve target specificity and efficiency for the GGTA1 gene using the CRISPR/Cas9 gene editing system. PLoS ONE 2019, 14, e0226107. [Google Scholar] [CrossRef]
- Sentmanat, M.F.; Peters, S.T.; Florian, C.P.; Connelly, J.P.; Pruett-Miller, S.M. A Survey of Validation Strategies for CRISPR-Cas9 Editing. Sci. Rep. 2018, 8, 888. [Google Scholar] [CrossRef]
- Wang, H.-Y.; Song, J.K.; Shin, S.; Choi, K.M.; Kim, H. One-Tube Nested Real-Time PCR Assay for Rapid Screening of Porcine Cytomegalovirus in Clinical Samples. Front. Vet. Sci. 2020, 7, 586045. [Google Scholar] [CrossRef]
- Mettenleiter, T.C.; Ehlers, B.; Müller, T.; Yoon, K.-J.; Teifke, J.P. Herpesviruses. In Diseases of Swine; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2019; pp. 548–575. [Google Scholar]
- Ma, X.; Zeng, W.; Wang, L.; Cheng, R.; Zhao, Z.; Huang, C.; Sun, Z.; Tao, P.; Wang, T.; Zhang, J.; et al. Validation of reliable safe harbor locus for efficient porcine transgenesis. Funct. Integr. Genom. 2022, 22, 553–563. [Google Scholar] [CrossRef]
- Guo, X.; Geng, L.; Jiang, C.; Yao, W.; Jin, J.; Liu, Z.; Mu, Y. Multiplexed genome engineering for porcine fetal fibroblasts with gRNA-tRNA arrays based on CRISPR/Cas9. Anim. Biotechnol. 2023, 34, 4703–4712. [Google Scholar] [CrossRef]
- Thierry, E.; Brennich, M.; Round, A.; Buisson, M.; Burmeister, W.P.; Hutin, S. Production and characterisation of Epstein–Barr virus helicase–primase complex and its accessory protein BBLF2/3. Virus Genes 2015, 51, 171–181. [Google Scholar] [CrossRef]
- Bonavita, C.M.; White, T.M.; Francis, J.; Farrell, H.E.; Davis-Poynter, N.J.; Cardin, R.D. The Viral G-Protein-Coupled Receptor Homologs M33 and US28 Promote Cardiac Dysfunction during Murine Cytomegalovirus Infection. Viruses 2023, 15, 711. [Google Scholar] [CrossRef]
- Nashan, B. Porcine cytomegalovirus in xenotransplantation: The new frontier in human transplantation? Health Care Sci. 2022, 1, 11–13. [Google Scholar] [CrossRef]
- Mueller, N.J. Porcine cytomegalovirus: A very unwelcome stowaway. Xenotransplantation 2022, 29, e12769. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mao, H.; Li, J.; Gao, M.; Liu, X.; Zhang, H.; Zhuang, Y.; He, T.; Zuo, W.; Bai, L.; Bao, J. Targeted Integration of siRNA against Porcine Cytomegalovirus (PCMV) Enhances the Resistance of Porcine Cells to PCMV. Microorganisms 2024, 12, 837. https://doi.org/10.3390/microorganisms12040837
Mao H, Li J, Gao M, Liu X, Zhang H, Zhuang Y, He T, Zuo W, Bai L, Bao J. Targeted Integration of siRNA against Porcine Cytomegalovirus (PCMV) Enhances the Resistance of Porcine Cells to PCMV. Microorganisms. 2024; 12(4):837. https://doi.org/10.3390/microorganisms12040837
Chicago/Turabian StyleMao, Hongzhen, Jinyang Li, Mengyu Gao, Xinmei Liu, Haohan Zhang, Yijia Zhuang, Tianyi He, Wei Zuo, Lang Bai, and Ji Bao. 2024. "Targeted Integration of siRNA against Porcine Cytomegalovirus (PCMV) Enhances the Resistance of Porcine Cells to PCMV" Microorganisms 12, no. 4: 837. https://doi.org/10.3390/microorganisms12040837