
A Comparative Transcriptome Analysis Unveils the Mechanisms of Response in Feather Degradation by Pseudomonas aeruginosa Gxun-7
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Strains, Chemicals, and Media
2.2. Determination of Keratinase Activity and Feather Degradation Rate and Amino Acid
2.3. Bacterial Growth and Sample Preparation
2.4. RNA Extraction and Sequencing
2.5. Analysis of RNA-Seq Data and Feather Degradation Mechanism
2.6. Analysis by qRT–PCR and Determination of Sulfur-Containing Compound Contents
2.7. Statistical Analysis
3. Results
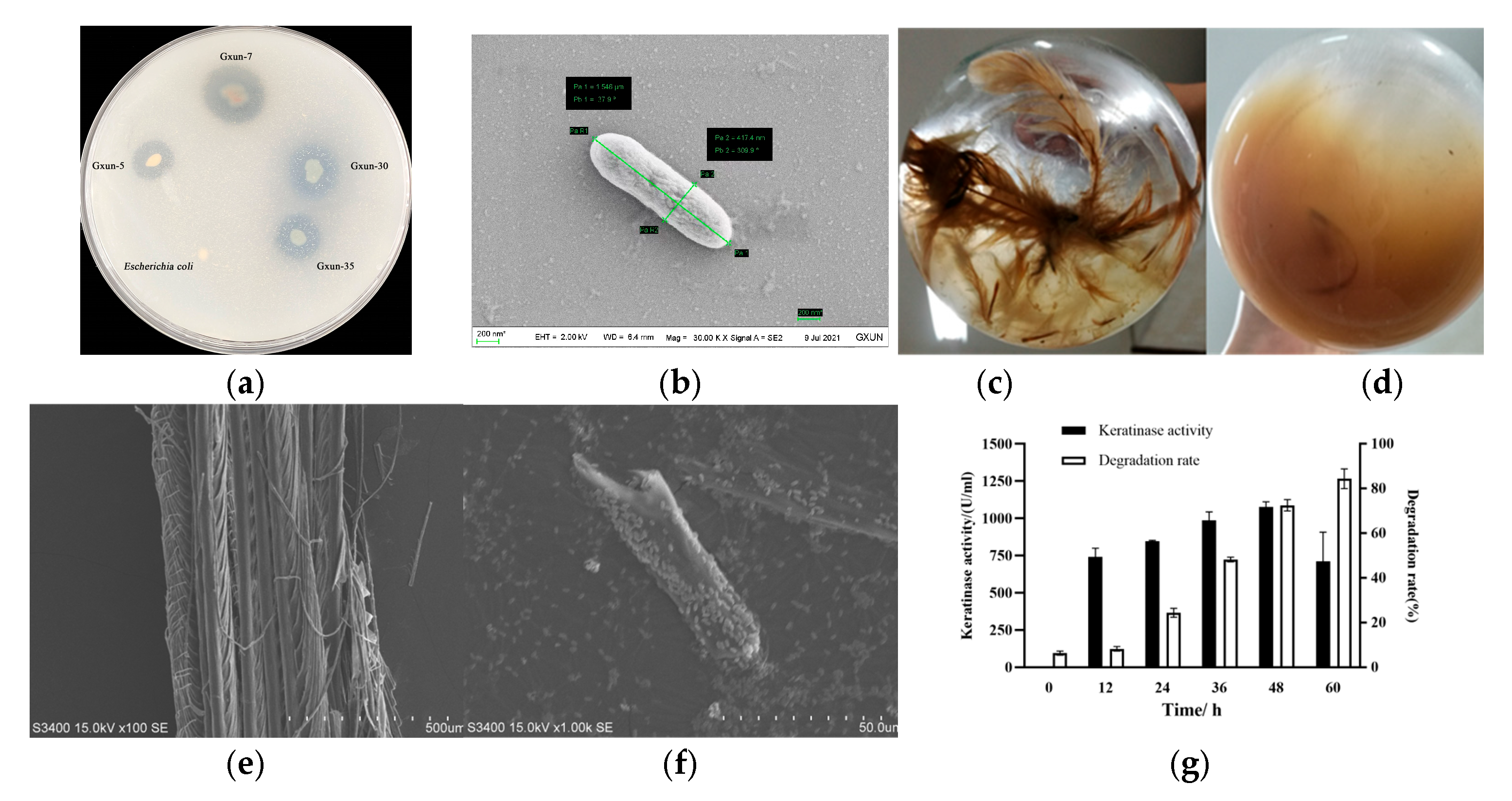
3.1. Effects of P. aeruginosa Gxun-7 on Growth and Feather Degradation
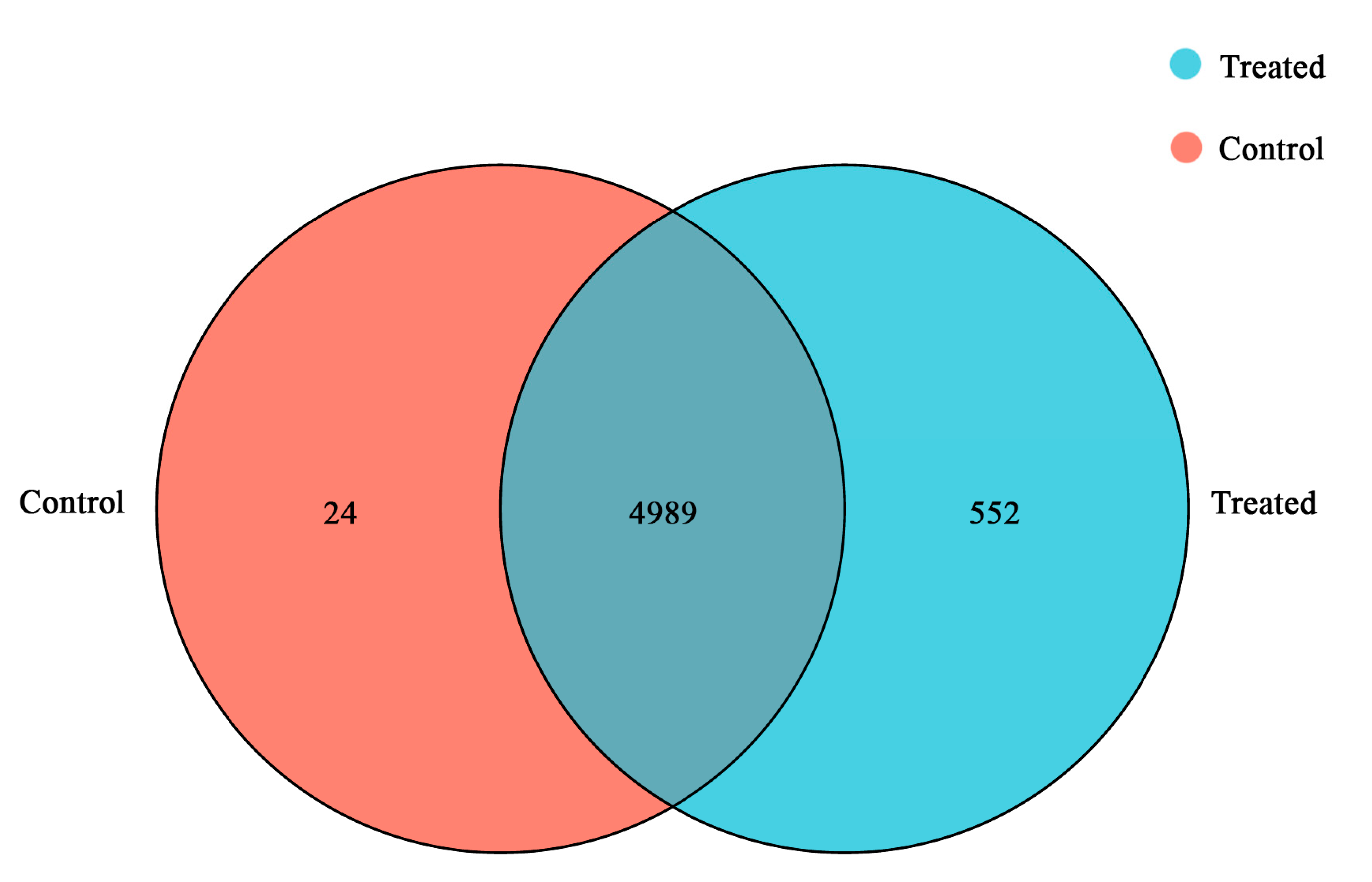
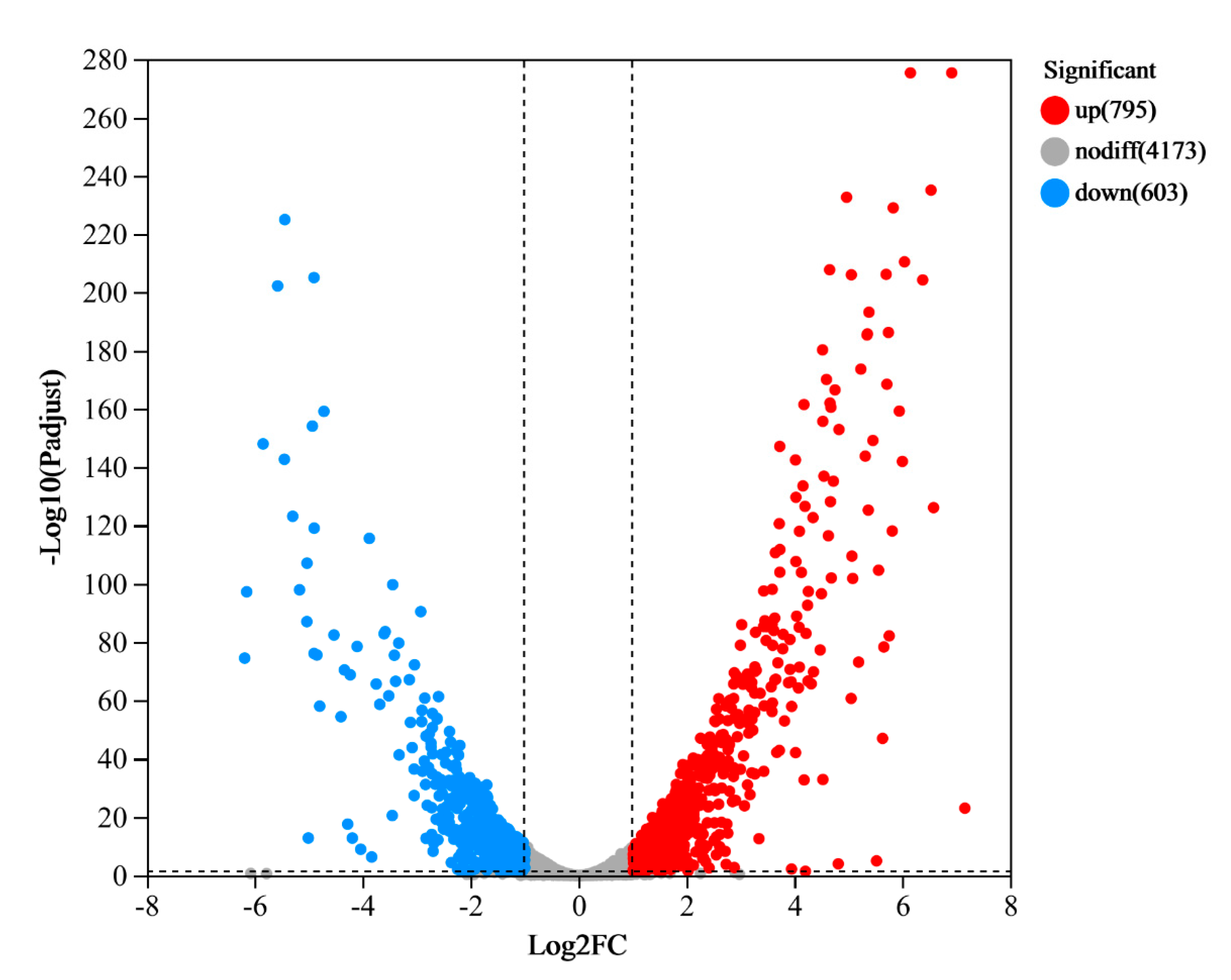
3.2. RNA-seq Analysis
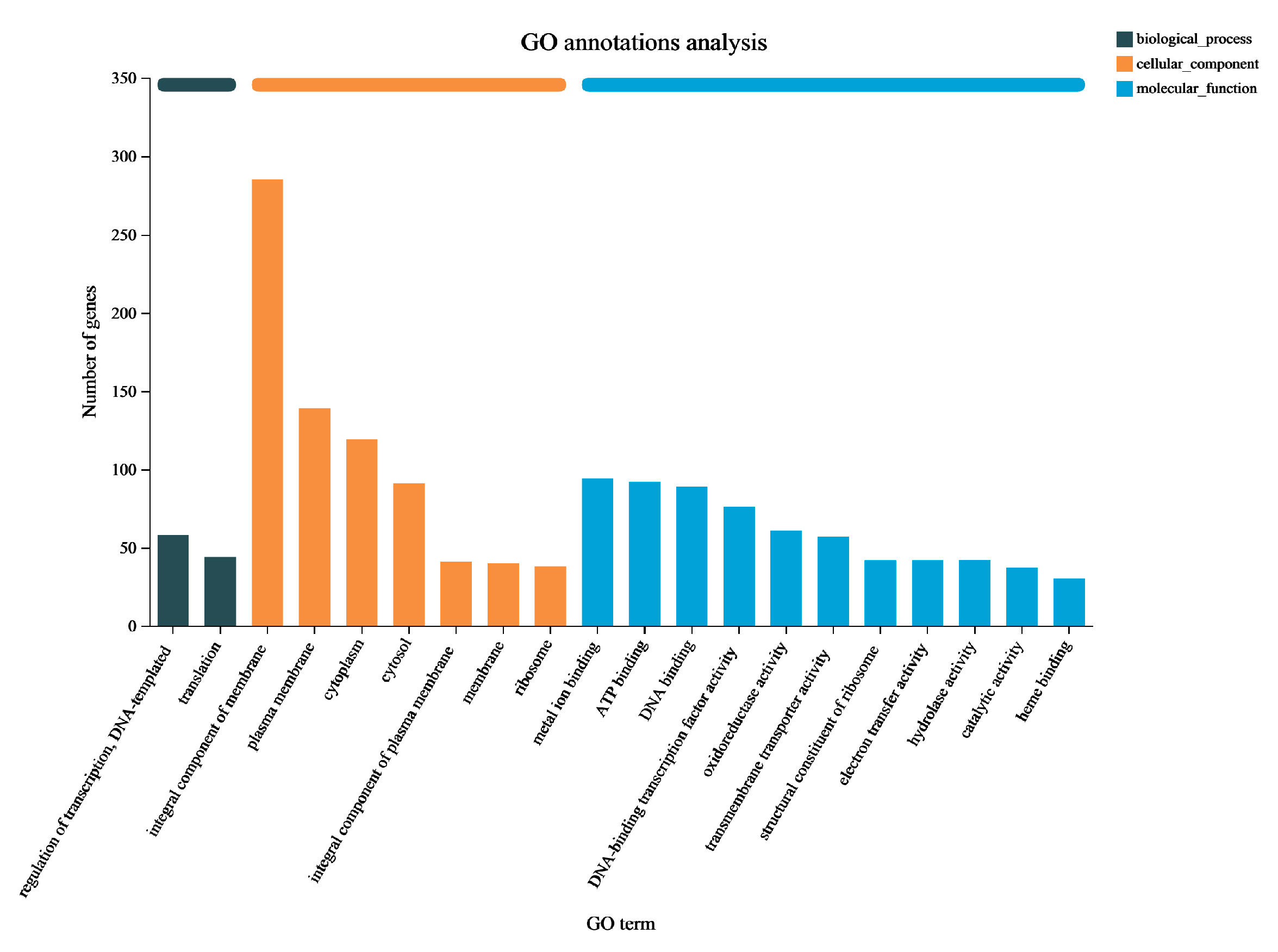
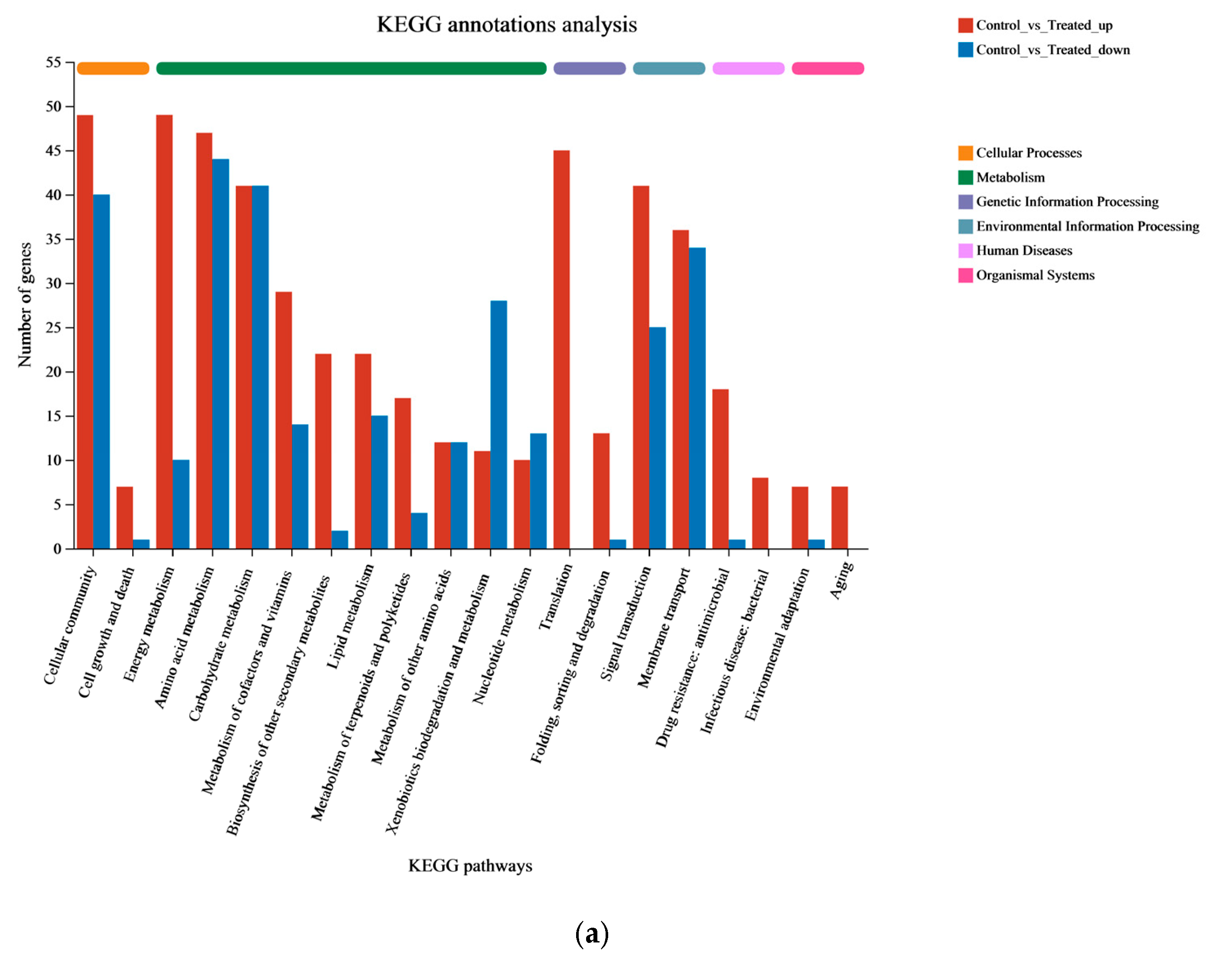
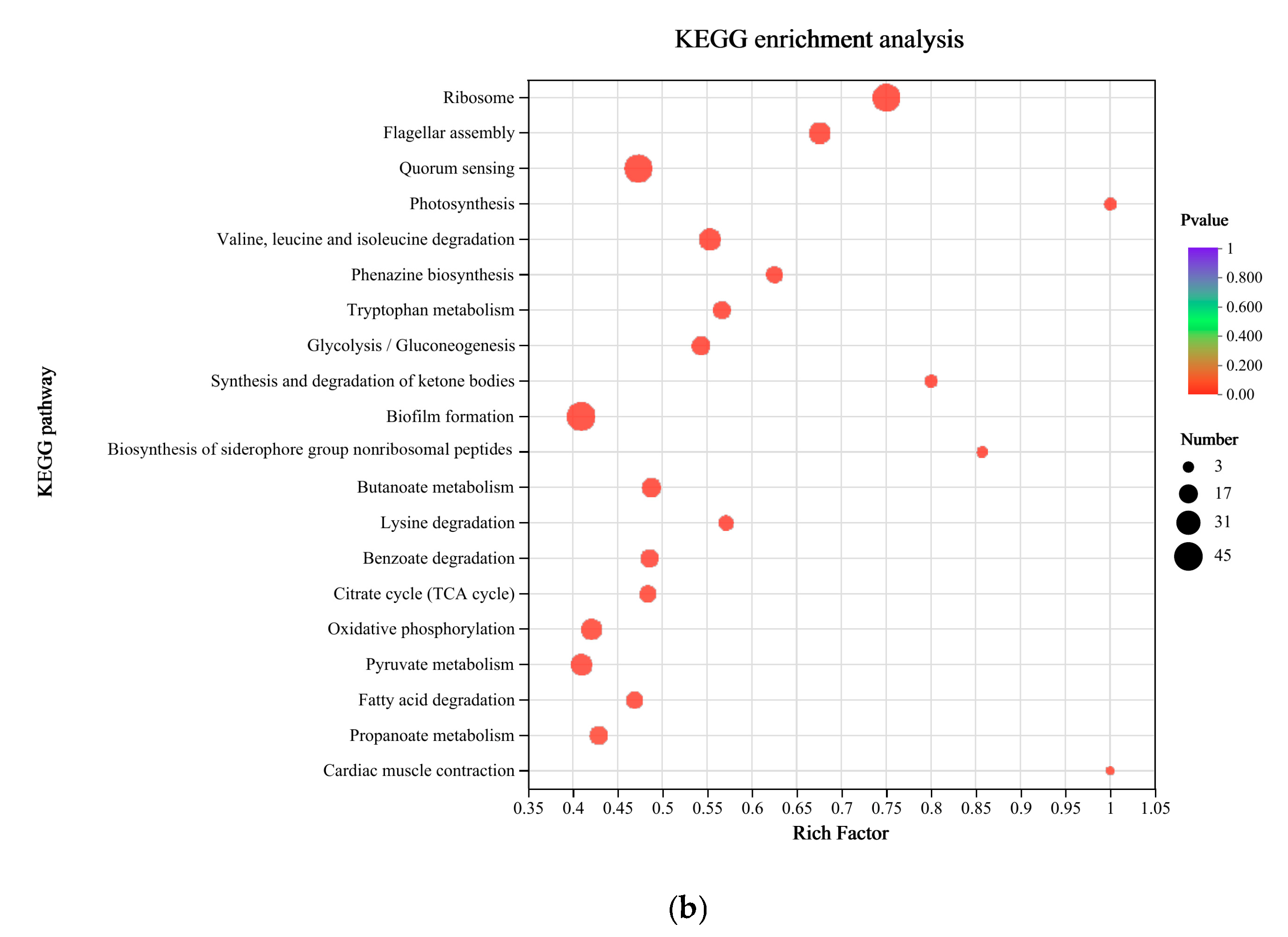
3.3. Functional Enrichment Analysis of DEGs in P. aeruginosa Gxun-7
3.4. Transcriptome Analysis of the Gene Expression Differences in Cells of CG and TG
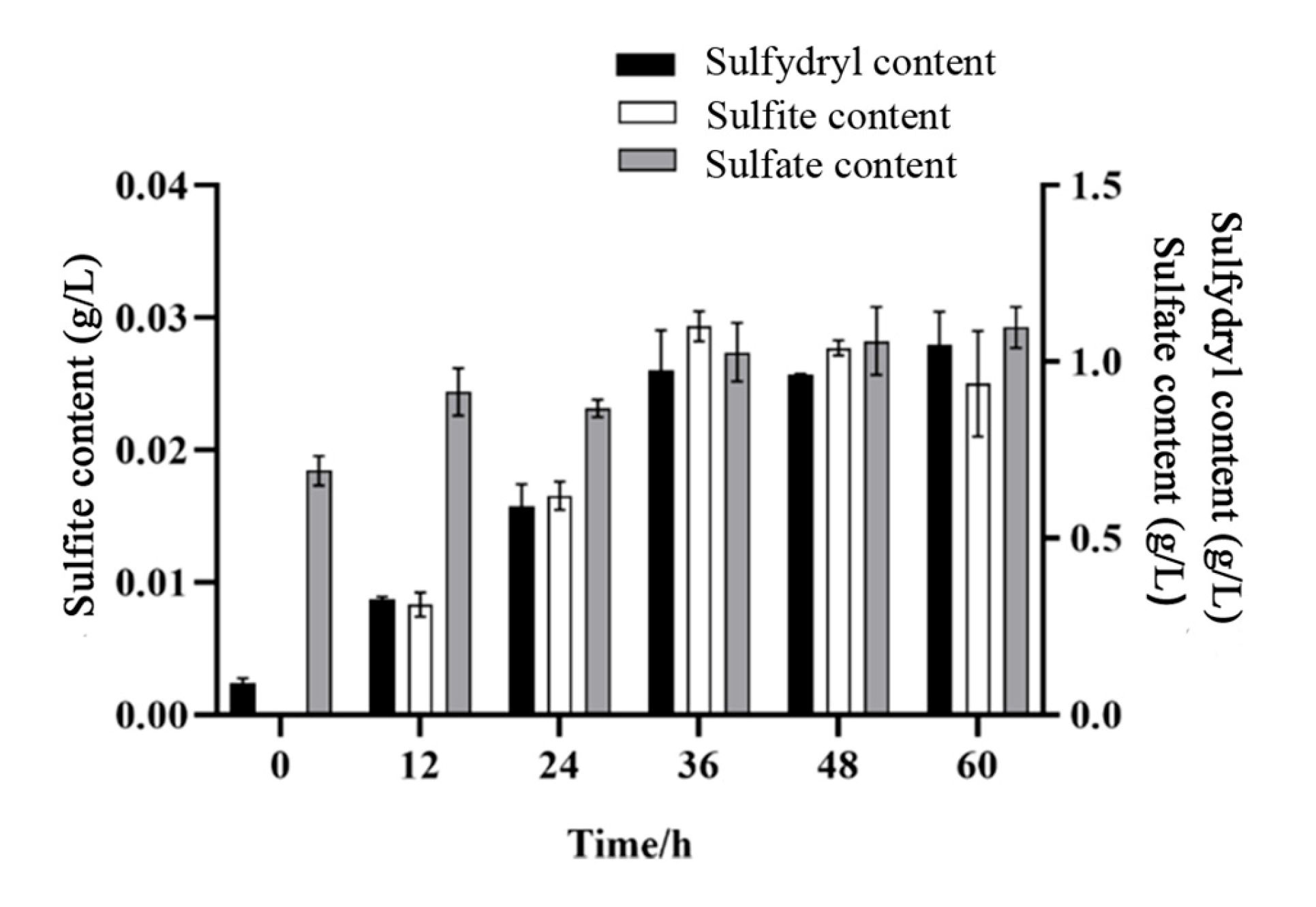
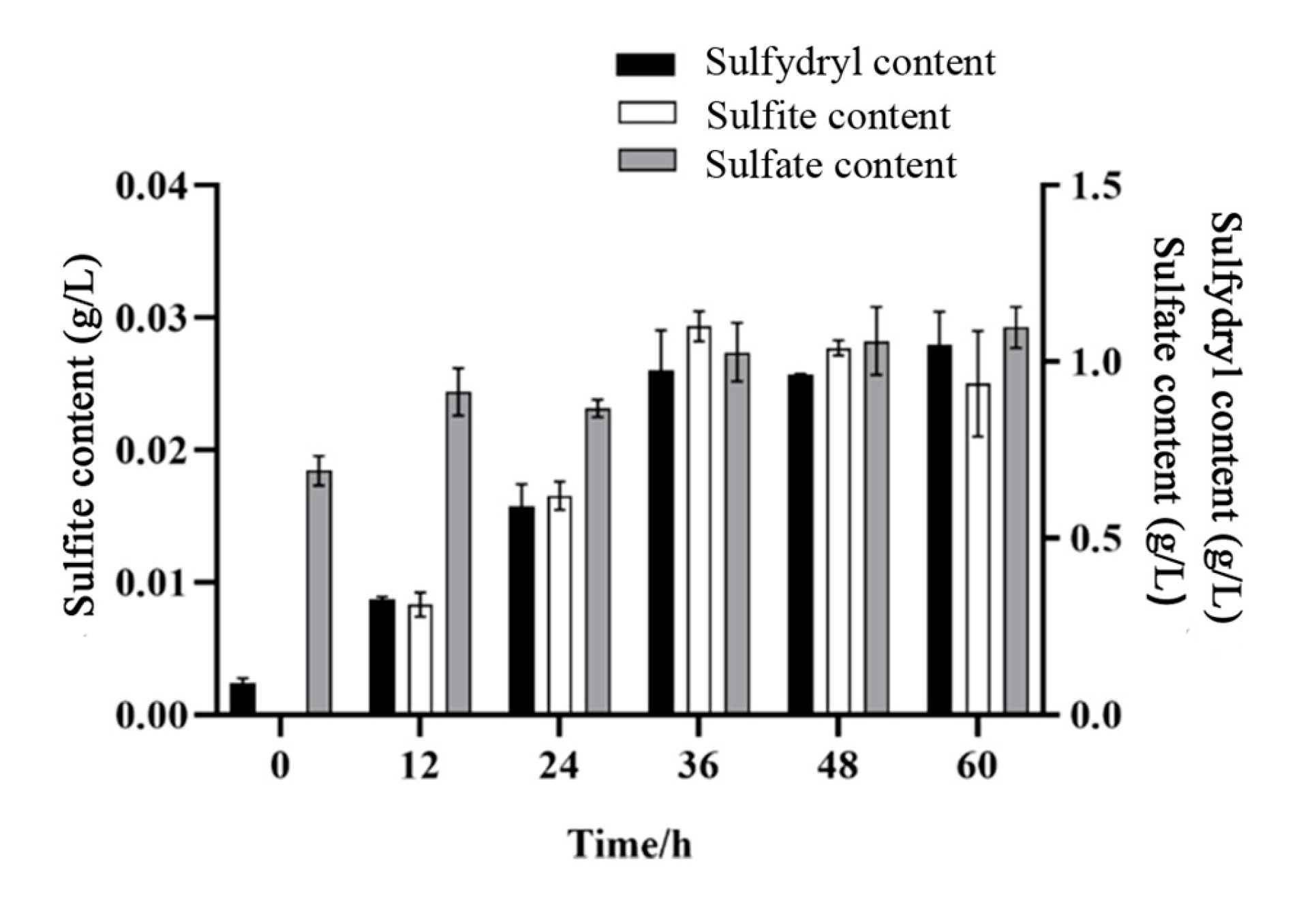
3.5. Results of Sulfate, Sulfite, and Sulfur-Containing Compounds
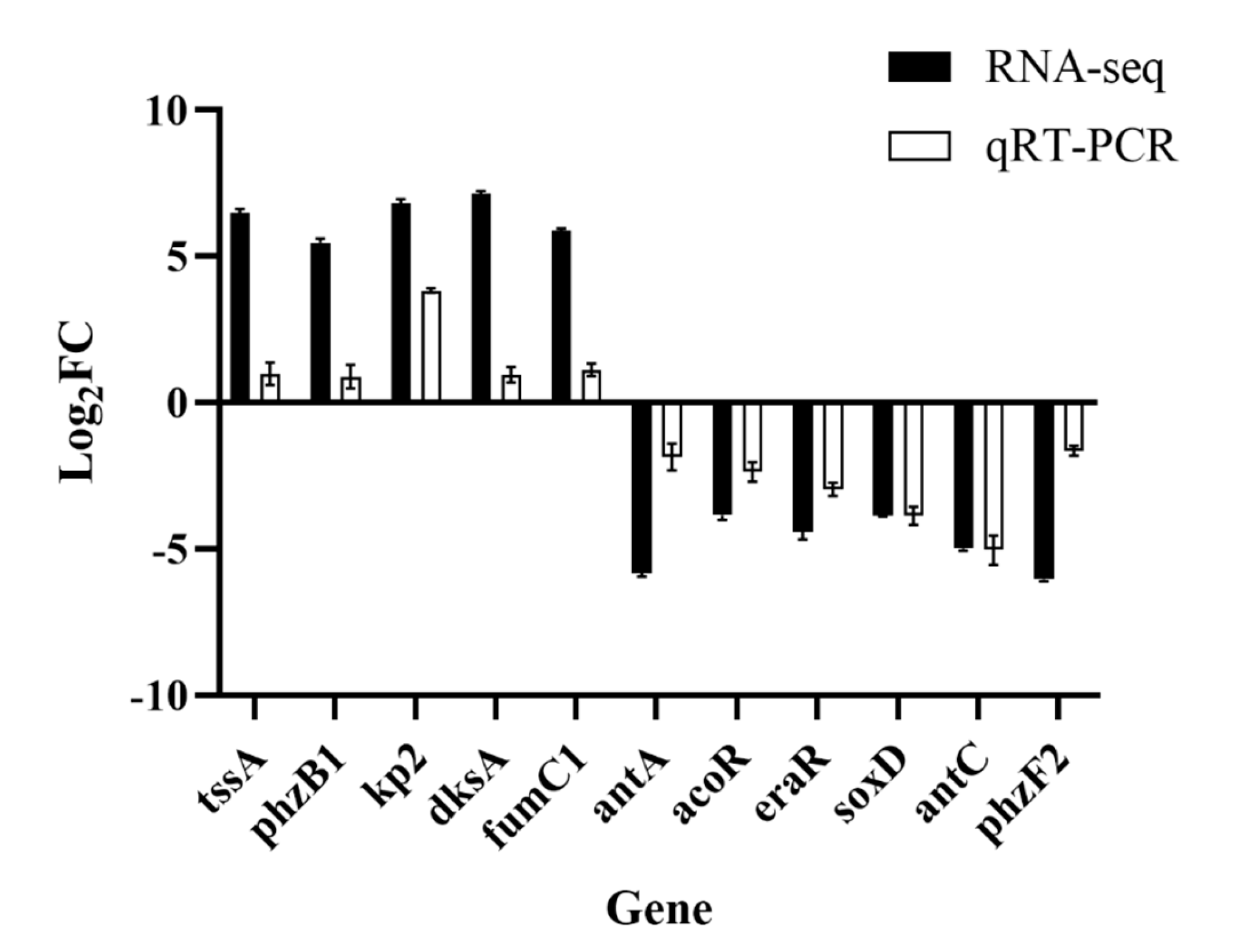
3.6. Validation by qRT–PCR
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Yamamura, S.; Morita, Y.; Hasan, Q.; Yokoyama, K.; Tamiya, E. Keratin Degradation: A Cooperative Action of Two Enzymes from Stenotrophomonas Sp. Biochem. Biophys. Res. Commun. 2002, 294, 1138–1143. [Google Scholar] [CrossRef]
- Fraser, R.D.B.; Parry, D.A.D. The Structural Basis of the Filament-Matrix Texture in the Avian/Reptilian Group of Hard β-Keratins. J. Struct. Biol. 2011, 173, 391–405. [Google Scholar] [CrossRef] [PubMed]
- Yusuf, I.; Ahmad, S.A.; Phang, L.Y.; Syed, M.A.; Shamaan, N.A.; Abdul Khalil, K.; Dahalan, F.A.; Shukor, M.Y. Keratinase Production and Biodegradation of Polluted Secondary Chicken Feather Wastes by a Newly Isolated Multi Heavy Metal Tolerant Bacterium-Alcaligenes Sp. AQ05-001. J. Environ. Manag. 2016, 183, 182–195. [Google Scholar] [CrossRef]
- Akram, F.; Aqeel, A.; Shoaib, M.; Haq, I.U.; Shah, F.I. Multifarious Revolutionary Aspects of Microbial Keratinases: An Efficient Green Technology for Future Generation with Prospective Applications. Environ. Sci. Pollut. Res. 2022, 29, 86913–86932. [Google Scholar] [CrossRef] [PubMed]
- Chukwunonso Ossai, I.; Shahul Hamid, F.; Hassan, A. Valorisation of Keratinous Wastes: A Sustainable Approach towards a Circular Economy. Waste Manag. 2022, 151, 81–104. [Google Scholar] [CrossRef] [PubMed]
- Callegaro, K.; Welter, N.; Daroit, D.J. Feathers as Bioresource: Microbial Conversion into Bioactive Protein Hydrolysates. Process Biochem. 2018, 75, 1–9. [Google Scholar] [CrossRef]
- Li, Q. Progress in Microbial Degradation of Feather Waste. Front. Microbiol. 2019, 10, 2717. [Google Scholar] [CrossRef] [PubMed]
- Lai, Y.; Wu, X.; Zheng, X.; Li, W.; Wang, L. Insights into the Keratin Efficient Degradation Mechanism Mediated by Bacillus Sp. CN2 Based on Integrating Functional Degradomics. Biotechnol. Biofuels 2023, 16, 59. [Google Scholar] [CrossRef] [PubMed]
- Yahaya, R.S.R.; Phang, L.Y.; Normi, Y.M.; Abdullah, J.O.; Ahmad, S.A.; Sabri, S. Feather-Degrading Bacillus Cereus HD1: Genomic Analysis and Its Optimization for Keratinase Production and Feather Degradation. Curr. Microbiol. 2022, 79, 166. [Google Scholar] [CrossRef] [PubMed]
- Brandelli, A. Bacterial Keratinases: Useful Enzymes for Bioprocessing Agroindustrial Wastes and Beyond. Food Bioprocess Technol. 2008, 1, 105–116. [Google Scholar] [CrossRef]
- Daroit, D.J.; Brandelli, A. A Current Assessment on the Production of Bacterial Keratinases. Crit. Rev. Biotechnol. 2014, 34, 372–384. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; Mao, X.; Zhang, J.; Du, G.; Chen, J. Biotransformation of Keratin Waste to Amino Acids and Active Peptides Based on Cell-Free Catalysis. Biotechnol. Biofuels 2020, 13, 61. [Google Scholar] [CrossRef] [PubMed]
- Ramnani, P.; Singh, R.; Gupta, R. Keratinolytic Potential of Bacillus Licheniformis RG1: Structural and Biochemical Mechanism of Feather Degradation. Can. J. Microbiol. 2005, 51, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Nnolim, N.E.; Okoh, A.I.; Nwodo, U.U. Bacillus Sp. FPF-1 Produced Keratinase with High Potential for Chicken Feather Degradation. Molecules 2020, 25, 1505. [Google Scholar] [CrossRef] [PubMed]
- Hamiche, S.; Mechri, S.; Khelouia, L.; Annane, R.; El Hattab, M.; Badis, A.; Jaouadi, B. Purification and Biochemical Characterization of Two Keratinases from Bacillus Amyloliquefaciens S13 Isolated from Marine Brown Alga Zonaria Tournefortii with Potential Keratin-Biodegradation and Hide-Unhairing Activities. Int. J. Biol. Macromol. 2019, 122, 758–769. [Google Scholar] [CrossRef] [PubMed]
- Li, Q. Structure, Application, and Biochemistry of Microbial Keratinases. Front. Microbiol. 2021, 12, 674345. [Google Scholar] [CrossRef] [PubMed]
- Abd El-Aziz, N.M.; Khalil, B.E.; Ibrahim, H.F. Enhancement of Feather Degrading Keratinase of Streptomyces Swerraensis KN23, Applying Mutagenesis and Statistical Optimization to Improve Keratinase Activity. BMC Microbiol. 2023, 23, 158. [Google Scholar] [CrossRef] [PubMed]
- Garg, S.K.; Alam, M.S.; Soni, V.; Radha Kishan, K.V.; Agrawal, P. Characterization of Mycobacterium Tuberculosis WhiB1/Rv3219 as a Protein Disulfide Reductase. Protein Expr. Purif. 2007, 52, 422–432. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; Zhang, J.; Du, G.; Chen, J. Keratin Waste Recycling Based on Microbial Degradation: Mechanisms and Prospects. ACS Sustain. Chem. Eng. 2019, 7, 9727–9736. [Google Scholar] [CrossRef]
- González, V.; Vargas-Straube, M.J.; Beys-da-Silva, W.O.; Santi, L.; Valencia, P.; Beltrametti, F.; Cámara, B. Enzyme Bioprospection of Marine-Derived Actinobacteria from the Chilean Coast and New Insight in the Mechanism of Keratin Degradation in Streptomyces Sp. G11C. Mar. Drugs 2020, 18, 537. [Google Scholar] [CrossRef]
- Wang, Z.; Chen, Y.; Yan, M.; Li, K.; Okoye, C.O.; Fang, Z.; Ni, Z.; Chen, H. Research Progress on the Degradation Mechanism and Modification of Keratinase. Appl. Microbiol. Biotechnol. 2023, 107, 1003–1017. [Google Scholar] [CrossRef] [PubMed]
- Shen, N.; Yang, M.; Xie, C.; Pan, J.; Pang, K.; Zhang, H.; Wang, Y.; Jiang, M. Isolation and Identification of a Feather Degrading Bacillus Tropicus Strain Gxun-17 from Marine Environment and Its Enzyme Characteristics. BMC Biotechnol. 2022, 22, 11. [Google Scholar] [CrossRef] [PubMed]
- Aktayeva, S.; Baltin, K.; Kiribayeva, A.; Akishev, Z.; Silayev, D.; Ramankulov, Y.; Khassenov, B. Isolation of Bacillus Sp. A5.3 Strain with Keratinolytic Activity. Biology 2022, 11, 244. [Google Scholar] [CrossRef] [PubMed]
- Peng, S.; Li, H.; Zhang, S.; Zhang, R.; Cheng, X.; Li, K. Isolation of a Novel Feather-Degrading Ectobacillus Sp. JY-23 Strain and Characterization of a New Keratinase in the M4 Metalloprotease Family. Microbiol. Res. 2023, 274, 127439. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Ma, J.; Li, S.; Chen, F.; Song, C.; Zhang, H.; Jiang, M.; Shen, N. Comparative Transcriptome Analysis Unravels the Response Mechanisms of Fusarium oxysporum f.sp. cubense to a Biocontrol Agent, Pseudomonas aeruginosa Gxun-2. Int. J. Mol. Sci. 2022, 23, 15432. [Google Scholar] [CrossRef] [PubMed]
- Arocho, A.; Chen, B.; Ladanyi, M.; Pan, Q. Validation of the 2-DeltaDeltaCt Calculation as an Alternate Method of Data Analysis for Quantitative PCR of BCR-ABL P210 Transcripts. Diagn. Mol. Pathol. 2006, 15, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhao, M. Simple Methods for Rapid Determination of Sulfite in Food Products. Food Control 2006, 17, 975–980. [Google Scholar] [CrossRef]
- Sakuragawa, A.; Nakayama, S.; Okutani, T. Flow-Injection Spectrophotometric Determination of Micro Amounts of Sulfate Ion in Surface- and Sea-Water Samples with a Barium Chromate Reaction Column. Anal. Sci. 1994, 10, 77–81. [Google Scholar] [CrossRef]
- Ellman, G. Tissue Sulfhydryl Groups. Arch. Biochem. Biophys. 1959, 82, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Veremeenko, E.G.; Maksimova, N.P. Activation of the Antioxidant Complex in Pseudomonas Aurantiaca—Producer of Phenazine Antibiotics. Microbiology 2010, 79, 439–444. [Google Scholar] [CrossRef]
- Ramakrishna Reddy, M.; Sathi Reddy, K.; Ranjita Chouhan, Y.; Bee, H.; Reddy, G. Effective Feather Degradation and Keratinase Production by Bacillus Pumilus GRK for Its Application as Bio-Detergent Additive. Bioresour. Technol. 2017, 243, 254–263. [Google Scholar] [CrossRef] [PubMed]
- Grumbt, M.; Monod, M.; Yamada, T.; Hertweck, C.; Kunert, J.; Staib, P. Keratin Degradation by Dermatophytes Relies on Cysteine Dioxygenase and a Sulfite Efflux Pump. J. Investig. Dermatol. 2013, 133, 1550–1555. [Google Scholar] [CrossRef] [PubMed]
- Hennicke, F.; Grumbt, M.; Lermann, U.; Ueberschaar, N.; Palige, K.; Böttcher, B.; Jacobsen, I.D.; Staib, C.; Morschhäuser, J.; Monod, M.; et al. Factors Supporting Cysteine Tolerance and Sulfite Production in Candida Albicans. Eukaryot. Cell 2013, 12, 604–613. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; Xu, P.; Song, Y.; Du, G.; Zhang, J.; Chen, J. Cysteine-Mediated Cyclic Metabolism Drives the Microbial Degradation of Keratin. ACS Sustain. Chem. Eng. 2021, 9, 9861–9870. [Google Scholar] [CrossRef]
- Kshetri, P.; Roy, S.S.; Sharma, S.K.; Singh, T.S.; Ansari, M.A.; Prakash, N.; Ngachan, S.V. Transforming Chicken Feather Waste into Feather Protein Hydrolysate Using a Newly Isolated Multifaceted Keratinolytic Bacterium Chryseobacterium Sediminis RCM-SSR-7. Waste Biomass Valoriz. 2019, 10, 1–11. [Google Scholar] [CrossRef]
- Sharma, R.; Gupta, R. Extracellular Expression of Keratinase Ker P from Pseudomonas Aeruginosa in E. Coli. Biotechnol. Lett. 2010, 32, 1863–1868. [Google Scholar] [CrossRef] [PubMed]
- Kunutsor, S.K. Gamma-Glutamyltransferase-Friend or Foe Within? Liver Int. 2016, 36, 1723–1734. [Google Scholar] [CrossRef]
- Li, Z.-W.; Liang, S.; Ke, Y.; Deng, J.-J.; Zhang, M.-S.; Lu, D.-L.; Li, J.-Z.; Luo, X.-C. The Feather Degradation Mechanisms of a New Streptomyces Sp. Isolate SCUT-3. Commun. Biol. 2020, 3, 191. [Google Scholar] [CrossRef] [PubMed]
- Inada, S.; Watanabe, K. Draft Genome Sequence of Meiothermus Ruber H328, Which Degrades Chicken Feathers, and Identification of Proteases and Peptidases Responsible for Degradation. Genome Announc. 2013, 1, e00176-13. [Google Scholar] [CrossRef] [PubMed]
- Mentel, M.; Ahuja, E.G.; Mavrodi, D.V.; Breinbauer, R.; Thomashow, L.S.; Blankenfeldt, W. Of Two Make One: The Biosynthesis of Phenazines. ChemBioChem 2009, 10, 2295–2304. [Google Scholar] [CrossRef] [PubMed]
- Mishra, S.; Imlay, J. Why Do Bacteria Use so Many Enzymes to Scavenge Hydrogen Peroxide? Arch. Biochem. Biophys. 2012, 525, 145–160. [Google Scholar] [CrossRef]
- Lalaouna, D.; Baude, J.; Wu, Z.; Tomasini, A.; Chicher, J.; Marzi, S.; Vandenesch, F.; Romby, P.; Caldelari, I.; Moreau, K. RsaC sRNA Modulates the Oxidative Stress Response of Staphylococcus Aureus during Manganese Starvation. Nucleic Acids Res. 2019, 47, 9871–9887. [Google Scholar] [CrossRef] [PubMed]
- Queiroux, C.; Bonnet, M.; Saraoui, T.; Delpech, P.; Veisseire, P.; Rifa, E.; Moussard, C.; Gagne, G.; Delbès, C.; Bornes, S. Dialogue between Staphylococcus Aureus SA15 and Lactococcus Garvieae Strains Experiencing Oxidative Stress. BMC Microbiol. 2018, 18, 193. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Si, M.; Song, Y.; Zhu, W.; Gao, F.; Wang, Y.; Zhang, L.; Zhang, W.; Wei, G.; Luo, Z.-Q.; et al. Type VI Secretion System Transports Zn2+ to Combat Multiple Stresses and Host Immunity. PLoS Pathog. 2015, 11, e1005020. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.-J.; Kuo, T.-Y.; Hsieh, F.-C.; Chen, P.-Y.; Wang, C.-S.; Shih, Y.-L.; Lai, Y.-M.; Liu, J.-R.; Yang, Y.-L.; Shih, M.-C. Involvement of Type VI Secretion System in Secretion of Iron Chelator Pyoverdine in Pseudomonas Taiwanensis. Sci. Rep. 2016, 6, 32950. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Zhang, W.; Cheng, J.; Yang, X.; Zhu, K.; Wang, Y.; Wei, G.; Qian, P.-Y.; Luo, Z.-Q.; Shen, X. A Pseudomonas T6SS Effector Recruits PQS-Containing Outer Membrane Vesicles for Iron Acquisition. Nat. Commun. 2017, 8, 14888. [Google Scholar] [CrossRef]
- Si, M.; Zhao, C.; Burkinshaw, B.; Zhang, B.; Wei, D.; Wang, Y.; Dong, T.G.; Shen, X. Manganese Scavenging and Oxidative Stress Response Mediated by Type VI Secretion System in Burkholderia Thailandensis. Proc. Natl. Acad. Sci. USA 2017, 114, E2233–E2242. [Google Scholar] [CrossRef] [PubMed]
- Filloux, A. The Type VI Secretion System: A Tubular Story. EMBO J. 2009, 28, 309–310. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Zou, Y.; She, P.; Wu, Y. Composition, Function, and Regulation of T6SS in Pseudomonas Aeruginosa. Microbiol. Res. 2015, 172, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Miranda, S.W.; Asfahl, K.L.; Dandekar, A.A.; Greenberg, E.P. Pseudomonas Aeruginosa Quorum Sensing. Adv. Exp. Med. Biol. 2022, 1386, 95–115. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Gene Description | Log2FC |
|---|---|---|
| tssA | Type Ⅵ secretion system protein | 7.15 |
| kp2 | Keratinase KP2 | 6.91 |
| ahpF | Alkyl hydroperoxide reductase subunit F | 6.15 |
| phzB1 | Phenazine biosynthesis protein | 5.76 |
| dhcB | Dehydrocarnitine CoA transferase C subunit B | 5.71 |
| phzA1 | Phenazine biosynthesis protein | 5.63 |
| phzC2 | Phenazine biosynthesis protein | 5.52 |
| dhcA | Dehydrocarnitine CoA transferase C subunit A | 5.34 |
| pchD | Pyochelin biosynthesis protein | 4.97 |
| sodM | Superoxide dismutase | 4.82 |
| phzF1 | Phenazine biosynthesis protein | 4.81 |
| phzM | Phenazine-specific methyltransferase | 4.72 |
| katA | Catalase | 4.68 |
| pchE | Dihydroaeruginoic acid synthetase | 4.65 |
| phzD1 | Phenazine biosynthesis protein | 4.53 |
| katB | Catalase | 4.21 |
| chiC | Chitinase | 4.12 |
| lasA | Protease precursor | 2.87 |
| sir2 | Sulfite reductase | 1.61 |
| gor | Glutathione reductase | 1.18 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, C.; Liu, R.; Yin, D.; Xie, C.; Liang, Y.; Yang, D.; Jiang, M.; Zhang, H.; Shen, N. A Comparative Transcriptome Analysis Unveils the Mechanisms of Response in Feather Degradation by Pseudomonas aeruginosa Gxun-7. Microorganisms 2024, 12, 841. https://doi.org/10.3390/microorganisms12040841
Song C, Liu R, Yin D, Xie C, Liang Y, Yang D, Jiang M, Zhang H, Shen N. A Comparative Transcriptome Analysis Unveils the Mechanisms of Response in Feather Degradation by Pseudomonas aeruginosa Gxun-7. Microorganisms. 2024; 12(4):841. https://doi.org/10.3390/microorganisms12040841
Chicago/Turabian StyleSong, Chaodong, Rui Liu, Doudou Yin, Chenjie Xie, Ying Liang, Dengfeng Yang, Mingguo Jiang, Hongyan Zhang, and Naikun Shen. 2024. "A Comparative Transcriptome Analysis Unveils the Mechanisms of Response in Feather Degradation by Pseudomonas aeruginosa Gxun-7" Microorganisms 12, no. 4: 841. https://doi.org/10.3390/microorganisms12040841