
Genomic Characterization and Molecular Detection of Rehmannia Allexivirus Virus, a Novel Allexivirus Infecting Rehmannia glutinosa

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Material
2.2. High-Throughput Sequencing and Data Analysis
2.3. Amplification of the Full-Length ReAV Genome
2.4. Genome End Sequence Amplification
2.5. Recombination Analysis of Nine ReAV Isolates
2.6. RT-PCR Detection of R. glutinosa Samples
2.7. Sequence Assembly and ReAV Full Sequence Analysis
3. Results
3.1. HTS Data Analysis
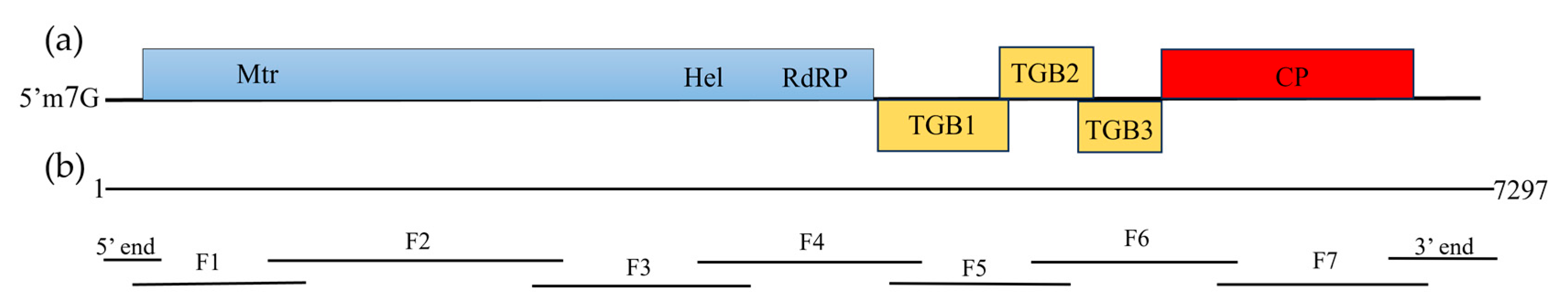
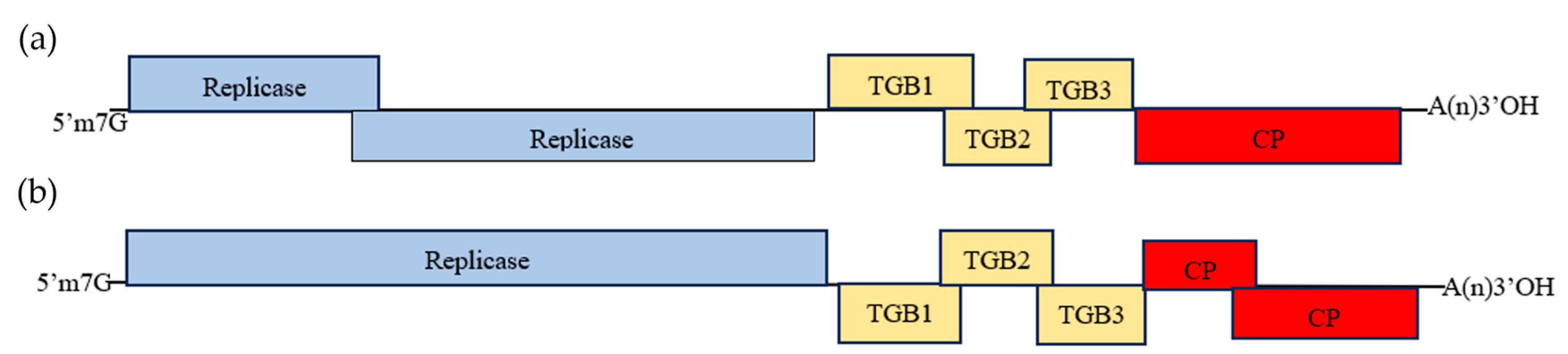
3.2. Amplification and Analysis of the Complete Genome Sequence of the New Virus
3.3. Molecular Variation of ReAV Genome Sequences
3.4. Recombination Analysis of ReAV Genome
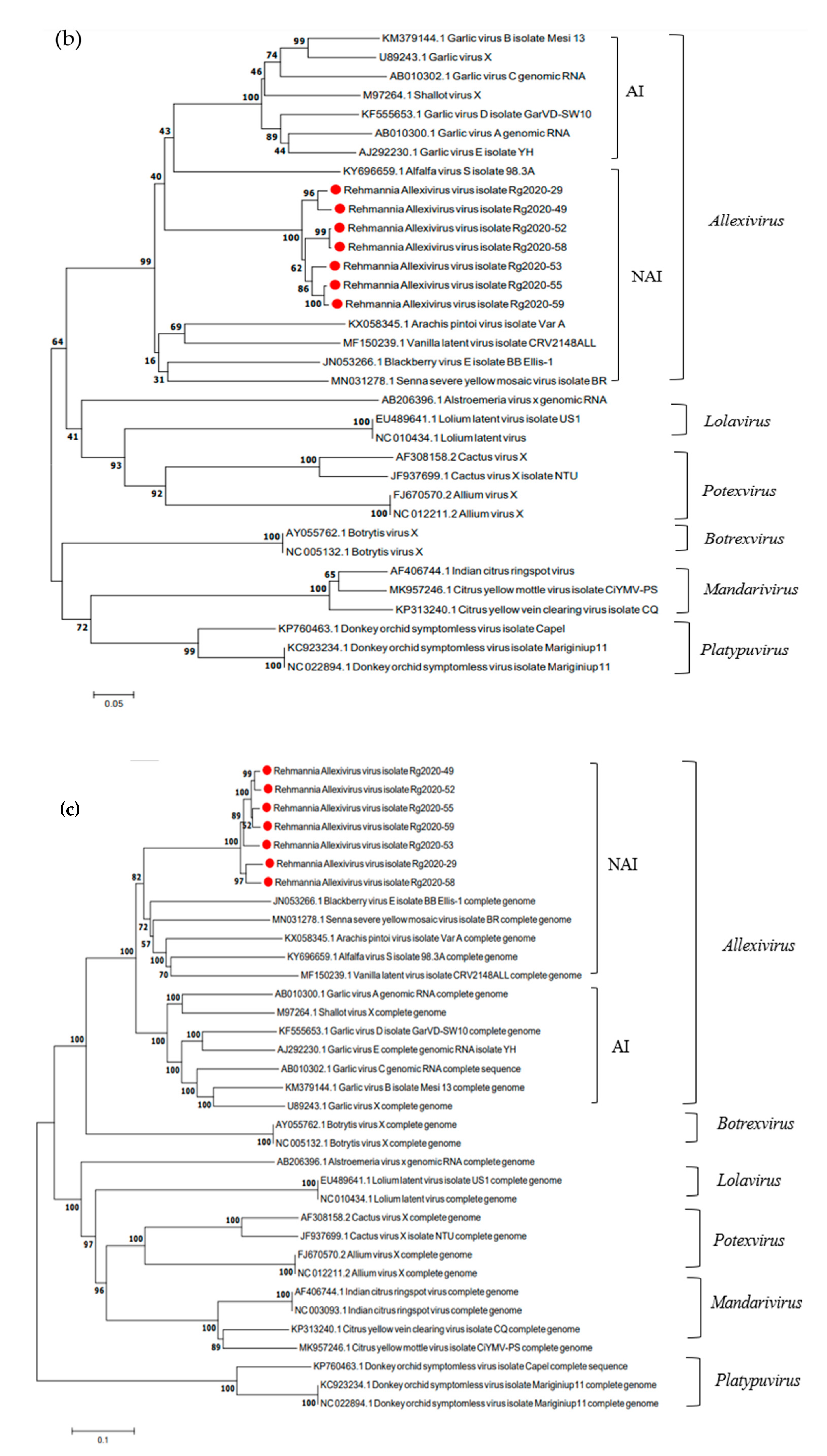
3.5. Phylogenetic Analysis of ReAV Isolates and Other Allexivirus Species
3.6. RT-PCR Detection of ReAV in R. glutinosa Samples
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhang, B.; Li, X.; Wang, F.; Li, M.; Zhang, J.; Gu, L.; Zhang, L.; Tu, W.; Zhang, Z. Assaying the potential autotoxins and microbial community associated with Rehmannia glutinosa replant problems based on its ‘autotoxic circle’. Plant Soil 2016, 407, 307–322. [Google Scholar] [CrossRef]
- Du, J.F.; Nian, W.K.; Zhou, Z.J.; Dou, T.; Song, G.H.; Gu, L.; Liu, X.Y. Leaf Spot Disease Caused by Alternaria alternata on Rehmannia glutinosa in China. Plant Dis. 2020, 104, 3059. [Google Scholar] [CrossRef]
- Yoshie, Y.; Ando, H.; Yoshihara, K.; Fukuda, K.; Sasaki, Y. Study on morphological and genetic diversity of Rehmannia glutinosa cultivated in Japan. J. Nat. Med. 2022, 76, 352–366. [Google Scholar] [CrossRef] [PubMed]
- Ahn, S.W.; Kim, Y.G.; Kim, M.H.; Lee, H.Y.; Seong, N.S. Comparison of biological activities on Rehmannia radix and R. radix Preparata produced in Korea. Korean J. Med. Crop Sci. 1999, 7, 257–262. [Google Scholar]
- Lim, S.; Zhao, F.; Yoo, R.H.; Igori, D.; Jeong, J.C.; Lee, H.S.; Kwak, S.S.; Moon, J.S. Complete genome sequence of Rehmannia mosaic virus infecting Rehmannia glutinosa in South Korea. Genome Announc. 2016, 4, 1110–1128. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Zhuang, X.; Guo, X.; Xu, H.; He, Z.; Chen, J. Cucurbit chlorotic yellows virus infecting Rehmannia glutinosa was detected in China. Plant Dis. 2021, 105, 3310. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.H.; Wang, F.; Lu, C.T.; Wang, F.L.; Wen, Y.; Liu, Y.X.; Gao, S.X.; Qi, W.P.; Li, X.M.; Yang, J. First Report of Tobacco Mild Green Mosaic Virus Infecting Rehmannia glutinosa in China. Plant Dis. 2022, 106, 3004. [Google Scholar] [CrossRef] [PubMed]
- Kwon, S.J.; Kim, Y.B.; Back, C.K.; Chung, B.N. First report of a mixed infection of Youcai mosaic virus and Rehmannia mosaic virus in Rehmannia glutinosa in Korea. Plant Dis. 2018, 102, 462. [Google Scholar] [CrossRef]
- Kwak, H.R.; Kim, M.; Kim, J.; Choi, H.S.; Seo, J.K.; Ko, S.J.; Kim, J.S. First report of Plantago asiatica mosaic virus in Rehmannia glutinosa in Korea. Plant Dis. 2018, 102, 1046. [Google Scholar] [CrossRef]
- Qin, Y.H.; Wen, Y.; Gao, S.X.; Zhang, D.S.; Liu, Y.X.; Liu, Y.K.; Li, S.; Zhao, Z.W.; Wang, F.L.; Wang, F.; et al. Identification of the pathogens of Rehmannia glutinosa viral disease and molecular variation analysis of the major viruses. Acta Phytopathol. Sin. 2023, 1–9. [Google Scholar]
- Matsumoto, M.; Shoyama, Y.; Nishioka, I.; Iwai, H.; Wakimoto, S. Identification of viruses infected in Rehmannia glutinosa Libosch. var purpurea Makino and effect of virus infection on root yield and iridoid glycoside contents. Plant Cell Rep. 1989, 7, 636–638. [Google Scholar] [CrossRef] [PubMed]
- Martelli, G.P.; Adams, M.J.; Kreuze, J.F.; Dolja, V.V. Family Flexiviridae: A case study in virion and genome plasticity. Annu. Rev. Phytopathol. 2007, 45, 73–100. [Google Scholar] [CrossRef]
- Kreuze, J.F.; Vaira, A.M.; Menzel, W.; Candresse, T.; Zavriev, S.K.; Hammond, J.; Ryu, K.H. ICTV virus taxonomy profile: Alphaflexiviridae. J. Gen. Virol. 2020, 101, 699–700. [Google Scholar] [CrossRef] [PubMed]
- Morozov, S.Y.; Solovyev, A.G. Phylogenetic relationship of some “accessory” helicases of plant positive-stranded RNA viruses: Toward understanding the evolution of triple gene block. Front. Microbiol. 2015, 6, 508. [Google Scholar] [CrossRef] [PubMed]
- Sumi, S.; Matsumi, T.; Tsuneyoshi, T. Complete nucleotide sequences of garlic viruses A and C, members of the newly ratified genus Allexivirus. Arch. Virol. 1999, 144, 1819–1826. [Google Scholar] [CrossRef] [PubMed]
- Grisoni, M.; Marais, A.; Filloux, D.; Saison, A.; Faure, C.; Julian, C.; Theil, S.; Contreras, S.; Teycheney, P.-Y.; Roumagnac, P.; et al. Two novel Alphaflexiviridae members revealed by deep sequencing of the Vanilla (Orchidaceae) virome. Arch. Virol. 2017, 162, 3855–3861. [Google Scholar] [CrossRef] [PubMed]
- Razvjazkina, G.M. Das Zwiebelmosaikvirus und seine Verbreitung im Freiland. Tagungs-Berichte Der Dtsch. Akad. Der Landwirtschaftswissenschaften 1971, 115, 69–76. [Google Scholar]
- Van Dijk, P.; Verbeek, M.; Bos, I. Mite-borne virus isolates from cultivated Allium species, and their classification into two new rymoviruses in the family Potyviridae. Neth. J. Plant Pathol. 1991, 97, 381–399. [Google Scholar] [CrossRef]
- Fidan, H.; Çağlar, B.K.; Baloğlu, S.; Yılmaz, M.A. Urginea maritime (L.) is a new host of Allexivirus group on onion and garlic plants in Turkey. In Proceedings of the XI International Symposium on Flower Bulbs and Herbaceous Perennials, Antalya, Turkey, 28 March–1 April 2012; Volume 1002, pp. 309–312. [Google Scholar]
- Sabanadzovic, S.; Abou Ghanem-Sabanadzovic, N.; Tzanetakis, I.E. Blackberry virus E: An unusual flexivirus. Arch. Virol. 2011, 156, 1665–1669. [Google Scholar] [CrossRef]
- Alves, T.M.; de Novaes, Q.S.; de Paula, A.; Camelo-García, V.M.; Nagata, T.; Silva, J.M.F.; Rezende, J.A.M.; Kitajima, E.W. Near-complete genome sequence and biological properties of an allexivirus found in Senna rizzinii in Brazil. Arch. Virol. 2020, 165, 1463–1467. [Google Scholar] [CrossRef]
- Nemchinov, L.G.; Grinstead, S.C.; Mollov, D.S. Alfalfa virus S, a new species in the family Alphaflexiviridae. PLoS ONE 2017, 12, e0178222. [Google Scholar] [CrossRef]
- Sánchez, P.A.G.; Mesa, H.J.; Montoya, M.M. Next generation sequence analysis of the forage peanut (Arachis pintoi) virome. Rev. Fac. Nac. De Agron. Medellín 2016, 69, 7881–7891. [Google Scholar] [CrossRef]
- Zhang, Z.C.; Zhang, L.F.; Qiao, Q.; Wang, Y.J.; Jin, X.L. Identification of viral pathogens of Rehmannia glutinosa disease in Henan Province. Acta Phytopathol. Sin. 2004, 34, 395–399. [Google Scholar]
- Beris, D.; Malandraki, I.; Kektsidou, O.; Varveri, C. First Report of Eggplant Mottled Crinkle Virus Infecting Eggplant in Greece. Plant Dis. 2021, 105, 3769. [Google Scholar] [CrossRef]
- Minicka, J.; Taberska, A.; Borodynko-Filas, N.; Kaźmińska, K.; Bartoszewski, G.; Hasiów-Jaroszewska, B. Viruses infecting Capsicum crops in Poland and molecular characterization of newly detected bell pepper alphaendornavirus (BPEV). Crop Prot. 2024, 176, 106478. [Google Scholar] [CrossRef]
- Qin, Y.H.; Wang, F.L.; Cai, L.; Gao, S.X.; Wen, Y.; Liu, Y.X.; Li, S.J.; Lu, C.T.; Wang, F. First Report of Youcai mosaic Virus Infecting Yam in China. Plant Dis. 2023, 107, 1247. [Google Scholar] [CrossRef]
- De Lima, M.O.; Ferro, M.M.; Ramos-Sobrinho, R.; Melo, F.L.; Nagata, T.; Assunção, I.P.; Lima, G.S.A.; Silva, S.J. DNA virome of Brazilian sugarcane germplasm via high-throughput sequencing reveals divergent badnavirus species. Trop. Plant Pathol. 2023, 48, 713–719. [Google Scholar] [CrossRef]
- Chirkov, S.; Sheveleva, A.; Tsygankova, S.; Slobodova, N.; Sharko, F.; Petrova, K.; Mitrofanova, I. Whole Genome Characterization of Prunus Necrotic Ringspot Virus and Prune Dwarf Virus Infecting Stone Fruits in Russia. Horticulturae 2023, 9, 941. [Google Scholar] [CrossRef]
- Diaz-Lara, A.; Stevens, K.; Aguilar-Molina, V.H.; Fernández-Cortés, J.M.; Chabacano León, V.M.; De Donato, M.; Sharma, A.; Erickson, T.M.; Al Rwahnih, M. High-Throughput Sequencing of Grapevine in Mexico Reveals a High Incidence of Viruses including a New Member of the Genus Enamovirus. Viruses 2023, 15, 1561. [Google Scholar] [CrossRef]
- Yan, D.; Han, K.; Chen, Y.; Ma, C.; Hu, S.; Zhao, W.; Wang, F. Complete genome sequence of triticum yellow stripe virus, a new polerovirus infecting wheat (Triticum aestivum) in China. Arch. Virol. 2023, 168, 146. [Google Scholar] [CrossRef]
- Tang, L.; Song, L.; Lin, C.; Wang, B.; Lin, J.; Gao, C.; Wang, A. Complete nucleotide sequence of a novel partitivirus from Brassica campestris L. ssp. Chinensis. Arch. Virol. 2021, 166, 1775–1778. [Google Scholar] [CrossRef] [PubMed]
- Lecoq, H.; Verdin, E.; Tepfer, M.; Wipf-Scheibel, C.; Millot, P.; Dafalla, G.; Desbiez, C. Characterization and occurrence of squash chlorotic leaf spot virus, a tentative new torradovirus infecting cucurbits in Sudan. Arch. Virol. 2016, 161, 1651–1655. [Google Scholar] [CrossRef] [PubMed]
- ICTV. Virus Taxonomy: 2018b Release. International Committee on Taxonomy of Viruses. 2018. Available online: https://talk.ictvonline.org/taxonomy/ (accessed on 8 May 2019).
- Syler, J. Facilitative and antagonistic interactions between plant viruses in mixed infections. Mol. Plant Pathol. 2012, 13, 204–216. [Google Scholar] [CrossRef] [PubMed]
- Rentería-Canett, I.; Xoconostle-Cázares, B.; Ruiz-Medrano, R.; Rivera-Bustamante, R.F. Geminivirus mixed infection on pepper plants: Synergistic interaction between PHYVV and PepGMV. Virol. J. 2011, 8, 104. [Google Scholar] [CrossRef] [PubMed]
- Goodman, R.M.; Ross, A.F. Enhancement by Potato virus Y of Potato virus X synthesis in doubly infected tobacco depends on the timing of invasion by the viruses. Virology 1974, 58, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Turina, M.; Medina, V.; Falk, B.W. Synergistic interaction between the Potyvirus, Turnip mosaic virus and the Crinivirus, Lettuce infectious yellows virus in plants and protoplasts. Virus Res. 2009, 144, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Karyeija, R.F.; Kreuze, J.F.; Gibson, R.W.; Valkonen, J.P.T. Synergistic interactions of a potyvirus and a phloem-limited crinivirus in sweet potato plants. Virology 2000, 269, 26–36. [Google Scholar] [CrossRef] [PubMed]
- Cafrune, E.E.; Perotto, M.C.; Conci, V.C. Effect of two Allexivirus isolates on garlic yield. Plant Dis. 2006, 90, 898–904. [Google Scholar] [CrossRef]
- Perotto, M.C.; Cafrune, E.E.; Conci, V.C. The effect of additional viral infections on garlic plants initially infected with Allexiviruses. Eur. J. Plant Pathol. 2010, 126, 489–495. [Google Scholar] [CrossRef]
- Celli, M.G.; Perotto, M.C.; Buraschi, D.; Conci, V.C. Biological and molecular characterization of Garlic virus D and its effects on yields of garlic. Acta Hortic. 2016, 1143, 193–200. [Google Scholar] [CrossRef]
- Conci, V.C.; Canavelli, A.; Lunello, P.; Di Rienzo, J.; Nome, S.F.; Zumelzu, G.; Italia, R. Yield losses associated with virus-infected garlic plants during five successive years. Plant Dis. 2003, 87, 1411–1415. [Google Scholar] [CrossRef] [PubMed]
- Mansouri, F.; Ryšánek, P. Allexivirus: Review and perspectives. Phytopathol. Mediterr. 2021, 60, 389–402. [Google Scholar] [CrossRef]
- Song, S.I.; Song, J.T.; Kim, C.H.; Lee, J.S.; Choi, Y.D. Molecular characterization of the garlic virus X genome. J. Gen. Virol. 1998, 79, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Lezzhov, A.A.; Gushchin, V.A.; Lazareva, E.A.; Vishnichenko, V.K.; Morozov, S.Y.; Solovyev, A.G. Translation of the Shallot virus X TGB3 gene depends on non-AUG initiation and leaky scanning. J. Gen. Virol. 2015, 96, 3159–3164. [Google Scholar] [CrossRef] [PubMed]
- Vishnichenko, V.K.; Stel’mashchuk, V.Y.; Zavriev, S.K. The 42K protein of shallot virus X participates in formation of virus particles. Mol. Biol. 2002, 36, 879–882. [Google Scholar] [CrossRef]
- Celli, M.G.; Perotto, M.C.; Luciani, C.E.; Pozzi, E.A.; Conci, V.C. Molecular characterization of the garlic virus B genome and evidence of allexivirus recombination. Eur. J. Plant Pathol. 2019, 153, 301–310. [Google Scholar] [CrossRef]
- Arkhipov, A.V.; Solovyev, A.G.; Vishnichenko, V.K. Reproduction of Shallot virus X in absence of its own active suppressor protein of RNA silencing. Russ. Agric. Sci. 2013, 39, 218–221. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Collection Time | Collection Region | Sample Size |
|---|---|---|
| 10 June 2020 | Wude Town, Wenxian County | 18 |
| 10 June2020 | Dafeng Town, Wuzhi County | 15 |
| 10 June2020 | Xitao Town, Wuzhi County | 5 |
| 8 July 2020 | Xiangyun Town, Wenxian County | 5 |
| 14 July 2020 | Zhangde Town, Yuzhou City | 17 |
| Primer Name | Sequence (5′-3′) | Size (nt) |
|---|---|---|
| ReAV-Rep-1F | ATGAGCACCCAGCAGGTAGTGAC | 507 |
| ReAV-Rep-1R | AAGTGATACGGCTTTGACGGAGA | |
| ReAV-Rep-2F | CGCCATCGCCCTGTTCAACAAAT | 781 |
| ReAV-Rep-2R | AAGCAAGCGGTCGCCCATTCTGT | |
| ReAV-CP-1F | AGGCTCGCAGTTCAATCAGGTCTTC | 624 |
| ReAV-CP-1R | AACTCAGCACATGCCCGTGAGTTT | |
| ReAV-CP-2F | TACAAACTCACGGGCATGTGCTGA | 518 |
| ReAV-CP-2R | TGCAATGTTGCTCCACTATGTCCTTC |
| Virus Name | GenBank No. | Complete Genome | Replicase | TGB1 | TGB2 | TGB3 | CP |
|---|---|---|---|---|---|---|---|
| garlic virus A | AB010300 | 49.9–50.9% | 61.2–62.1% | 48.0–49.9% | 51.0–52.6% | 36.7–39.5% | 50.1–51.7% |
| garlic virus B | KM379144 | 51.4–52.4% | 60.7–61.7% | 48.0–49.7% | 50.0–51.0% | 30.6–32.9% | 49.9–51.7% |
| garlic virus C | AB010302 | 52.2–-53.0% | 61.3–62.5% | 50.6–52.2% | 51.6–52.6% | 38.4–39.8% | 47.8–49.1% |
| garlic virus D | KF555653 | 51.5–52.2% | 60.1–60.9% | 49.9–51.3% | 49.0–50.0% | 35.2–36.1% | 50.0–52.2% |
| garlic virus E | AJ292230 | 52.8–53.5% | 61.2–62.4% | 49.0–50.5% | 52.9–54.2% | 36.1–40.3% | 51.3–53.3% |
| garlic virus X | U89243 | 49.6–50.9% | 59.7–61.1% | 49.5–51.2% | 48.6–50.8% | 36.7–37.7% | 51.3–52.2% |
| Shallot virus X | M97264 | 48.0–49.1% | 61.7–62.8% | 48.0–49.7% | 51.3–53.8% | 31.5–35.2% | 47.5–49.8% |
| Alfalfa virus S | KY696659 | 54.5–55.2% | 61.2–61.9% | 52.7–54.5% | 54.5–56.4% | 48.1–49.1% | 50.4–51.1% |
| Arachis pintoi virus | KX058345 | 55.1–55.5% | 64.5–65.2% | 48.2–49.7% | 49.7–50.6% | 33.2–38.4% | 50.2–53.3% |
| Blackberry virus E | JN053266 | 55.1–55.8% | 63.3–64.1% | 51.7–53.4% | 51.9–52.9% | 42.1–44.4% | 52.7–55.5% |
| Senna severe yellow mosaic virus | MN031278 | 54.9–55.5% | 63.2–64.0% | 49.3–50.5% | 51.8–53.4% | 47.2–57.0% | 50.1–53.5% |
| Vanilla latent virus | MF150239 | 53.6–54.4% | 59.9–60.8% | 39.0–41.0% | 46.4–49.7% | 36.1–39.8% | 50.2–51.4% |
| Complete Genome | |||||||
|---|---|---|---|---|---|---|---|
| Virus | ReAV-29 | ReAV-49 | ReAV-52 | ReAV-53 | ReAV-55 | ReAV-58 | ReAV-59 |
| ReAV-29 | - | - | - | - | - | - | |
| ReAV-49 | 88.7% | - | - | - | - | - | |
| ReAV-52 | 87.2% | 95.4% | - | - | - | - | |
| ReAV-53 | 92.4% | 90.6% | 90.9% | - | - | - | |
| ReAV-55 | 89.2% | 95.0% | 93.8% | 94.1% | - | - | |
| ReAV-58 | 91.0% | 91.8% | 91.6% | 90.2% | 91.6% | - | |
| ReAV-59 | 87.4% | 94.8% | 94.1% | 92.6% | 96.5% | 92.8% | |
| Replicase | |||||||
| virus | ReAV-29 | ReAV-49 | ReAV-52 | ReAV-53 | ReAV-55 | ReAV-58 | ReAV-59 |
| ReAV-29 | 92.4% | 91.9% | 97.6% | 94.9% | 93.9% | 92.7% | |
| ReAV-49 | 86.7% | 96.7% | 93.0% | 96.8% | 98.4% | 98.3% | |
| ReAV-52 | 86.8% | 95.5% | 92.5% | 94.7% | 95.4% | 96.2% | |
| ReAV-53 | 92.3% | 90.6% | 90.3% | 95.7% | 92.7% | 93.4% | |
| ReAV-55 | 90.5% | 95.2% | 92.2% | 94.1% | 96.4% | 96.6% | |
| ReAV-58 | 91.3% | 92.4% | 88.7% | 87.8% | 91.4% | 98.0% | |
| ReAV-59 | 87.6% | 95.6% | 93.4% | 92.1% | 94.9% | 94.0% | |
| TGB1 | |||||||
| virus | ReAV-29 | ReAV-49 | ReAV-52 | ReAV-53 | ReAV-55 | ReAV-58 | ReAV-59 |
| ReAV-29 | 90.9% | 91.3% | 97.1% | 90.9% | 94.2% | 90.5% | |
| ReAV-49 | 86.5% | 99.6% | 93.4% | 99.2% | 91.7% | 98.8% | |
| ReAV-52 | 86.5% | 99.6% | 93.8% | 99.6% | 92.1% | 99.2% | |
| ReAV-53 | 97.5% | 86.8% | 86.8% | 93.8% | 97.1% | 92.9% | |
| ReAV-55 | 86.5% | 99.4% | 99.6% | 86.8% | 91.7% | 98.8% | |
| ReAV-58 | 96.0% | 86.0% | 86.0% | 98.1% | 86.0% | 91.3% | |
| ReAV-59 | 86.1% | 98.3% | 98.8% | 86.4% | 98.3% | 85.5% | |
| TGB2 | |||||||
| virus | ReAV-29 | ReAV-49 | ReAV-52 | ReAV-53 | ReAV-55 | ReAV-58 | ReAV-59 |
| ReAV-29 | 94.2% | 95.2% | 100.0% | 95.2% | 95.2% | 96.2% | |
| ReAV-49 | 87.6% | 99.0% | 94.2% | 99.0% | 99.0% | 98.1% | |
| ReAV-52 | 88.3% | 98.7% | 95.2% | 100.0% | 100.0% | 99.0% | |
| ReAV-53 | 98.7% | 88.9% | 89.5% | 95.2% | 95.2% | 96.2% | |
| ReAV-55 | 87.6% | 99.0% | 99.0% | 88.9% | 100.0% | 99.0% | |
| ReAV-58 | 87.9% | 98.4% | 99.7% | 89.2% | 98.7% | 99.0% | |
| ReAV-59 | 87.9% | 98.4% | 99.0% | 89.2% | 98.7% | 98.7% | |
| TGB3 | |||||||
| virus | ReAV-29 | ReAV-49 | ReAV-52 | ReAV-53 | ReAV-55 | ReAV-58 | ReAV-59 |
| ReAV-29 | 87.3% | 71.8% | 81.7% | 87.3% | 88.7% | 88.7% | |
| ReAV-49 | 88.0% | 81.7% | 71.8% | 97.2% | 98.6% | 98.6% | |
| ReAV-52 | 87.0% | 97.7% | 90.1% | 81.7% | 83.1% | 83.1% | |
| ReAV-53 | 95.8% | 88.9% | 91.2% | 71.8% | 73.2% | 73.2% | |
| ReAV-55 | 88.0% | 99.1% | 97.7% | 88.9% | 98.6% | 98.6% | |
| ReAV-58 | 88.0% | 99.1% | 97.7% | 88.9% | 99.1% | 100.0% | |
| ReAV-59 | 88.4% | 98.6% | 97.2% | 89.4% | 98.6% | 98.6% | |
| Coat protein | |||||||
| virus | ReAV-29 | ReAV-49 | ReAV-52 | ReAV-53 | ReAV-55 | ReAV-58 | ReAV-59 |
| ReAV-29 | 88.2% | 79.8% | 80.3% | 80.3% | 82.7% | 79.2% | |
| ReAV-49 | 93.4% | 89.0% | 89.5% | 89.5% | 85.9% | 87.7% | |
| ReAV-52 | 87.0% | 91.6% | 91.4% | 91.3% | 95.4% | 89.4% | |
| ReAV-53 | 88.5% | 91.1% | 93.0% | 99.1% | 88.2% | 96.2% | |
| ReAV-55 | 87.1% | 90.8% | 92.9% | 97.9% | 88.2% | 96.4% | |
| ReAV-58 | 88.5% | 90.5% | 98.0% | 91.9% | 91.8% | 86.6% | |
| ReAV-59 | 86.7% | 89.9% | 92.1% | 96.5% | 98.3% | 91.1% | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qin, Y.; Lu, S.; Wen, Y.; Li, S.; Gao, S.; Liu, Y.; Li, X.; Yang, J.; Wang, F.; Wang, F.; et al. Genomic Characterization and Molecular Detection of Rehmannia Allexivirus Virus, a Novel Allexivirus Infecting Rehmannia glutinosa. Microorganisms 2024, 12, 844. https://doi.org/10.3390/microorganisms12050844
Qin Y, Lu S, Wen Y, Li S, Gao S, Liu Y, Li X, Yang J, Wang F, Wang F, et al. Genomic Characterization and Molecular Detection of Rehmannia Allexivirus Virus, a Novel Allexivirus Infecting Rehmannia glutinosa. Microorganisms. 2024; 12(5):844. https://doi.org/10.3390/microorganisms12050844
Chicago/Turabian StyleQin, Yanhong, Shuhao Lu, Yi Wen, Shaojian Li, Suxia Gao, Yuxia Liu, Xuemeng Li, Jin Yang, Fengli Wang, Fei Wang, and et al. 2024. "Genomic Characterization and Molecular Detection of Rehmannia Allexivirus Virus, a Novel Allexivirus Infecting Rehmannia glutinosa" Microorganisms 12, no. 5: 844. https://doi.org/10.3390/microorganisms12050844