
Evaluation of the Increased Genetic Resolution and Utility for Source Tracking of a Recently Developed Method for Genotyping Cyclospora cayetanensis
 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Clinical Specimens
2.2. Raspberry Sample Preparation
2.3. DNA Extraction and Real-Time PCR
2.4. Targeted Amplicon Sequencing
2.5. Multi-Locus Sequencing Typing
2.6. Bioinformatic Analyses
3. Results
3.1. Genotyping Clinical Specimens
3.2. Genotyping without Hybridization Capture Enrichment
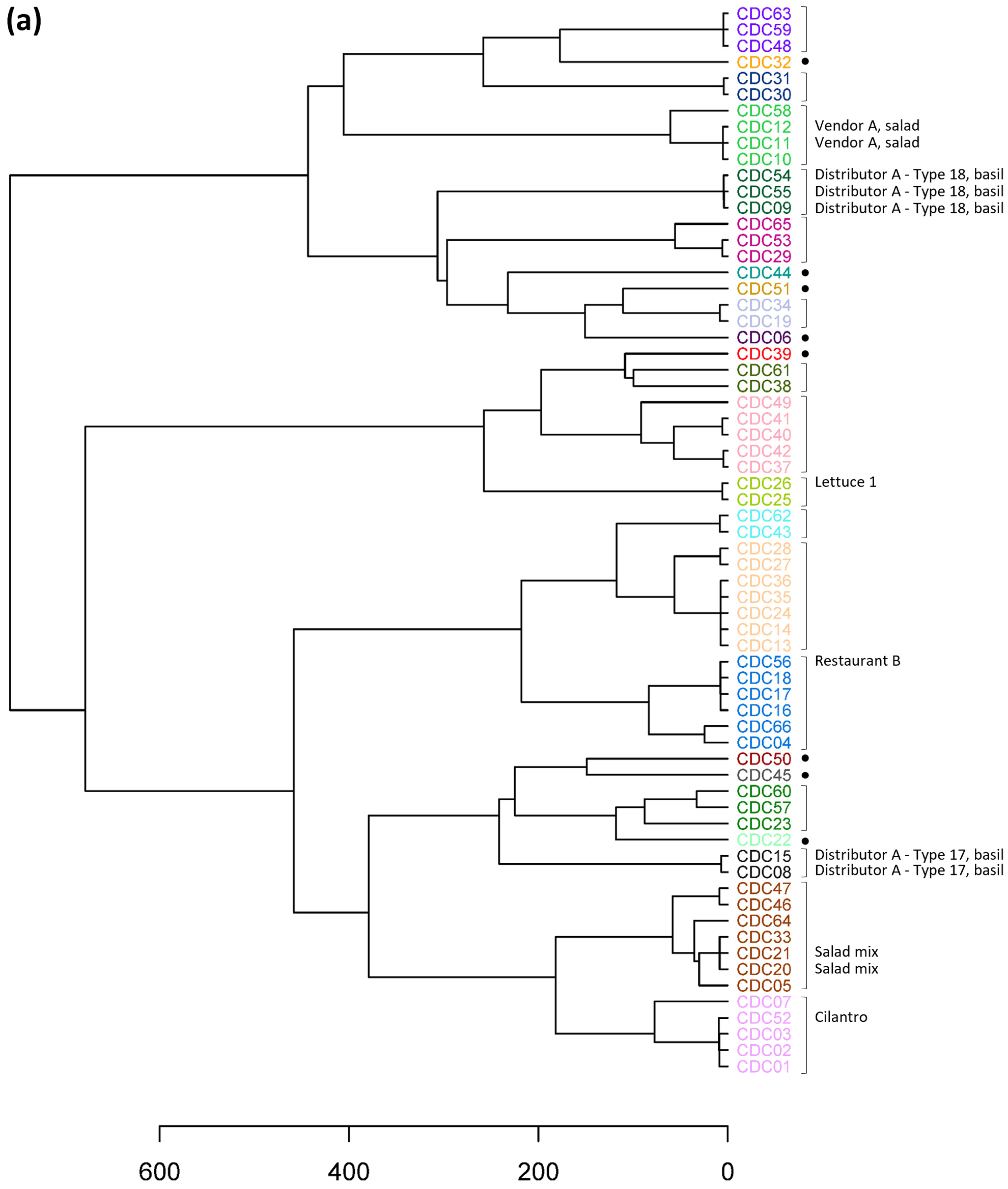
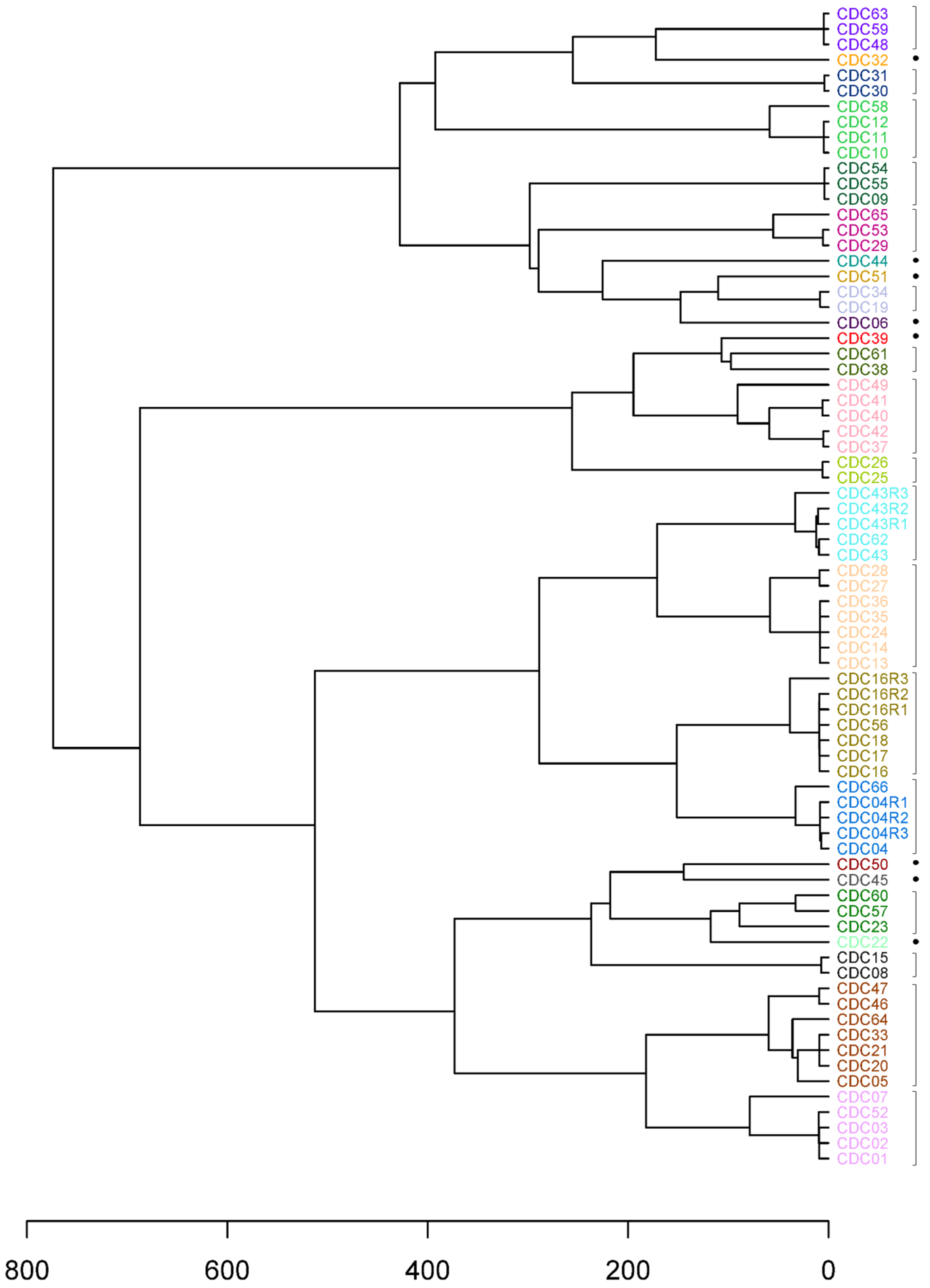
3.3. Genetic Cluster Resolution
3.4. Application of Epidemiologic Information
3.5. Assessment of Traceback Utility
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mathison, B.A.; Pritt, B.S. Cyclosporiasis-Updates on Clinical Presentation, Pathology, Clinical Diagnosis, and Treatment. Microorganisms 2021, 9, 1863. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Qin, Z.; Li, J.; Xiao, L.; Zhang, L. The global prevalence of Cyclospora cayetanensis infection: A systematic review, meta-analysis, and meta-regression. Acta Trop. 2024, 253, 107175. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, R.; Chen, Y.; Xiao, L.; Zhang, L. Cyclospora cayetanensis infection in humans: Biological characteristics, clinical features, epidemiology, detection method and treatment. Parasitology 2020, 147, 160–170. [Google Scholar] [CrossRef] [PubMed]
- Barratt, J.L.N.; Shen, J.; Houghton, K.; Richins, T.; Sapp, S.G.H.; Cama, V.; Arrowood, M.J.; Straily, A.; Qvarnstrom, Y. Cyclospora cayetanensis comprises at least 3 species that cause human cyclosporiasis. Parasitology 2023, 150, 269–285. [Google Scholar] [CrossRef] [PubMed]
- Ortega, Y.R.; Sanchez, R. Update on Cyclospora cayetanensis, a food-borne and waterborne parasite. Clin. Microbiol. Rev. 2010, 23, 218–234. [Google Scholar] [CrossRef] [PubMed]
- Giangaspero, A.; Marangi, M.; Koehler, A.V.; Papini, R.; Normanno, G.; Lacasella, V.; Lonigro, A.; Gasser, R.B. Molecular detection of Cyclospora in water, soil, vegetables and humans in southern Italy signals a need for improved monitoring by health authorities. Int. J. Food Microbiol. 2015, 211, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Chacin-Bonilla, L.; Santin, M. Cyclospora cayetanensis Infection in Developed Countries: Potential Endemic Foci? Microorganisms 2023, 11, 540. [Google Scholar] [CrossRef] [PubMed]
- Hadjilouka, A.; Tsaltas, D. Cyclospora Cayetanensis-Major Outbreaks from Ready to Eat Fresh Fruits and Vegetables. Foods 2020, 9, 1703. [Google Scholar] [CrossRef] [PubMed]
- Casillas, S.M.; Bennett, C.; Straily, A. Multiple Cyclosporiasis Outbreaks—United States, 2018. Mmwr-Morbid Mortal W 2018, 67, 1101–1102. [Google Scholar] [CrossRef] [PubMed]
- Morton, V.; Meghnath, K.; Gheorghe, M.; Fitzgerald-Husek, A.; Hobbs, J.; Honish, L.; David, S. Use of a case-control study and control bank to investigate an outbreak of locally acquired cyclosporiasis in Canada, 2016. Can. Commun. Dis. Rep. 2019, 45, 225–229. [Google Scholar] [CrossRef] [PubMed]
- Kozak, G.K.; MacDonald, D.; Landry, L.; Farber, J.M. Foodborne outbreaks in Canada linked to produce: 2001 through 2009. J. Food Prot. 2013, 76, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Yanta, C.A.; Pollo, S.M.J.; Barta, J.R.; Reiling, S.J.; Wasmuth, J.D.; Dixon, B.R.; Guy, R.A. Draft Hybrid Genome Assembly of a Canadian Cyclospora cayetanensis Isolate. Microbiol. Resour. Announc. 2022, 11, e0107221. [Google Scholar] [CrossRef] [PubMed]
- Qvarnstrom, Y.; Wei-Pridgeon, Y.; Van Roey, E.; Park, S.; Srinivasamoorthy, G.; Nascimento, F.S.; Moss, D.M.; Talundzic, E.; Arrowood, M.J. Purification of Cyclospora cayetanensis oocysts obtained from human stool specimens for whole genome sequencing. Gut Pathog. 2018, 10, 45. [Google Scholar] [CrossRef] [PubMed]
- Nascimento, F.S.; Barratt, J.; Houghton, K.; Plucinski, M.; Kelley, J.; Casillas, S.; Bennett, C.; Snider, C.; Tuladhar, R.; Zhang, J.; et al. Evaluation of an ensemble-based distance statistic for clustering MLST datasets using epidemiologically defined clusters of cyclosporiasis. Epidemiol. Infect. 2020, 148, e172. [Google Scholar] [CrossRef] [PubMed]
- Barratt, J.L.N.; Park, S.; Nascimento, F.S.; Hofstetter, J.; Plucinski, M.; Casillas, S.; Bradbury, R.S.; Arrowood, M.J.; Qvarnstrom, Y.; Talundzic, E. Genotyping genetically heterogeneous Cyclospora cayetanensis infections to complement epidemiological case linkage. Parasitology 2019, 146, 1275–1283. [Google Scholar] [CrossRef] [PubMed]
- Houghton, K.A.; Lomsadze, A.; Park, S.; Nascimento, F.S.; Barratt, J.; Arrowood, M.J.; VanRoey, E.; Talundzic, E.; Borodovsky, M.; Qvarnstrom, Y. Development of a workflow for identification of nuclear genotyping markers for Cyclospora cayetanensis. Parasite 2020, 27, 24. [Google Scholar] [CrossRef] [PubMed]
- Nascimento, F.S.; Barta, J.R.; Whale, J.; Hofstetter, J.N.; Casillas, S.; Barratt, J.; Talundzic, E.; Arrowood, M.J.; Qvarnstrom, Y. Mitochondrial Junction Region as Genotyping Marker for Cyclospora cayetanensis. Emerg. Infect. Dis. 2019, 25, 1314–1319. [Google Scholar] [CrossRef] [PubMed]
- Barratt, J.; Houghton, K.; Richins, T.; Straily, A.; Threlkel, R.; Bera, B.; Kenneally, J.; Clemons, B.; Madison-Antenucci, S.; Cebelinski, E.; et al. Investigation of US Cyclospora cayetanensis outbreaks in 2019 and evaluation of an improved Cyclospora genotyping system against 2019 cyclosporiasis outbreak clusters. Epidemiol. Infect. 2021, 149, e214. [Google Scholar] [CrossRef] [PubMed]
- Ahart, L.; Jacobson, D.; Rice, M.; Richins, T.; Peterson, A.; Zheng, Y.L.; Barratt, J.; Cama, V.; Qvarnstrom, Y.; Montgomery, S.; et al. Retrospective evaluation of an integrated molecular-epidemiological approach to cyclosporiasis outbreak investigations—United States, 2021. Epidemiol. Infect. 2023, 151, e131. [Google Scholar] [CrossRef] [PubMed]
- Rehme, P. Notes from the Field: Doubling of Cyclosporiasis Cases Partially Attributable to a Salad Kit-Florida, 2021–2022. MMWR Morb. Mortal Wkly. Rep. 2023, 72, 751–752. [Google Scholar] [CrossRef] [PubMed]
- Yanta, C.A.; Barta, J.R.; Corbeil, A.; Menan, H.; Thivierge, K.; Needle, R.; Morshed, M.; Dixon, B.R.; Wasmuth, J.D.; Guy, R.A. Genotyping Canadian Cyclospora cayetanensis Isolates to Supplement Cyclosporiasis Outbreak Investigations. Microorganisms 2022, 10, 447. [Google Scholar] [CrossRef] [PubMed]
- Barratt, J.; Ahart, L.; Rice, M.; Houghton, K.; Richins, T.; Cama, V.; Arrowood, M.; Qvarnstrom, Y.; Straily, A. Genotyping Cyclospora cayetanensis From Multiple Outbreak Clusters With An Emphasis on a Cluster Linked to Bagged Salad Mix-United States, 2020. J. Infect. Dis. 2022, 225, 2176–2180. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Xu, F.; Karim, M.R.; Zhang, L. Review on Cyclosporiasis Outbreaks and Potential Molecular Markers for Tracing Back Investigations. Foodborne Pathog. Dis. 2022, 19, 796–805. [Google Scholar] [CrossRef] [PubMed]
- Leonard, S.R.; Mammel, M.K.; Gharizadeh, B.; Almeria, S.; Ma, Z.; Lipman, D.J.; Torrence, M.E.; Wang, C.; Musser, S.M. Development of a targeted amplicon sequencing method for genotyping Cyclospora cayetanensis from fresh produce and clinical samples with enhanced genomic resolution and sensitivity. Front. Microbiol. 2023, 14, 1212863. [Google Scholar] [CrossRef] [PubMed]
- Pightling, A.W.; Pettengill, J.B.; Luo, Y.; Baugher, J.D.; Rand, H.; Strain, E. Interpreting Whole-Genome Sequence Analyses of Foodborne Bacteria for Regulatory Applications and Outbreak Investigations. Front. Microbiol. 2018, 9, 1482. [Google Scholar] [CrossRef] [PubMed]
- Barratt, J.L.N.; Plucinski, M.M. Epidemiologic utility of a framework for partition number selection when dissecting hierarchically clustered genetic data evaluated on the intestinal parasite Cyclospora cayetanensis. Am. J. Epidemiol. 2023, 192, 772–781. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, D.K.; Low, R.; Plucinski, M.M.; Barratt, J.L.N. An improved framework for detecting discrete epidemiologically meaningful partitions in hierarchically clustered genetic data. Bioinform. Adv. 2023, 3, vbad118. [Google Scholar] [CrossRef] [PubMed]
- Qvarnstrom, Y.; Benedict, T.; Marcet, P.L.; Wiegand, R.E.; Herwaldt, B.L.; da Silva, A.J. Molecular detection of Cyclospora cayetanensis in human stool specimens using UNEX-based DNA extraction and real-time PCR. Parasitology 2018, 145, 865–870. [Google Scholar] [CrossRef] [PubMed]
- Assurian, A.; Murphy, H.; Ewing, L.; Cinar, H.N.; da Silva, A.; Almeria, S. Evaluation of the U.S. Food and Drug Administration validated molecular method for detection of Cyclospora cayetanensis oocysts on fresh and frozen berries. Food Microbiol. 2020, 87, 103397. [Google Scholar] [CrossRef] [PubMed]
- Balan, K.V.; Mammel, M.; Lipman, D.; Babu, U.; Harrison, L.M.; Almeria, S.; Durigan, M.; Leonard, S.R.; Hyein, J.; Gebru, S.; et al. Development and Single Laboratory Evaluation of a Refined and Specific Real-time PCR Detection Method, Using Mitochondrial Primers (Mit1C), for Detection of Cyclospora cayetanensis in Produce. J. Food Prot. 2023, 86, 100037. [Google Scholar] [CrossRef] [PubMed]
- Maechler, M.; Rousseeuw, P.; Struyf, A.; Hubert, M.; Hornik, K. Cluster: Cluster Analysis Basics and Extensions, R package Version 2.1.4; R Foundation: Vienna, Austria, 2022. [Google Scholar]
- Bholowalia, P.; Kumar, A. EBK-Means: A Clustering Technique based on Elbow Method and K-Means in WSN. Int. J. Comput. Appl. 2014, 105, 17–24. [Google Scholar] [CrossRef]
- Rousseeuw, P.J. Silhouettes—A Graphical Aid to the Interpretation and Validation of Cluster-Analysis. J. Comput. Appl. Math. 1987, 20, 53–65. [Google Scholar] [CrossRef]
- Charrad, M.; Ghazzali, N.; Boiteau, V.; Niknafs, A. Nbclust: An R Package for Determining the Relevant Number of Clusters in a Data Set. J. Stat. Softw. 2014, 61, 1–36. [Google Scholar] [CrossRef]
- Balaban, M.; Moshiri, N.; Mai, U.; Jia, X.; Mirarab, S. TreeCluster: Clustering biological sequences using phylogenetic trees. PLoS ONE 2019, 14, e0221068. [Google Scholar] [CrossRef] [PubMed]
- Markin, A.; Wagle, S.; Grover, S.; Vincent Baker, A.L.; Eulenstein, O.; Anderson, T.K. PARNAS: Objectively Selecting the Most Representative Taxa on a Phylogeny. Syst. Biol. 2023, 72, 1052–1063. [Google Scholar] [CrossRef] [PubMed]
- Korbie, D.J.; Mattick, J.S. Touchdown PCR for increased specificity and sensitivity in PCR amplification. Nat. Protoc. 2008, 3, 1452–1456. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Wang, J.; Deng, F.; Yan, Z.; Xia, Y.; Wang, Z.; Ye, J.; Deng, Y.; Zhang, Z.; Qiao, M.; et al. TqPCR: A Touchdown qPCR Assay with Significantly Improved Detection Sensitivity and Amplification Efficiency of SYBR Green qPCR. PLoS ONE 2015, 10, e0132666. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Gomez, J.P.; Lozano-Aguirre, L.F.; Medrano-Felix, J.A.; Chaidez, C.; Gerba, C.P.; Betancourt, W.Q.; Castro-Del Campo, N. Evaluation of nuclear and mitochondrial phylogenetics for the subtyping of Cyclospora cayetanensis. Parasitol. Res. 2023, 122, 2641–2650. [Google Scholar] [CrossRef] [PubMed]
- Weitzel, T.; Brown, A.; Libman, M.; Perret, C.; Huits, R.; Chen, L.; Leung, D.; Leder, K.; Connor, B.A.; Menendez, M.D.; et al. Intestinal protozoa in returning travellers: A GeoSentinel analysis from 2007 to 2019. J. Travel. Med. 2024. [Google Scholar] [CrossRef] [PubMed]
- Herwaldt, B.L.; Ackers, M.L. An outbreak in 1996 of cyclosporiasis associated with imported raspberries. The Cyclospora Working Group. N. Engl. J. Med. 1997, 336, 1548–1556. [Google Scholar] [CrossRef] [PubMed]
- Temesgen, T.T.; Stigum, V.M.; Robertson, L.J. Surveillance of berries sold on the Norwegian market for parasite contamination using molecular methods. Food Microbiol. 2022, 104, 103980. [Google Scholar] [CrossRef] [PubMed]
- Ho, A.Y.; Lopez, A.S.; Eberhart, M.G.; Levenson, R.; Finkel, B.S.; da Silva, A.J.; Roberts, J.M.; Orlandi, P.A.; Johnson, C.C.; Herwaldt, B.L. Outbreak of cyclosporiasis associated with imported raspberries, Philadelphia, Pennsylvania, 2000. Emerg. Infect. Dis. 2002, 8, 783–788. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Mesonero, L.; Soler, L.; Amoros, I.; Moreno, Y.; Ferrus, M.A.; Alonso, J.L. Protozoan parasites and free-living amoebae contamination in organic leafy green vegetables and strawberries from Spain. Food Waterborne Parasitol. 2023, 32, e00200. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Ramirez, L.C.; Djabayan-Djibeyan, P.; Prato, J.G.; Garcia Rios, C.A.; Carrero, J.C.; Trelis, M.; Fuentes, M.V. Field study of parasitic contamination of fruits, vegetables and leafy greens in the Ecuadorian Andes. F1000Research 2023, 12, 532. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Sample | CT | Collection Date | Epidemiologic Information |
|---|---|---|---|
| CDC01 | 25.83 | 17 August 2020 | Unknown |
| CDC02 | 24.89 | 18 August 2020 | Unknown |
| CDC03 | 27.20 | 10 August 2020 | Unknown |
| CDC04 | 26.05 | 6 August 2020 | Unknown |
| CDC05 | 24.47 | 21 July 2020 | Unknown |
| CDC06 | N/A | 1 September 2020 | Unknown |
| CDC07 | 19.58 | 26 August 2020 | Unknown |
| CDC08 | 21.62 | 26 June 2019 | Distributor A—Type 17, basil |
| CDC09 | 25.91 | 2 July 2019 | Distributor A—Type 18, basil |
| CDC10 | N/A | 2018 | Unknown |
| CDC11 | N/A | 2018 | Vendor A, salad |
| CDC12 | N/A | 2018 | Vendor A, salad |
| CDC13 | 21.33 | Unknown | Unknown |
| CDC14 | 30.51 | Unknown | Unknown |
| CDC15 | 23.24 | 31 July 2019 | Distributor A—Type 17, basil |
| CDC16 | 23.46 | 25 July 2019 | Unknown |
| CDC17 | 21.96 | 19 July 2019 | Unknown |
| CDC18 | 22.79 | 19 July 2019 | Unknown |
| CDC19 | 21.65 | 13 July 2019 | Unknown |
| CDC20 | 30.86 | 26 June 2020 | Prepackaged salad mix |
| CDC21 | 30.42 | 29 June 2020 | Prepackaged salad mix |
| CDC22 | 25.66 | Unknown | Unknown |
| CDC23 | 23.62 | 20 July 2020 | Unknown |
| CDC24 | 22.61 | Unknown | Unknown |
| CDC25 | 22.86 | 15 July 2021 | Unknown |
| CDC26 | 25.42 | 9 July 2021 | Lettuce 1 |
| CDC27 | 23.75 | 13 July 2021 | Unknown |
| CDC28 | 20.48 | 13 July 2021 | Unknown |
| CDC29 | 27.60 | 30 July 2021 | Unknown |
| CDC30 | 20.58 | 6 July 2019 | Unknown |
| CDC31 | N/A | 2018 | Unknown |
| CDC32 | 19.99 | 12 August 2021 | Unknown |
| CDC33 | 23.50 | 29 July 2022 | Unknown |
| CDC34 | 27.56 | 29 July 2022 | Unknown |
| CDC35 | 19.34 | 29 July 2022 | Unknown |
| CDC36 | 23.93 | 19 July 2022 | Unknown |
| CDC37 | 20.77 | 10 August 2022 | Unknown |
| CDC38 | 22.76 | 14 June 2022 | Unknown |
| CDC39 | 21.49 | 20 July 2022 | Unknown |
| CDC40 | 20.31 | 2022 | Unknown |
| CDC41 | 25.16 | 21 July 2022 | Unknown |
| CDC42 | 22.94 | 13 July 2022 | Unknown |
| CDC43 | 19.78 | 3 August 2022 | Unknown |
| CDC44 | 23.68 | 24 June 2022 | Unknown |
| CDC45 | 25.46 | 8 August 2022 | Unknown |
| CDC46 | 27.03 | 15 July 2022 | Unknown |
| CDC47 | 21.91 | 2022 | Unknown |
| CDC48 | 21.61 | 5 July 2022 | Unknown |
| CDC49 | 22.23 | 8 August 2022 | Unknown |
| CDC50 | 27.96 | 12 April 2022 | Unknown |
| CDC51 | 21.29 | 12 August 2020 | Unknown |
| CDC52 | 28.25 | 21 August 2020 | TN/GA/VA Mexican-style restaurant, cilantro |
| CDC53 | 23.94 | 9 July 2021 | Unknown |
| CDC54 | 26.09 | 16 July 2019 | Distributor A—Type 18, basil |
| CDC55 | 25.03 | 9 July 2019 | Distributor A—Type 18, basil |
| CDC56 | 26.46 | 3 July 2019 | Restaurant B, Romaine lettuce or basil |
| CDC57 | 22.09 | 25 June 2022 | Unknown |
| CDC58 | 28.06 | 23 July 2022 | Unknown |
| CDC59 | 26.53 | 5 July 2022 | Unknown |
| CDC60 | 28.38 | 27 June 2022 | Unknown |
| CDC61 | 23.74 | 1 July 2022 | Unknown |
| CDC62 | 27.62 | 31 July 2022 | Unknown |
| CDC63 | 26.93 | 20 July 2022 | Unknown |
| CDC64 | 31.76 | 14 August 2022 | Unknown |
| CDC65 | 18.17 | 19 July 2022 | Unknown |
| CDC66 | 23.51 | 8 August 2022 | Unknown |
| Baits | No Baits | No Baits, Touchdown PCR | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Sample | %Ccay 1 | Markers 2 | SNPs 2 | %Ccay 1 | Markers 2 | SNPs 2 | %Ccay 1 | Markers 2 | SNPs 2 |
| CDC04 | 80.7% | 51 | 396 | 32.0% | 52 | 396 | 23.0% | 51 | 388 |
| CDC07 | 81.3% | 52 | 396 | 37.3% | 52 | 396 | 45.2% | 51 | 388 |
| CDC08 | 82.8% | 51 | 396 | 27.6% | 52 | 396 | 39.2% | 51 | 388 |
| CDC11 | 80.9% | 51 | 396 | 2.45% | 51 | 388 | 4.88% | 50 | 375 |
| CDC12 | 82.0% | 51 | 396 | 6.93% | 51 | 388 | 13.3% | 51 | 388 |
| CDC16 | 88.7% | 51 | 396 | 1.64% | 50 | 375 | 4.39% | 51 | 388 |
| CDC22 | 88.4% | 51 | 396 | 2.71% | 50 | 375 | 6.64% | 51 | 388 |
| CDC36 | 91.7% | 52 | 396 | 7.44% | 51 | 388 | 14.3% | 51 | 388 |
| CDC40 | 91.4% | 51 | 396 | 13.9% | 51 | 388 | 26.4% | 51 | 388 |
| CDC43 | 91.4% | 51 | 396 | 29.3% | 51 | 388 | 37.1% | 51 | 388 |
| CDC44 | 96.6% | 50 | 380 | 7.44% | 50 | 372 | 11.9% | 50 | 372 |
| CDC46 | 93.7% | 51 | 396 | 0.727% | 47 | 356 | 1.60% | 50 | 375 |
| Sample | Inoculation | CT | %Ccay 1 | Markers 2 | SNPs 2 |
|---|---|---|---|---|---|
| CDC04 | NA | 19.5 ± 0.02 | 80.7% | 51 | 396 |
| CDC04R1 | 50 uL undiluted | 24.3 ± 0.1 | 73.0% | 51 | 396 |
| CDC04R2 | 50 uL of 1:10 | 25.3 ± 0.1 | 60.8% | 51 | 396 |
| CDC04R3 | 5 µL of 1:100 | 34.8 ± 0.3 | 64.8% | 29 | 196 |
| CDC16 | NA | 23.6 ± 0.05 | 88.7% | 51 | 396 |
| CDC16R1 | 50 uL undiluted | 28.0 ± 0.4 | 72.2% | 51 | 396 |
| CDC16R2 | 50 uL of 1:10 | 28.4 ± 0.3 | 69.6% | 51 | 396 |
| CDC16R3 | 5 µL of 1:100 | 35.7 ± 1.5 | 73.6% | 21 | 188 |
| CDC43 | NA | 18.9 ± 0.1 | 91.4% | 51 | 396 |
| CDC43R1 | 50 uL undiluted | 23.5 ± 0.1 | 68.9% | 52 | 396 |
| CDC43R2 | 50 uL of 1:10 | 23.6 ± 0.3 | 67.0% | 52 | 396 |
| CDC43R3 | 5 µL of 1:100 | 30.6 ± 0.2 | 73.8% | 50 | 389 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leonard, S.R.; Mammel, M.K.; Almeria, S.; Gebru, S.T.; Jacobson, D.K.; Peterson, A.C.; Barratt, J.L.N.; Musser, S.M. Evaluation of the Increased Genetic Resolution and Utility for Source Tracking of a Recently Developed Method for Genotyping Cyclospora cayetanensis. Microorganisms 2024, 12, 848. https://doi.org/10.3390/microorganisms12050848
Leonard SR, Mammel MK, Almeria S, Gebru ST, Jacobson DK, Peterson AC, Barratt JLN, Musser SM. Evaluation of the Increased Genetic Resolution and Utility for Source Tracking of a Recently Developed Method for Genotyping Cyclospora cayetanensis. Microorganisms. 2024; 12(5):848. https://doi.org/10.3390/microorganisms12050848
Chicago/Turabian StyleLeonard, Susan R., Mark K. Mammel, Sonia Almeria, Solomon T. Gebru, David K. Jacobson, Anna C. Peterson, Joel L. N. Barratt, and Steven M. Musser. 2024. "Evaluation of the Increased Genetic Resolution and Utility for Source Tracking of a Recently Developed Method for Genotyping Cyclospora cayetanensis" Microorganisms 12, no. 5: 848. https://doi.org/10.3390/microorganisms12050848