Substrate-Induced Response in Biogas Process Performance and Microbial Community Relates Back to Inoculum Source



Abstract
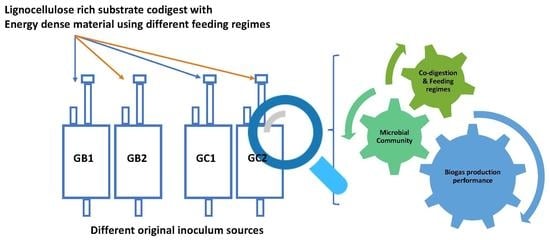
1. Introduction
2. Materials and Methods
2.1. Laboratory-Scale Semi-Continuous Anaerobic Reactors
2.2. Anaerobic Batch Test
2.3. Residual Methane Emissions Measurement
2.4. Analytical Methods
2.5. DNA Extraction and Microbial Community Analysis
3. Results and Discussion
3.1. BMP Test
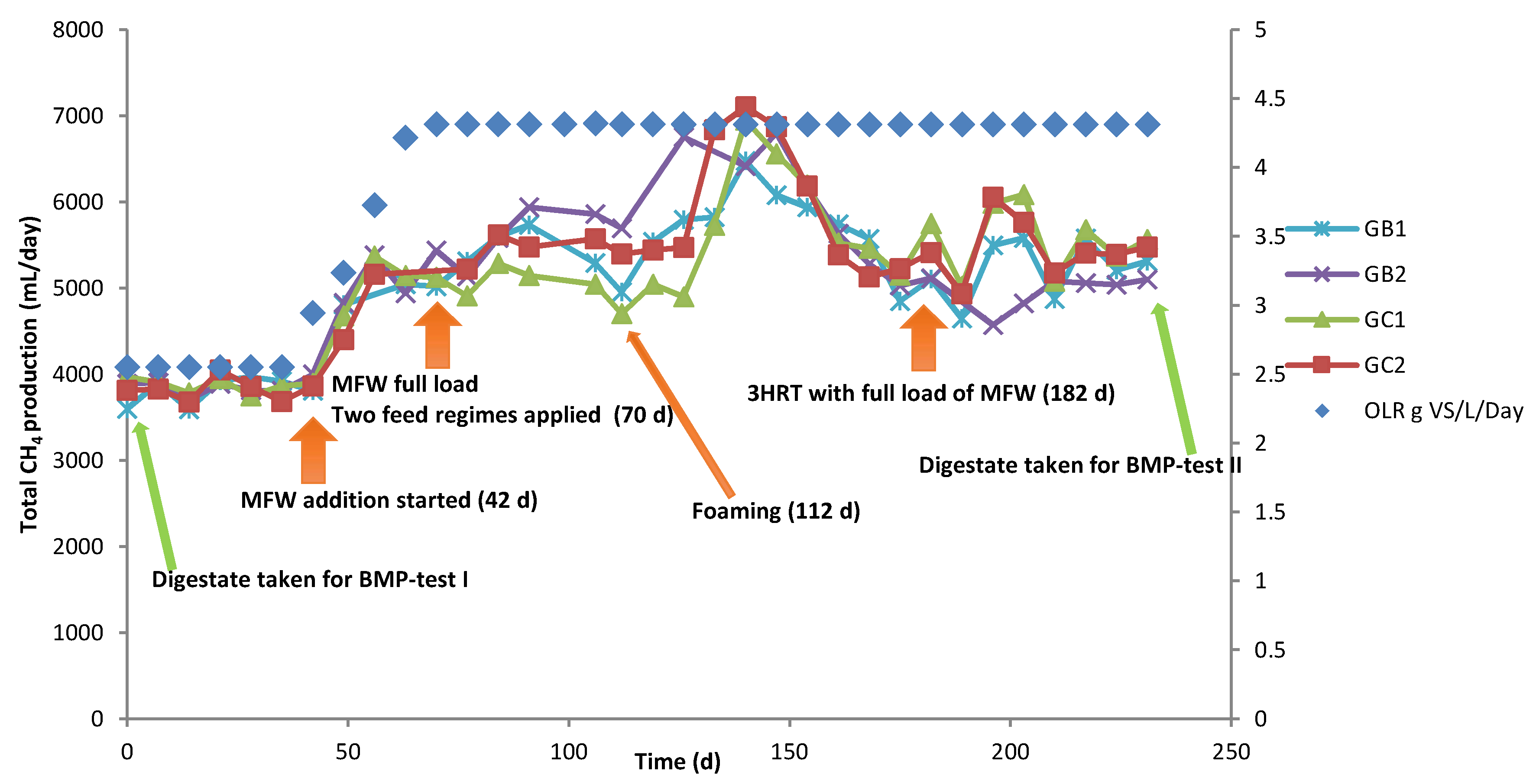
3.2. CSTR Processes
3.3. Residual Methane Potential
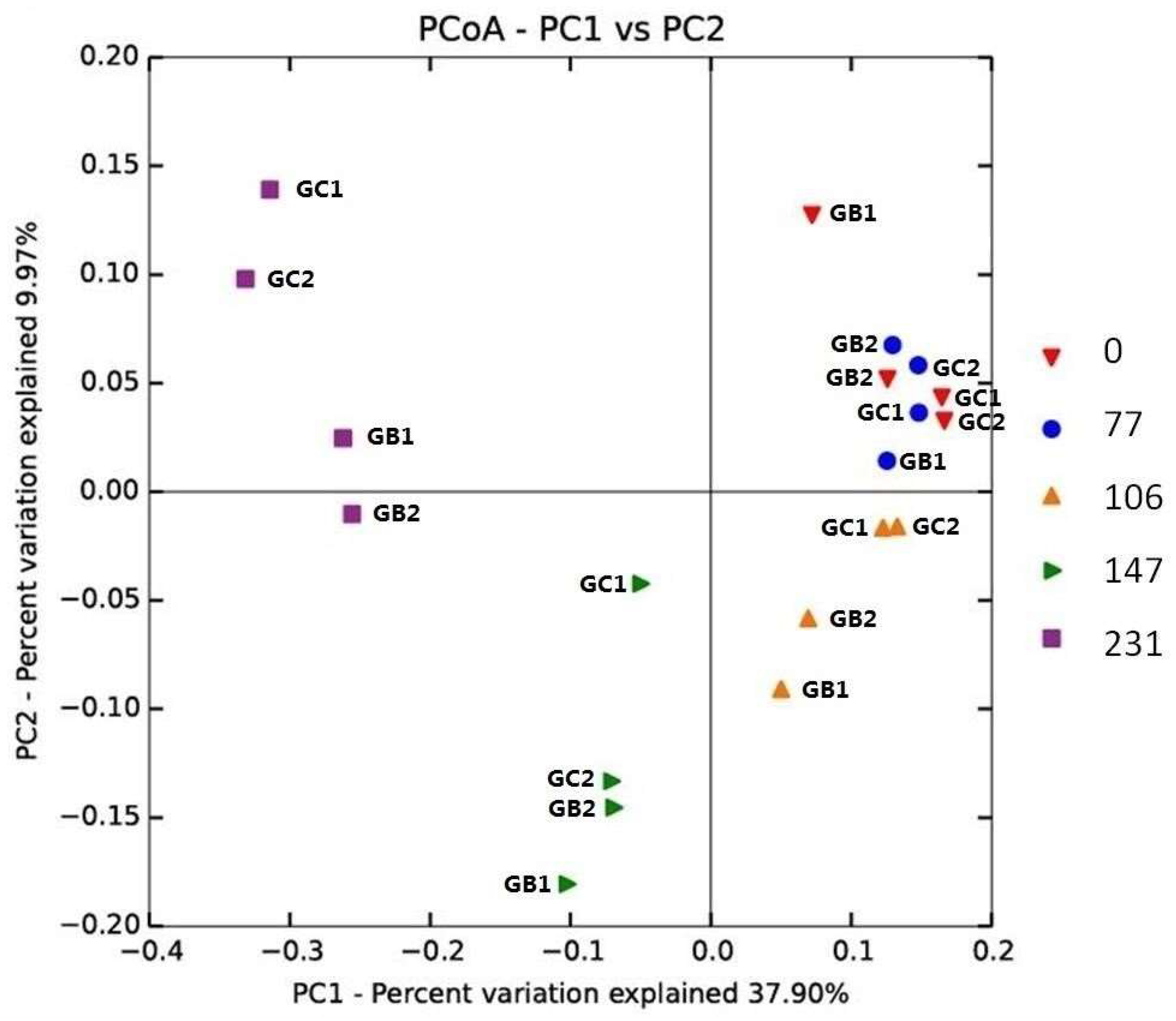
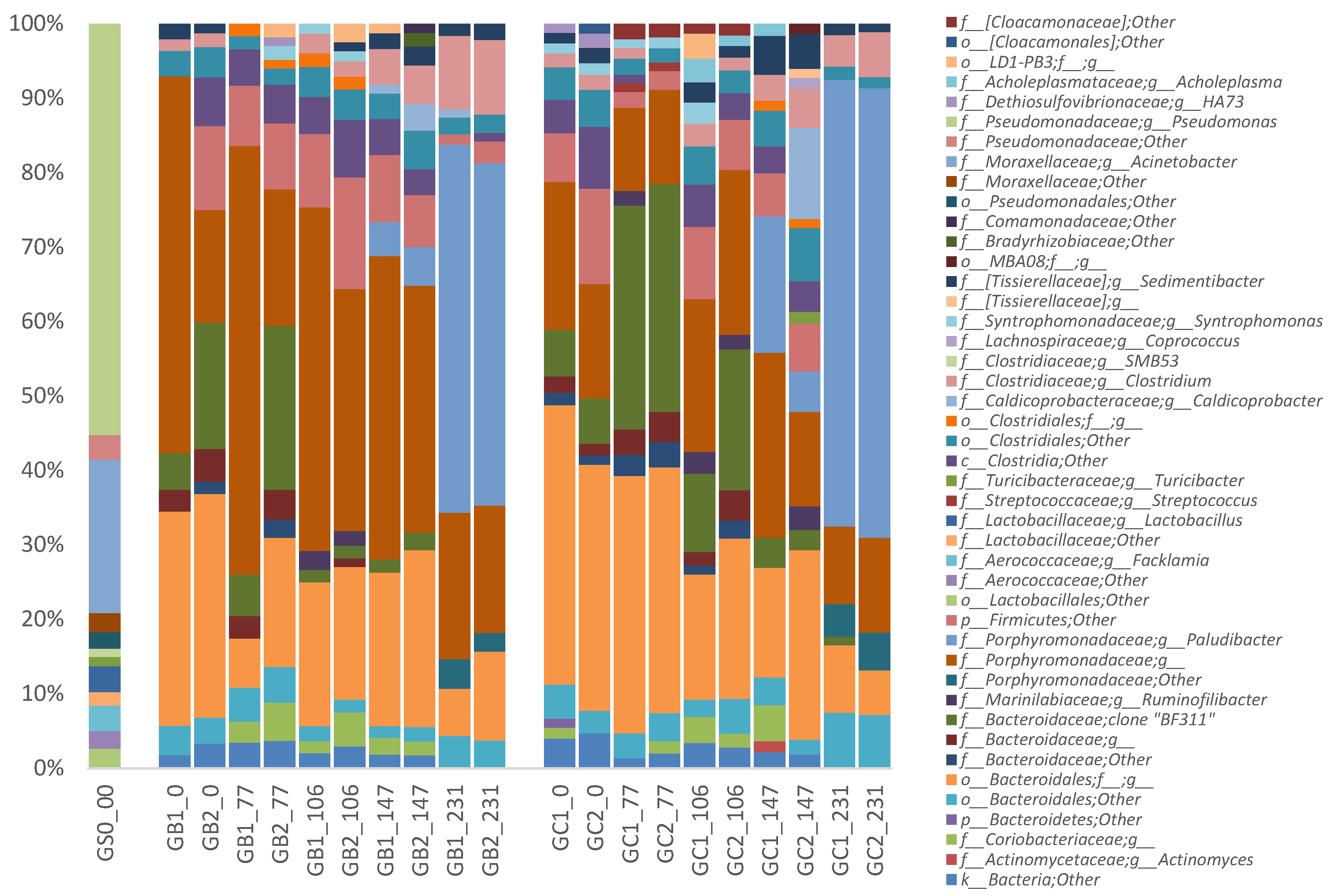
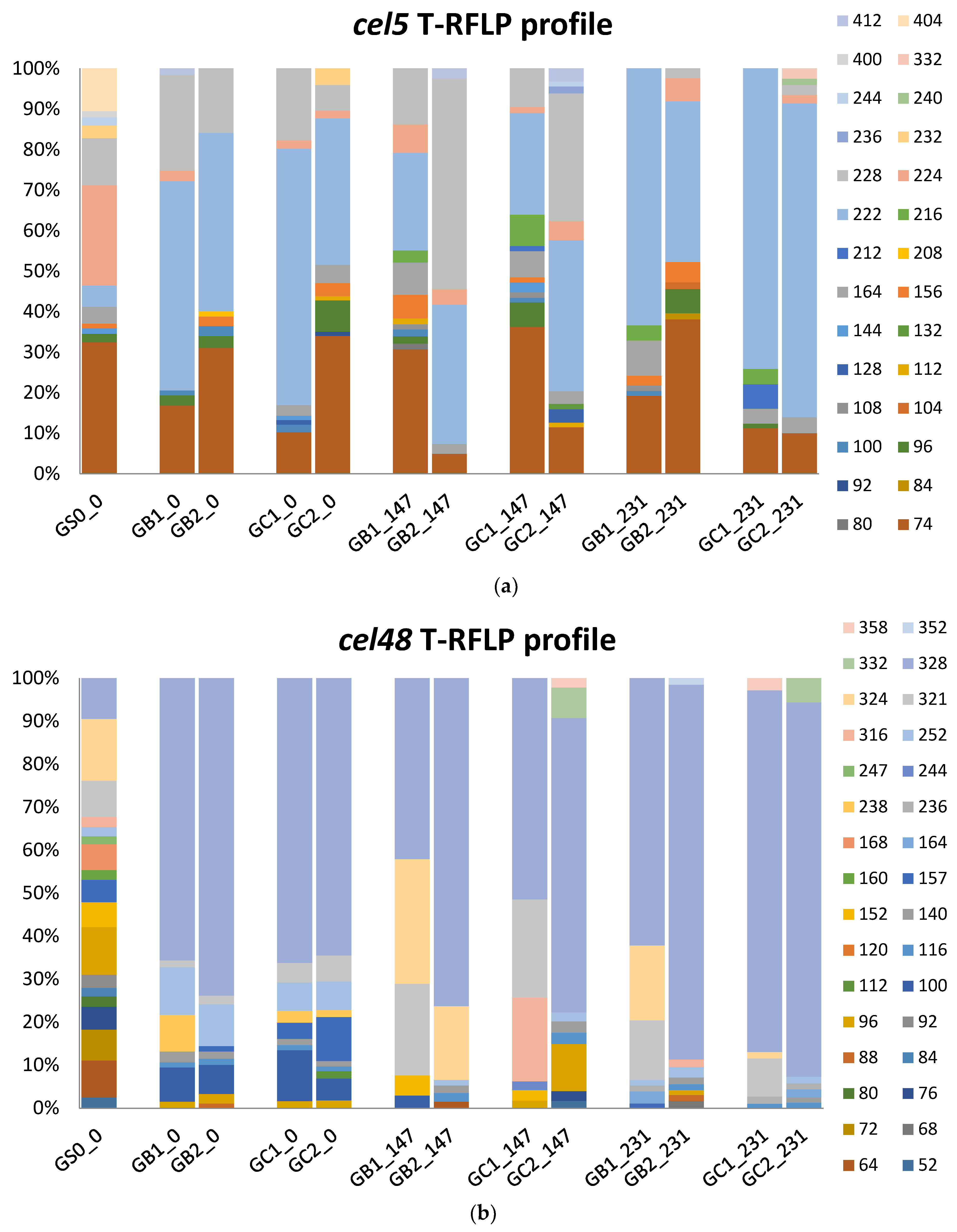
3.4. Microbial Communities
3.4.1. Diversity Indices
3.4.2. Phylogenetic Analysis
3.4.3. T-RFLP
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Meyer-Aurich, A.; Lochmann, Y.; Klauss, H.; Prochnow, A. Comparative advantage of maize-and grass-silage based feedstock for biogas production with respect to greenhouse gas mitigation. Sustainability 2016, 8, 617. [Google Scholar] [CrossRef]
- Azman, S.; Khadem, A.F.; Lier, J.B.; Zeeman, G.; Plugge, C.M. Presence and role of anaerobic hydrolytic microbes in conversion of lignocellulosic biomass for biogas production. Crit. Rev. Environ. Sci. Technol. 2015, 45, 2523–2564. [Google Scholar] [CrossRef]
- Lynd, L.R.; Weimer, P.J.; Van Zyl, W.H.; Pretorius, I.S. Microbial cellulose utilization: Fundamentals and biotechnology. Microbiol. Mol. Biol. Rev. 2002, 66, 506–577. [Google Scholar] [CrossRef]
- Ahlberg-Eliasson, K.; Liu, T.; Nadeau, E.; Schnürer, A. Forage types and origin of manure in co-digestion affect methane yield and microbial community structure. Grass Forage Sci. 2018. [Google Scholar] [CrossRef]
- Monlau, F.; Barakat, A.; Trably, E.; Dumas, C.; Steyer, J.-P.; Carrère, H. Lignocellulosic materials into biohydrogen and biomethane: Impact of structural features and pretreatment. Crit. Rev. Environ. Sci. Technol. 2013, 43, 260–322. [Google Scholar] [CrossRef]
- Neshat, S.A.; Mohammadi, M.; Najafpour, G.D.; Lahijani, P. Anaerobic co-digestion of animal manures and lignocellulosic residues as a potent approach for sustainable biogas production. Renew. Sustain. Energy Rev. 2017, 79, 308–322. [Google Scholar] [CrossRef]
- Mata-Alvarez, J.; Dosta, J.; Romero-Güiza, M.; Fonoll, X.; Peces, M.; Astals, S. A critical review on anaerobic co-digestion achievements between 2010 and 2013. Renew. Sustain. Energy Rev. 2014, 36, 412–427. [Google Scholar] [CrossRef]
- Schnürer, A. Biogas production: Microbiology and technology. Adv. Biochem. Eng. Biotechnol. 2016, 156, 195–234. [Google Scholar] [CrossRef]
- Mulat, D.G.; Jacobi, H.F.; Feilberg, A.; Adamsen, A.P.S.; Richnow, H.-H.; Nikolausz, M. Changing feeding regimes to demonstrate flexible biogas production: Effects on process performance, microbial community structure, and methanogenesis pathways. Appl. Environ. Microbiol. 2016, 82, 438–449. [Google Scholar] [CrossRef] [PubMed]
- Pap, B.; Maróti, G. Diversity of microbial communities in biogas reactors. Curr. Biochem. Eng. 2016, 3, 177–187. [Google Scholar] [CrossRef]
- Zhao, C.; Mu, H.; Zhao, Y.; Wang, L.; Zuo, B. Microbial characteristics analysis and kinetic studies on substrate composition to methane after microbial and nutritional regulation of fruit and vegetable wastes anaerobic digestion. Bioresour. Technol. 2018, 249, 315–321. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Liu, T.; Müller, B.; Schnürer, A. The microbial community structure in industrial biogas plants influences the degradation rate of straw and cellulose in batch tests. Biotechnol. Biofuels 2016, 9, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Vrieze, J.; Pinto, A.; T Sloan, W.; Zeeshan Ijaz, U. The active microbial community more accurately reflects the anaerobic digestion process: 16s rRNA (gene) sequencing as a predictive tool. Microbiome 2018, 6, 63. [Google Scholar] [CrossRef] [PubMed]
- De Vrieze, J.; Gildemyn, S.; Vilchez-Vargas, R.; Jáuregui, R.; Pieper, D.H.; Verstraete, W.; Boon, N. Inoculum selection is crucial to ensure operational stability in anaerobic digestion. Appl. Microbiol. Biotechnol. 2015, 99, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Sun, L.; Schnürer, B.M.A. Importance of inoculum source and initial community structure for biogas production from agricultural substrates. Bioresour. Technol. 2017, 245, 768–777. [Google Scholar] [CrossRef] [PubMed]
- Moestedt, J.; Muller, B.; Westerholm, M.; Schnurer, A. Ammonia threshold for inhibition of anaerobic digestion of thin stillage and the importance of organic loading rate. Microb. Biotechnol. 2016, 9, 180–194. [Google Scholar] [CrossRef] [PubMed]
- Müller, B.; Sun, L.; Westerholm, M.; Schnürer, A. Bacterial community composition and fhs profiles of low-and high-ammonia biogas digesters reveal novel syntrophic acetate-oxidising bacteria. Biotechnol. Biofuels 2016, 9, 1. [Google Scholar] [CrossRef] [PubMed]
- Ziels, R.M.; Beck, D.A.; Martí, M.; Gough, H.L.; Stensel, H.D.; Svensson, B.H. Monitoring the dynamics of syntrophic β-oxidizing bacteria during anaerobic degradation of oleic acid by quantitative PCR. FEMS Microbiol. Ecol. 2015, 91. [Google Scholar] [CrossRef] [PubMed]
- De Vrieze, J.; Verstraete, W.; Boon, N. Repeated pulse feeding induces functional stability in anaerobic digestion. Microb. Biotechnol. 2013, 6, 414–424. [Google Scholar] [CrossRef] [PubMed]
- Lv, Z.; Hu, M.; Harms, H.; Richnow, H.H.; Liebetrau, J.; Nikolausz, M. Stable isotope composition of biogas allows early warning of complete process failure as a result of ammonia inhibition in anaerobic digesters. Bioresour. Technol. 2014, 167, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Ziels, R.M.; Beck, D.A.C.; Stensel, H.D. Long-chain fatty acid feeding frequency in anaerobic co-digestion impacts syntrophic community structure and biokinetics. Water Res. 2017, 117, 218–229. [Google Scholar] [CrossRef] [PubMed]
- Ziels, R.M.; Sousa, D.Z.; Stensel, H.D.; Beck, D.A.C. DNA-sip based genome-centric metagenomics identifies key long-chain fatty acid-degrading populations in anaerobic digesters with different feeding frequencies. ISME J. 2018, 12, 112–123. [Google Scholar] [CrossRef] [PubMed]
- Piao, Z.H.; Lee, J.; Kim, J.Y. Effect of substrate feeding frequencies on the methane production and microbial communities of laboratory-scale anaerobic digestion reactors. J. Mater. Cycles Waste 2018, 20, 147–154. [Google Scholar] [CrossRef]
- Mauky, E.; Jacobi, H.F.; Liebetrau, J.; Nelles, M. Flexible biogas production for demand-driven energy supply—Feeding strategies and types of substrates. Bioresour. Technol. 2015, 178, 262–269. [Google Scholar] [CrossRef] [PubMed]
- Strömberg, S.; Nistor, M.; Liu, J. Towards eliminating systematic errors caused by the experimental conditions in biochemical methane potential (bmp) tests. Waste Manag. 2014, 34, 1939–1948. [Google Scholar] [CrossRef] [PubMed]
- Dechrugsa, S.; Kantachote, D.; Chaiprapat, S. Effects of inoculum to substrate ratio, substrate mix ratio and inoculum source on batch co-digestion of grass and pig manure. Bioresour. Technol. 2013, 146, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Westerholm, M.; Hansson, M.; Schnurer, A. Improved biogas production from whole stillage by co-digestion with cattle manure. Bioresour Technol. 2012, 114, 314–319. [Google Scholar] [CrossRef] [PubMed]
- Hansen, K.H.; Angelidaki, I.; Ahring, B.K. Anaerobic digestion of swine manure: Inhibition by ammonia. Water Res. 1998, 32, 5–12. [Google Scholar] [CrossRef]
- Hugerth, L.W.; Wefer, H.A.; Lundin, S.; Jakobsson, H.E.; Lindberg, M.; Rodin, S.; Engstrand, L.; Andersson, A.F. Degeprime, a program for degenerate primer design for broad-taxonomic-range PCR in microbial ecology studies. Appl. Environ. Microbiol. 2014, 80, 5116–5123. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Pena, A.G.; Goodrich, J.K.; Gordon, J.I. Qiime allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335. [Google Scholar] [CrossRef] [PubMed]
- Rideout, J.R.; He, Y.; Navas-Molina, J.A.; Walters, W.A.; Ursell, L.K.; Gibbons, S.M.; Chase, J.; McDonald, D.; Gonzalez, A.; Robbins-Pianka, A. Subsampled open-reference clustering creates consistent, comprehensive otu definitions and scales to billions of sequences. PeerJ 2014, 2, e545. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. Search and clustering orders of magnitude faster than blast. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Bittinger, K.; Bushman, F.D.; DeSantis, T.Z.; Andersen, G.L.; Knight, R. Pynast: A flexible tool for aligning sequences to a template alignment. Bioinformatics 2009, 26, 266–267. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J.; Gevers, D.; Earl, A.M.; Feldgarden, M.; Ward, D.V.; Giannoukos, G.; Ciulla, D.; Tabbaa, D.; Highlander, S.K.; Sodergren, E. Chimeric 16s rRNA sequence formation and detection in sanger and 454-pyrosequenced PCR amplicons. Genome Res. 2011. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naive bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Müller, B.; Schnürer, A. Biogas production from wheat straw: Community structure of cellulose-degrading bacteria. Energy Sustain. Soc. 2013, 3, 1–11. [Google Scholar] [CrossRef]
- Ahlberg-Eliasson, K.; Nadeau, E.; Levén, L.; Schnürer, A. Production efficiency of swedish farm-scale biogas plants. Biomass Bioenergy 2017, 97, 27–37. [Google Scholar] [CrossRef]
- Schnürer, A.; Bohn, I.; Moestedt, J. Protocol for Start-Up and Operation of CSTR Biogas Processes. In Hydrocarbon and Lipid Microbiology Protocols: Bioproducts, Biofuels, Biocatalysts and Facilitating Tools; McGenity, T.J., Timmis, K.N., Nogales, B., Eds.; Springer: Berlin/Heidelberg, Germany, 2017; pp. 171–200. [Google Scholar]
- Lindorfer, H.; Demmig, C. Foam formation in biogas plants—A survey on causes and control strategies. Chem. Eng. Technol. 2016, 39, 620–626. [Google Scholar] [CrossRef]
- He, Q.; Li, L.; Zhao, X.; Qu, L.; Wu, D.; Peng, X. Investigation of foaming causes in three mesophilic food waste digesters: Reactor performance and microbial analysis. Sci. Rep. 2017, 7, 13701. [Google Scholar] [CrossRef] [PubMed]
- Westerholm, M.; Moestedt, J.; Schnürer, A. Biogas production through syntrophic acetate oxidation and deliberate operating strategies for improved digester performance. Appl. Energy 2016, 179, 124–135. [Google Scholar] [CrossRef]
- Ruile, S.; Schmitz, S.; Mönch-Tegeder, M.; Oechsner, H. Degradation efficiency of agricultural biogas plants—A full-scale study. Bioresour. Technol. 2015, 178, 341–349. [Google Scholar] [CrossRef] [PubMed]
- De Francisci, D.; Kougias, P.G.; Treu, L.; Campanaro, S.; Angelidaki, I. Microbial diversity and dynamicity of biogas reactors due to radical changes of feedstock composition. Bioresour. Technol. 2015, 176, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Wang, H.; Qiu, Y.; Ren, L.; Jiang, B. Microbial characteristics in anaerobic digestion process of food waste for methane production-review. Bioresour. Technol. 2018, 248, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Ye, L.; Hu, H.; Zhang, L.; Ding, L.; Ren, H. Determination and variation of core bacterial community in a two-stage full-scale anaerobic reactor treating high-strength pharmaceutical wastewater. J. Microbiol. Biotechnol. 2017, 27, 1808–1819. [Google Scholar] [CrossRef] [PubMed]
- Christy, P.M.; Gopinath, L.; Divya, D. A review on anaerobic decomposition and enhancement of biogas production through enzymes and microorganisms. Renew. Sustain. Energy Rev. 2014, 34, 167–173. [Google Scholar] [CrossRef]
- Ueki, A.; Akasaka, H.; Suzuki, D.; Ueki, K. Paludibacter propionicigenes gen. nov., sp. nov., a novel strictly anaerobic, Gram-negative, propionate-producing bacterium isolated from plant residue in irrigated rice-field soil in Japan. Int. J. Syst. Evol. Microbiol. 2006, 56, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Chang, S. Impact of temperatures on microbial community structures of sewage sludge biological hydrolysis. Bioresour. Technol. 2017, 245, 502–510. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, L.; Kong, X.; Zhen, F.; Wang, Z.; Sun, Y.; Dong, P.; Lv, P. Inhibition effect of sodium concentrations on the anaerobic digestion performance of Sargassum species. Energy Fuels 2017, 31, 7101–7109. [Google Scholar] [CrossRef]
- Girija, D.; Deepa, K.; Xavier, F.; Antony, I.; Shidhi, P. Analysis of cow dung microbiota—A metagenomic approach. Indian J. Biotechnol. 2013, 12, 372–378. [Google Scholar]
- Dai, Y.; Yan, Z.; Jia, L.; Zhang, S.; Gao, L.; Wei, X.; Mei, Z.; Liu, X. The composition, localization and function of low-temperature-adapted microbial communities involved in methanogenic degradations of cellulose and chitin from qinghai-tibetan plateau wetland soils. J. Appl. Microbiol. 2016, 121, 163–176. [Google Scholar] [CrossRef] [PubMed]
- El-Bery, H.; Tawfik, A.; Kumari, S.; Bux, F. Effect of thermal pre-treatment on inoculum sludge to enhance bio-hydrogen production from alkali hydrolysed rice straw in a mesophilic anaerobic baffled reactor. Environ. Technol. 2013, 34, 1965–1972. [Google Scholar] [CrossRef] [PubMed]
- Garg, R.; Srivastava, R.; Brahma, V.; Verma, L.; Karthikeyan, S.; Sahni, G. Biochemical and structural characterization of a novel halotolerant cellulase from soil metagenome. Sci. Rep. 2016, 6, 39634. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Meng, Q.; Ren, L.; Liu, W.; Zhang, X.; Huo, Y.; Zhou, Z. Effects of nitrate addition on rumen fermentation, bacterial biodiversity and abundance. Asian-Austral. J. Anim. 2015, 28, 1433–1441. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, K.A.; Kittelmann, S.; Rogers, C.W.; Gee, E.K.; Bolwell, C.F.; Bermingham, E.N.; Thomas, D.G. Faecal microbiota of forage-fed horses in New Zealand and the population dynamics of microbial communities following dietary change. PLoS ONE 2014, 9, e112846. [Google Scholar] [CrossRef] [PubMed]
- Klevenhusen, F.; Petri, R.M.; Kleefisch, M.-T.; Khiaosa-ard, R.; Metzler-Zebeli, B.U.; Zebeli, Q. Changes in fibre-adherent and fluid-associated microbial communities and fermentation profiles in the rumen of cattle fed diets differing in hay quality and concentrate amount. FEMS Microbiol. Ecol. 2017, 93. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, H.; Wagner, I.D.; Wiegel, J. Caldicoprobacter oshimai gen. Nov., sp. Nov., an anaerobic, xylanolytic, extremely thermophilic bacterium isolated from sheep faeces, and proposal of caldicoprobacteraceae fam. Nov. Int. J. Syst. Evol. Microbiol. 2010, 60, 67–71. [Google Scholar] [CrossRef] [PubMed]
- Bouanane-Darenfed, A.; Hania, W.B.; Hacene, H.; Cayol, J.-L.; Ollivier, B.; Fardeau, M.-L. Caldicoprobacter guelmensis sp. Nov., a thermophilic, anaerobic, xylanolytic bacterium isolated from a hot spring. Int. J. Syst. Evol. Microbiol. 2013, 63, 2049–2053. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Huang, Z.; Zhao, M.; Ruan, W.; Miao, H.; Ren, H. Effects of co-inoculating rice straw with ruminal microbiota and anaerobic sludge: Digestion performance and spatial distribution of microbial communities. Appl. Microbiol. Biotechnol. 2017, 101, 5937–5948. [Google Scholar] [CrossRef] [PubMed]
- Mulat, D.G.; Huerta, S.G.; Kalyani, D.; Horn, S.J. Enhancing methane production from lignocellulosic biomass by combined steam-explosion pretreatment and bioaugmentation with cellulolytic bacterium Caldicellulosiruptor bescii. Biotechnol. Biofuels 2018, 11, 19. [Google Scholar] [CrossRef] [PubMed]
- Poirier, S.; Desmond-Le Quéméner, E.; Madigou, C.; Bouchez, T.; Chapleur, O. Anaerobic digestion of biowaste under extreme ammonia concentration: Identification of key microbial phylotypes. Bioresour. Technol. 2016, 207, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Vos, P.; Garrity, G.; Jones, D.; Krieg, N.R.; Ludwig, W.; Rainey, F.A.; Schleifer, K.-H.; Whitman, W.B. Bergey’s Manual of Systematic Bacteriology: Volume 3: The Firmicutes; Springer Science & Business Media: Berlin, Germany, 2011. [Google Scholar]
- Poehlein, A.; Zverlov, V.V.; Daniel, R.; Schwarz, W.H.; Liebl, W. Complete genome sequence of Clostridium stercorarium subsp. stercorarium Strain DSM 8532, a Thermophilic Degrader of Plant Cell Wall Fibers. Genome Announc. 2013, 1, e00073-13. [Google Scholar]
- Wei, Y.; Zhou, H.; Zhang, J.; Zhang, L.; Geng, A.; Liu, F.; Zhao, G.; Wang, S.; Zhou, Z.; Yan, X. Insight into dominant cellulolytic bacteria from two biogas digesters and their glycoside hydrolase genes. PLoS ONE 2015, 10, e0129921. [Google Scholar] [CrossRef] [PubMed]
- Su, H.; Liu, L.; Wang, S.; Wang, Q.; Jiang, Y.; Hou, X.; Tan, T. Semi-continuous anaerobic digestion for biogas production: Influence of ammonium acetate supplement and structure of the microbial community. Biotechnol. Biofuels 2015, 8, 13. [Google Scholar] [CrossRef] [PubMed]
- Haroun, B.M.; Nakhla, G.; Hafez, H.; Velayutham, P.; Levin, D.B.; Derakhshani, H.; Khafipour, E.; Nasr, F.A. Significance of acclimatization for biohydrogen production from synthetic lignocellulose hydrolysate in continuous-flow systems. Int. J. Hydrogen Energy 2016, 41, 14003–14014. [Google Scholar] [CrossRef]
- Dewhirst, F.E.; Paster, B.J.; Tzellas, N.; Coleman, B.; Downes, J.; Spratt, D.A.; Wade, W.G. Characterization of novel human oral isolates and cloned 16S rDNA sequences that fall in the family Coriobacteriaceae: Description of Olsenella gen. Nov., reclassification of Lactobacillus uli as Olsenella uli comb. Nov. And description of Olsenella profusa sp. Nov. Int. J. Syst. Evol. Microbiol. 2001, 51, 1797–1804. [Google Scholar]
- Sundberg, C.; Al-Soud, W.A.; Larsson, M.; Alm, E.; Yekta, S.S.; Svensson, B.H.; Sørensen, S.J.; Karlsson, A. 454 pyrosequencing analyses of bacterial and archaeal richness in 21 full-scale biogas digesters. FEMS Microbiol. Ecol. 2013, 85, 612–626. [Google Scholar] [CrossRef] [PubMed]
- Pore, S.D.; Shetty, D.; Arora, P.; Maheshwari, S.; Dhakephalkar, P.K. Metagenome changes in the biogas producing community during anaerobic digestion of rice straw. Bioresour. Technol. 2016, 213, 50–53. [Google Scholar] [CrossRef] [PubMed]
- Goux, X.; Calusinska, M.; Lemaigre, S.; Marynowska, M.; Klocke, M.; Udelhoven, T.; Benizri, E.; Delfosse, P. Microbial community dynamics in replicate anaerobic digesters exposed sequentially to increasing organic loading rate, acidosis, and process recovery. Biotechnol. Biofuels 2015, 8, 122. [Google Scholar] [CrossRef] [PubMed]
- Sleat, R.; Mah, R.A.; Robinson, R. Isolation and characterization of an anaerobic, cellulolytic bacterium, Costridium cellulovorans sp. Nov. Appl. Environ. Microbiol. 1984, 48, 88–93. [Google Scholar]
- Renouf, M.; Hendrich, S. Bacteroides uniformis is a putative bacterial species associated with the degradation of the isoflavone genistein in human feces. J. Nutr. 2011, 141, 1120–1126. [Google Scholar] [CrossRef] [PubMed]
- Koeck, D.E.; Hahnke, S.; Zverlov, V.V. Herbinix luporum sp. Nov., a thermophilic cellulose-degrading bacterium isolated from a thermophilic biogas reactor. Int. J. Syst. Evol. Microbiol. 2016, 66, 4132–4137. [Google Scholar] [PubMed]
- Maus, I.; Bremges, A.; Stolze, Y.; Hahnke, S.; Cibis, K.G.; Koeck, D.E.; Kim, Y.S.; Kreubel, J.; Hassa, J.; Wibberg, D. Genomics and prevalence of bacterial and archaeal isolates from biogas-producing microbiomes. Biotechnol. Biofuels 2017, 10, 264. [Google Scholar] [CrossRef] [PubMed]
- Krieg, N.R.; Ludwig, W.; Euzéby, J.; Whitman, W.B. Phylum xiv. Bacteroidetes phyl. Nov. In Bergey’s manual® of systematic bacteriology: Volume four The Bacteroidetes, Spirochaetes, Tenericutes (Mollicutes), Acidobacteria, Fibrobacteres, Fusobacteria, Dictyoglomi, Gemmatimonadetes, Lentisphaerae, Verrucomicrobia, Chlamydiae, and Planctomycetes; Krieg, N.R., Staley, J.T., Brown, D.R., Hedlund, B.P., Paster, B.J., Ward, N.L., Ludwig, W., Whitman, W.B., Eds.; Springer: New York, NY, USA, 2010; pp. 25–469. [Google Scholar]
- Benítez-Páez, A.; Gómez del Pulgar, E.M.; Sanz, Y. The glycolytic versatility of Bacteroides uniformis CECT 7771 and its genome response to oligo and polysaccharides. Front. Cell. Infect. Microbiol. 2017, 7, 383. [Google Scholar] [CrossRef] [PubMed]
- Koeck, D.E.; Maus, I.; Wibberg, D.; Winkler, A.; Zverlov, V.V.; Liebl, W.; Pühler, A.; Schwarz, W.H.; Schlüter, A. Complete genome sequence of Herbinix luporum SD1D, a new cellulose-degrading bacterium isolated from a thermophilic biogas reactor. Genome Announc. 2016, 4, 00687-16. [Google Scholar] [CrossRef] [PubMed]
- Gold, N.D.; Martin, V.J.J. Global view of the Clostridium thermocellum cellulosome revealed by quantitative proteomic analysis. J. Bacterial. 2007, 189, 6787–6795. [Google Scholar] [CrossRef] [PubMed]
- Swamy, M.V.; Ram, M.S.; Seenayya, G. B-amylase from Clostridium thermocellum SS8—A thermophilic, anaerobic, cellulolytic bacterium. Lett. Appl. Microbiol. 1994, 18, 301–304. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| VS | Crude Protein | Starch | Crude Fat | Crude Fiber | Ash | |
|---|---|---|---|---|---|---|
| Manure-grass mixture | 10.2 | 2.1 | 0.4 | 4.9 | 26.2 | 9.0 |
| MFW | 84.0 | 16.0 | 28.0 | 5.0 | 6.5 | 3.5 |
| Cellulose | Grass-manure mixture | MFW | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Days to Reach % of Final Potential | Final Potential | Days to Reach % of Final Potential | Final Potential | Days to Reach % of Final Potential | Final Potential | ||||||||
| Test | Inoculum | 100% | 80% | 50% | 100% | 80% | 50% | 100% | 80% | 50% | |||
| I | GB0_0 | 28 | 14 | 9 | 261 ± 33 | 52 | 16 | 5 | 329 ± 27 | 28 | 14 | 4 | 282 ± 24 |
| GC0_0 | 28 | 14 | 9 | 281 ± 17 | 52 | 17 | 7 | 382 ± 71 | 28 | 13 | 5 | 307 ± 53 | |
| II | GB1_231 | 119 | 56 | 21 | 350 ± 43 | 47 | 17 | 9 | 252 ± 29 | 65 | 27 | 15 | 278 ± 13 |
| GB2_231 | 119 | 61 | 35 | 270 ± 72 | 119 | 55 | 8 | 289 ± 11 | 119 | 54 | 19 | 322 ± 51 | |
| GC1_231 | 119 | 61 | 35 | 308 ± 4 | 119 | 45 | 17 | 327 ± 46 | 119 | 54 | 25 | 333 ± 43 | |
| GC2_231 | 119 | 61 | 38 | 274 ± 30 | 119 | 37 | 15 | 379 ± 73 | 119 | 39 | 25 | 307 ± 14 | |
| Sample | Chao1 | Observed OTUs | Shannon | Simpson |
|---|---|---|---|---|
| GB1_0 | 1571 | 1302 | 6.178 | 0.950 |
| GB2_0 | 1619 | 1439 | 6.473 | 0.953 |
| GB1_77 | 1410 | 1159 | 5.291 | 0.898 |
| GB2_77 | 1485 | 1260 | 6.054 | 0.938 |
| GB1_106 | 1673 | 1364 | 5.749 | 0.921 |
| GB2_106 | 1773 | 1435 | 6.254 | 0.946 |
| GB1_147 | 1477 | 1232 | 6.181 | 0.943 |
| GB2_147 | 1544 | 1291 | 5.982 | 0.935 |
| GB1_231 | 1453 | 1104 | 5.410 | 0.887 |
| GB2_231 | 1449 | 1085 | 5.365 | 0.893 |
| GC1_0 | 1813 | 1664 | 7.210 | 0.959 |
| GC2_0 | 1872 | 1666 | 7.119 | 0.963 |
| GC1_77 | 1592 | 1299 | 5.583 | 0.891 |
| GC2_77 | 1650 | 1386 | 5.979 | 0.910 |
| GC1_106 | 1718 | 1529 | 7.073 | 0.970 |
| GC2_106 | 1744 | 1509 | 6.748 | 0.955 |
| GC1_147 | 1775 | 1448 | 6.681 | 0.962 |
| GC2_147 | 1675 | 1439 | 7.202 | 0.972 |
| GC1_231 | 1414 | 989 | 5.431 | 0.872 |
| GC2_231 | 1444 | 958 | 5.054 | 0.831 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, T.; Sun, L.; Nordberg, Å.; Schnürer, A. Substrate-Induced Response in Biogas Process Performance and Microbial Community Relates Back to Inoculum Source. Microorganisms 2018, 6, 80. https://doi.org/10.3390/microorganisms6030080
Liu T, Sun L, Nordberg Å, Schnürer A. Substrate-Induced Response in Biogas Process Performance and Microbial Community Relates Back to Inoculum Source. Microorganisms. 2018; 6(3):80. https://doi.org/10.3390/microorganisms6030080
Chicago/Turabian StyleLiu, Tong, Li Sun, Åke Nordberg, and Anna Schnürer. 2018. "Substrate-Induced Response in Biogas Process Performance and Microbial Community Relates Back to Inoculum Source" Microorganisms 6, no. 3: 80. https://doi.org/10.3390/microorganisms6030080
APA StyleLiu, T., Sun, L., Nordberg, Å., & Schnürer, A. (2018). Substrate-Induced Response in Biogas Process Performance and Microbial Community Relates Back to Inoculum Source. Microorganisms, 6(3), 80. https://doi.org/10.3390/microorganisms6030080