Effects of Vertical Water Mass Segregation on Bacterial Community Structure in the Beaufort Sea


Abstract
:1. Introduction
2. Materials and Methods
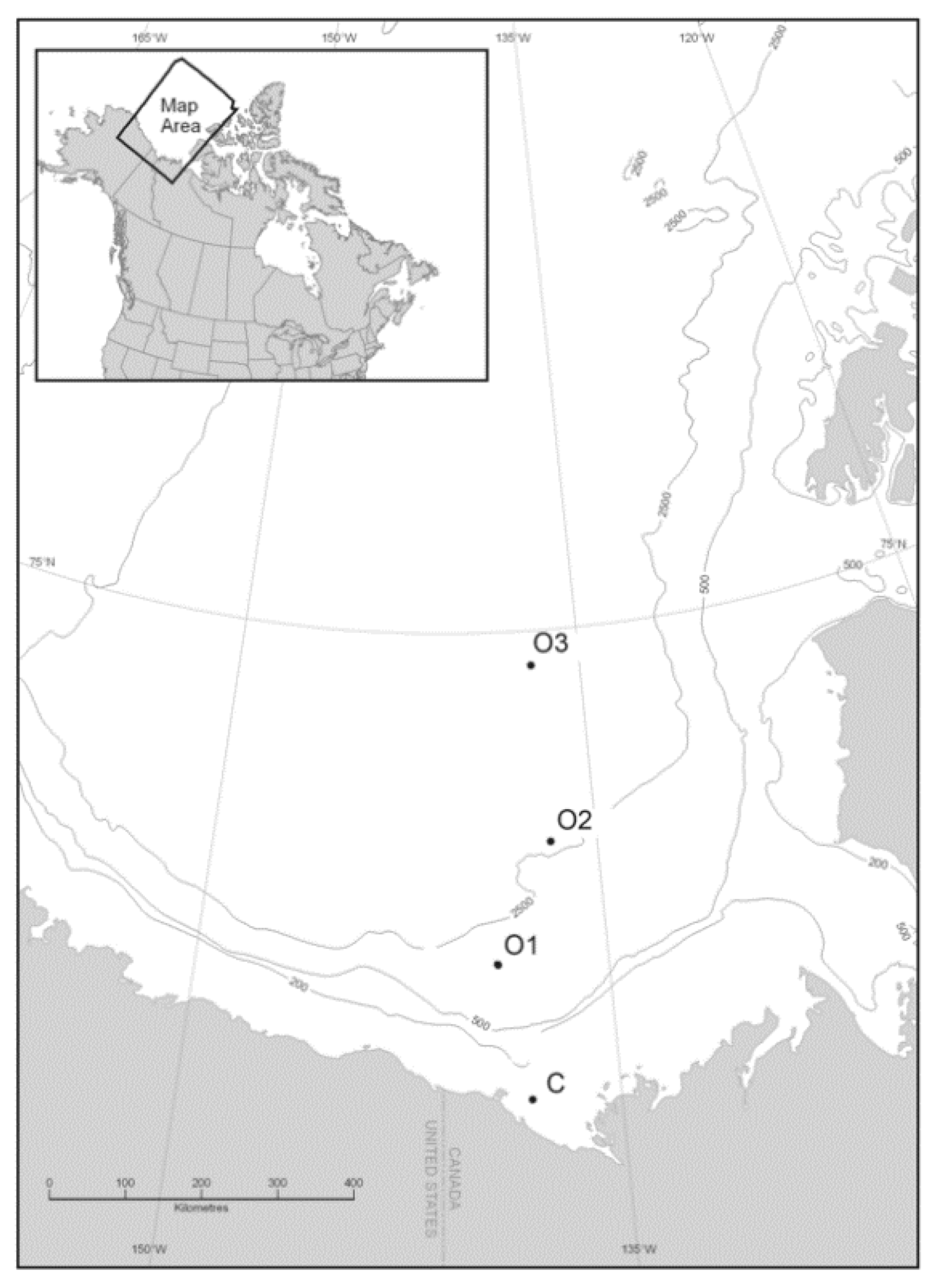
2.1. Sample Sites, Physicochemical Parameters, and Biological Properties
2.2. DNA Extraction, PCR Amplification and Sequencing
2.3. Sequence Analyses
2.4. Statistical Analyses of Bacterial Community Structure
3. Results
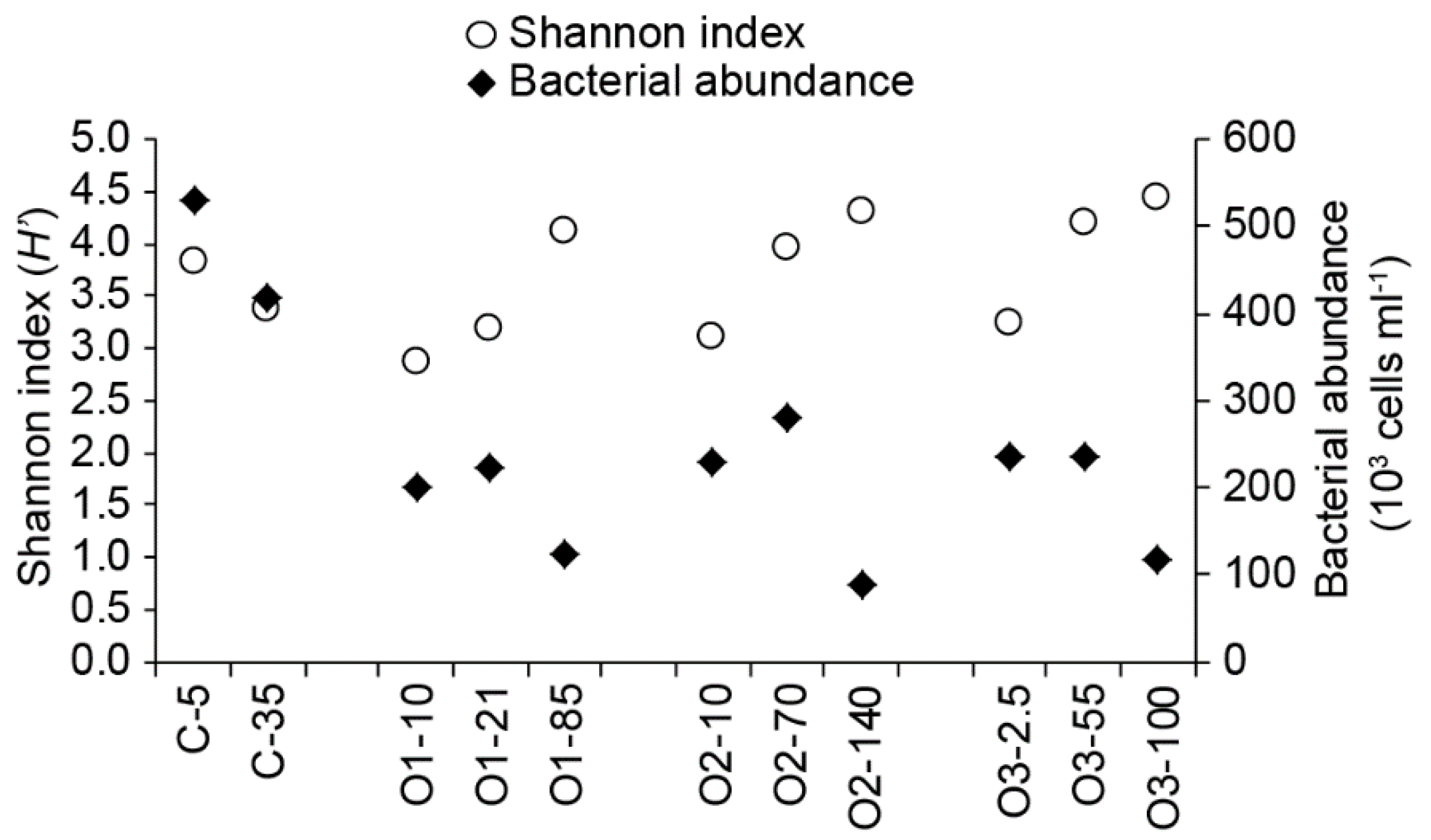
3.1. General Station and Sample Characteristics
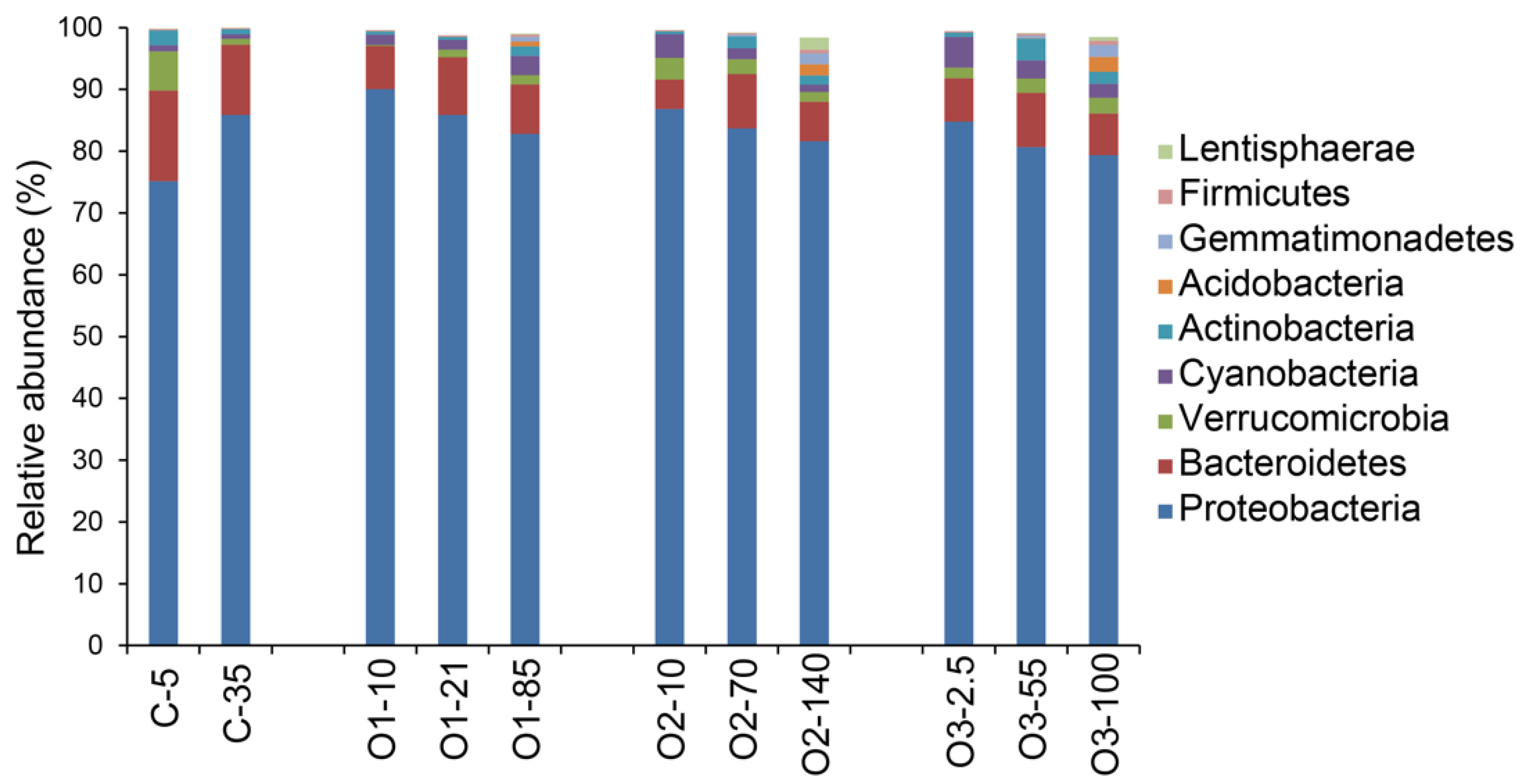
3.2. Bacterial Community Characterizations
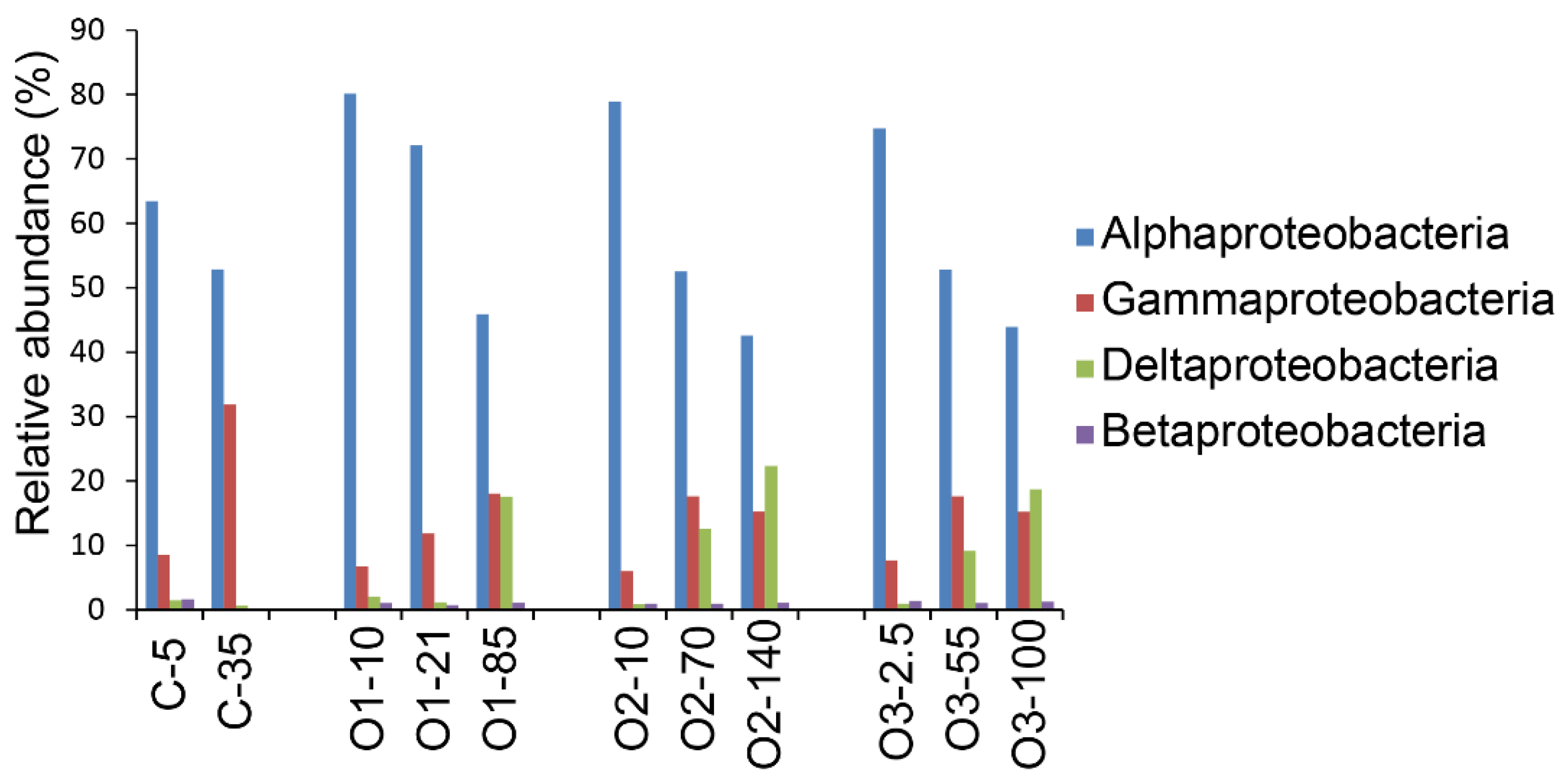
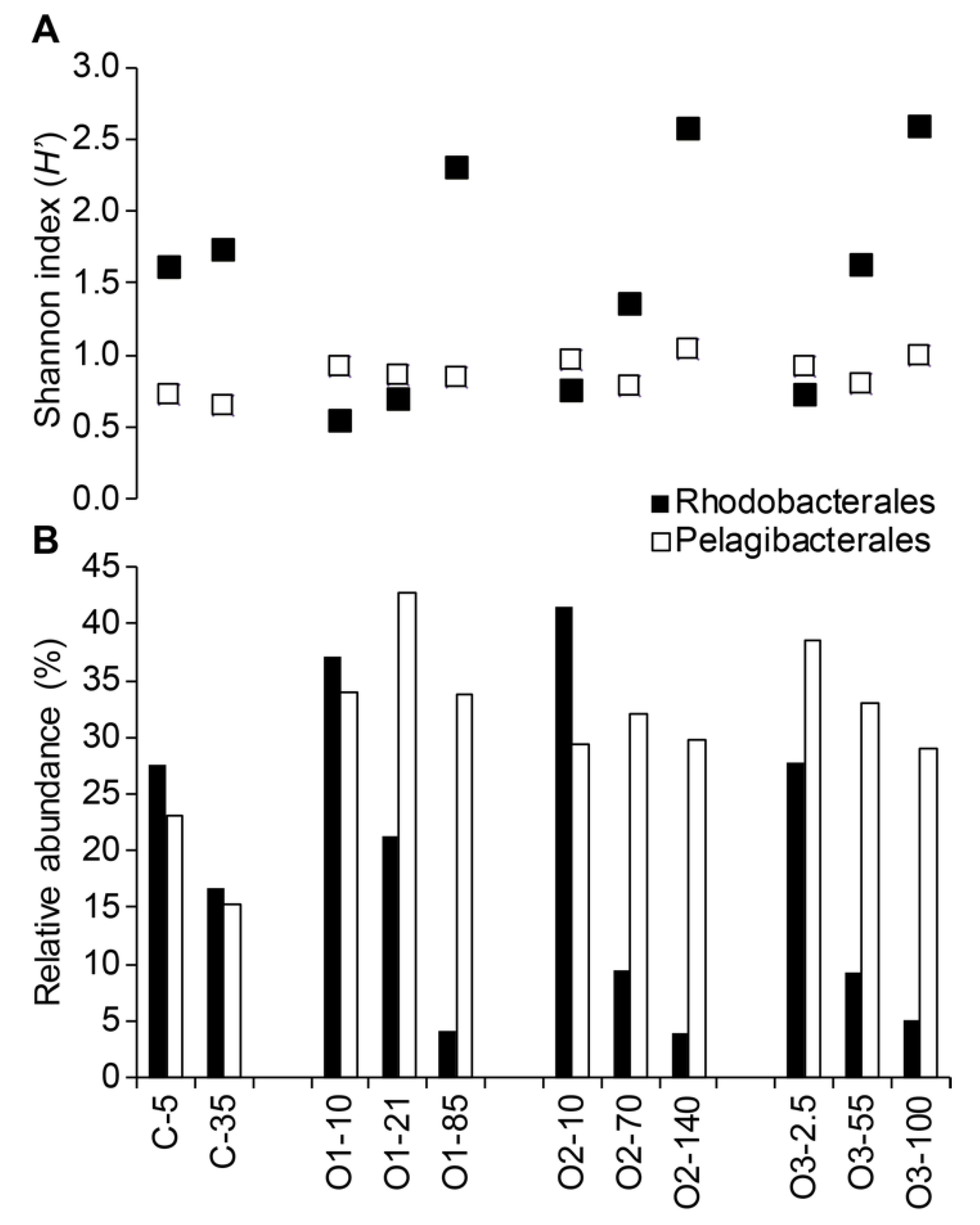
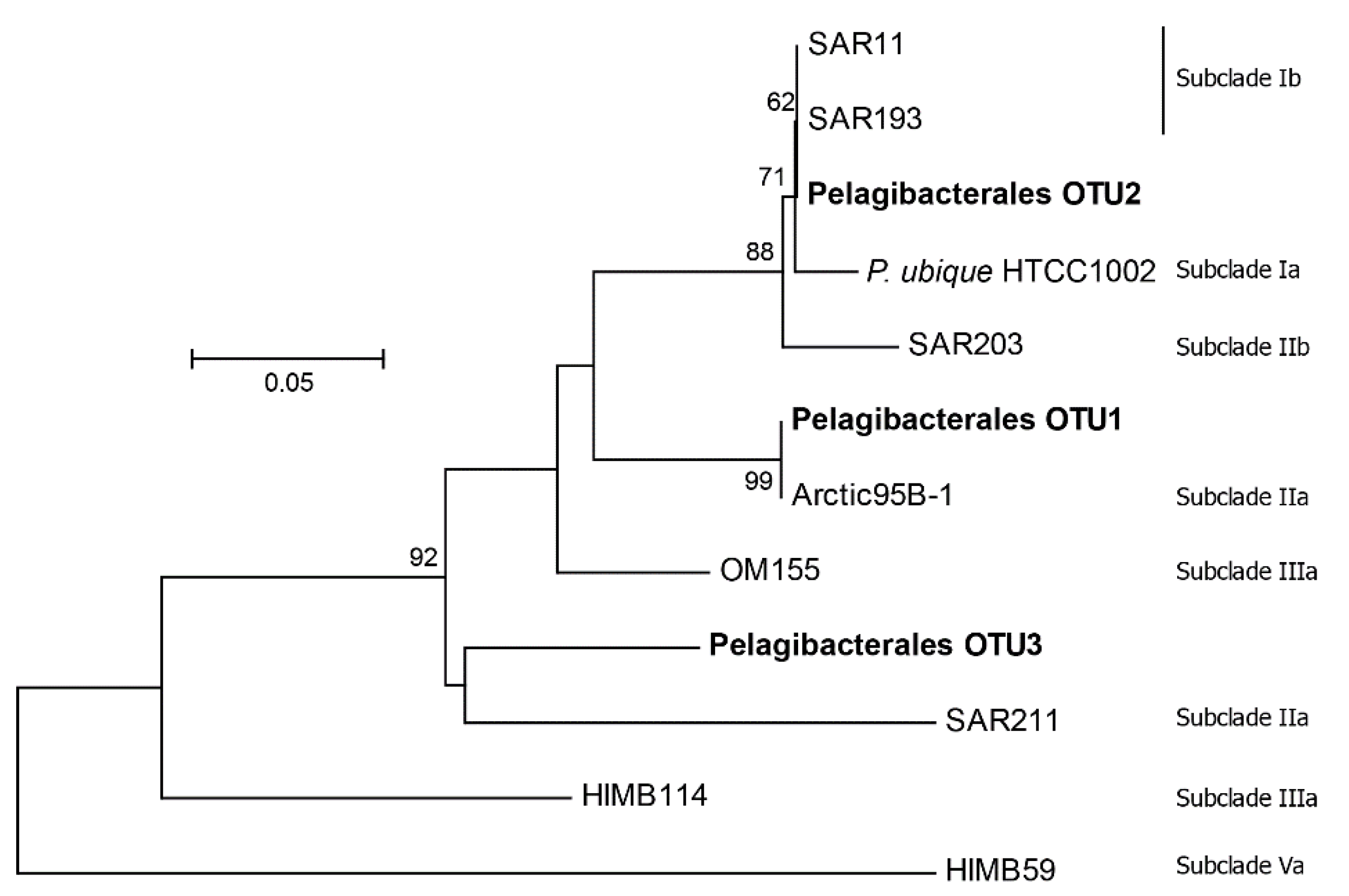
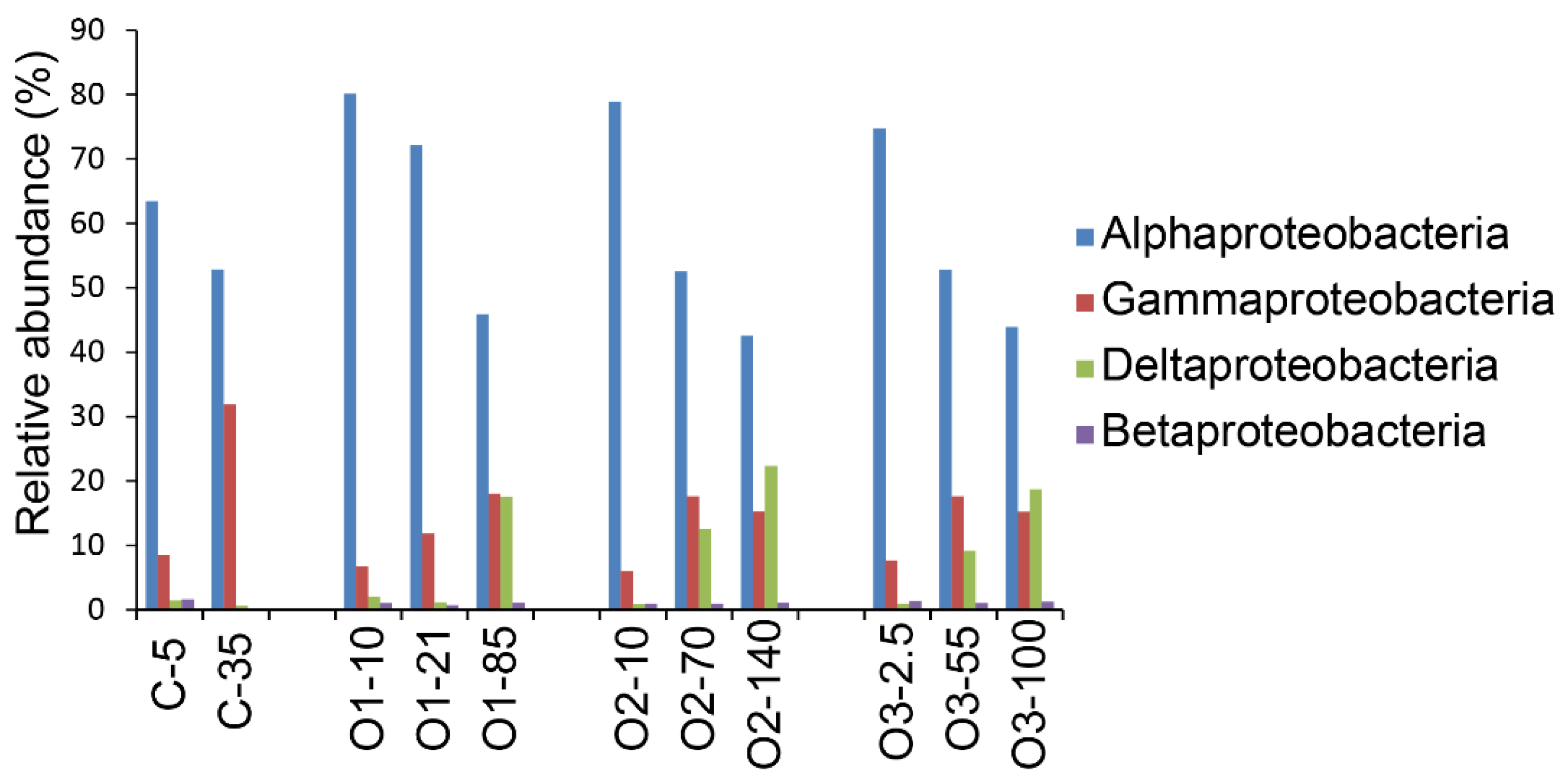
3.3. Patterns for the Alphaproteobacteria, Rhodobacterales, and Pelagibacterales
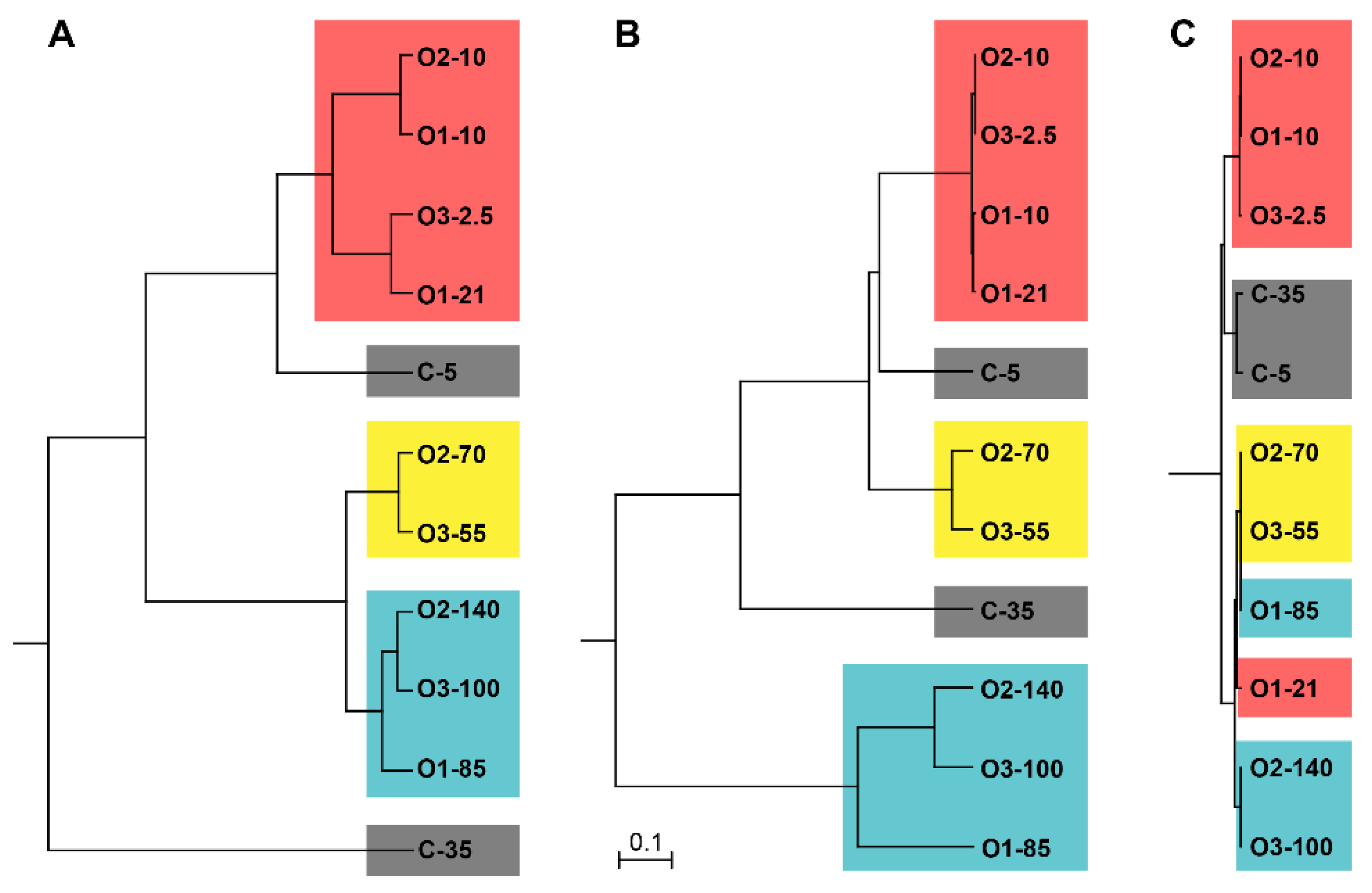
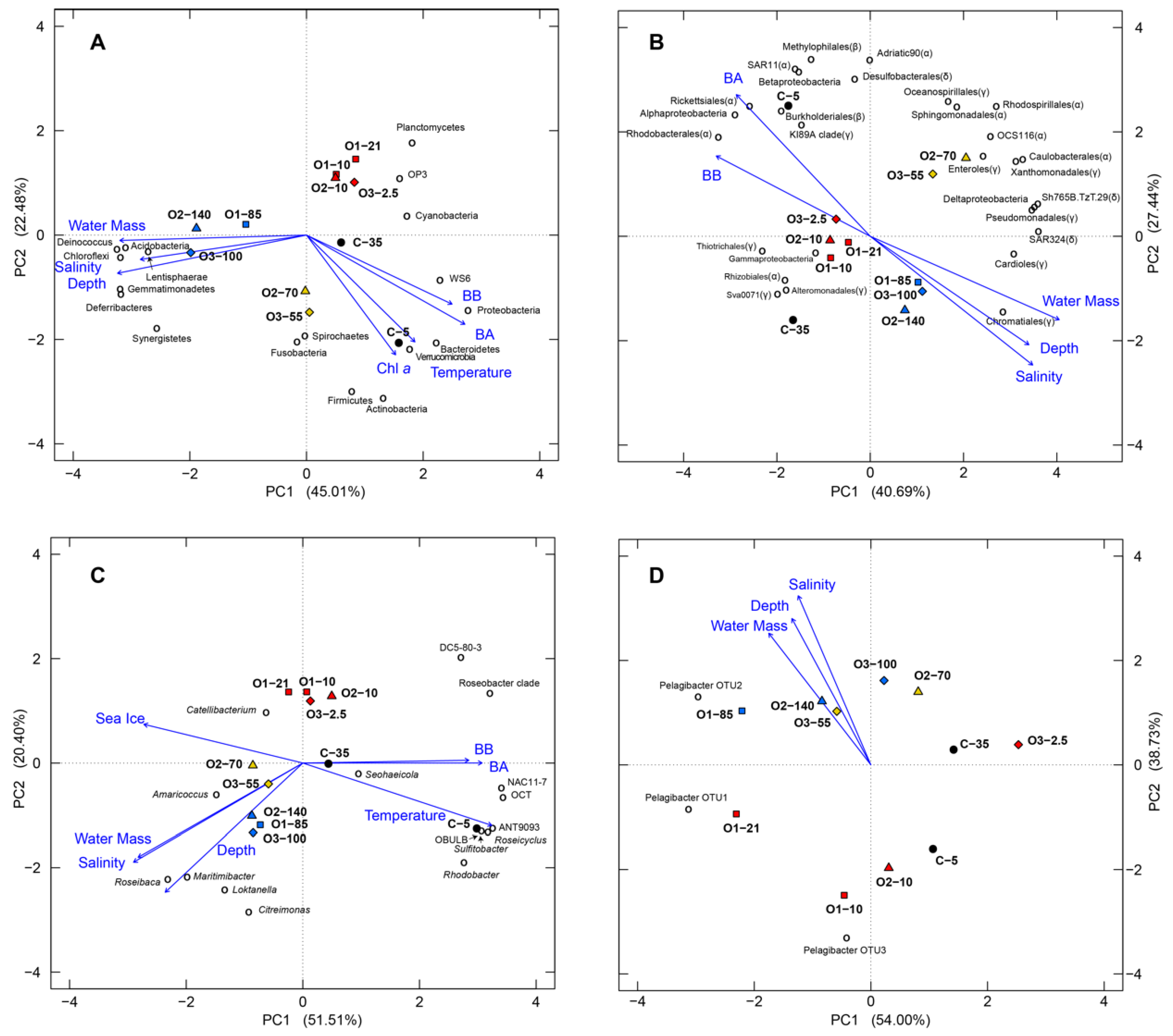
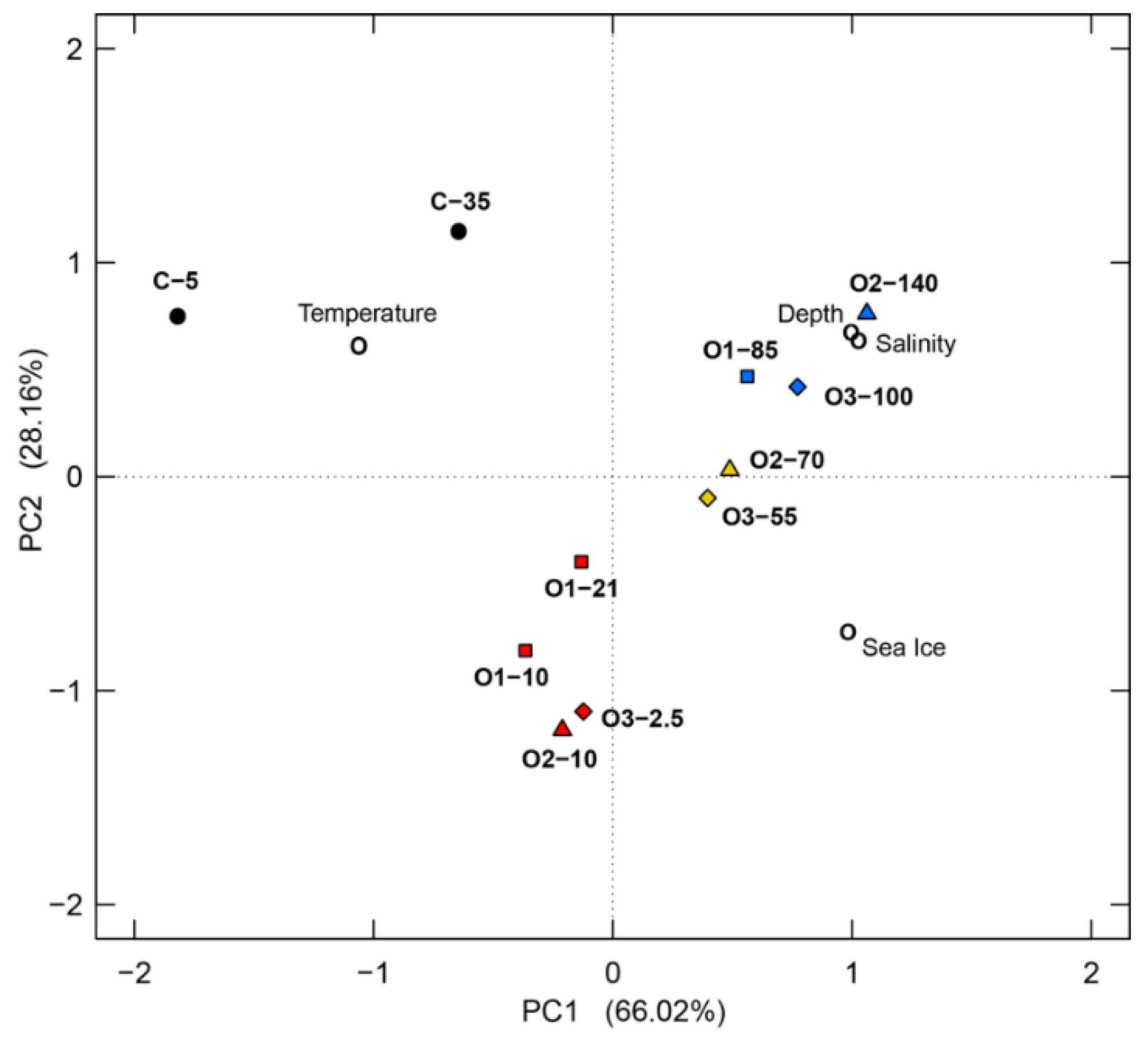
3.4. Bacterial Community Comparisons and the Effects of Environmental Parameters on Community Structure
4. Discussion
4.1. The Mackenzie River Plume
4.2. Water Mass Segregation and Bacterial Community Composition
4.3. Community Structure within Water Masses
4.4. Dominance of Alphaproteobacteria in the Beaufort Sea
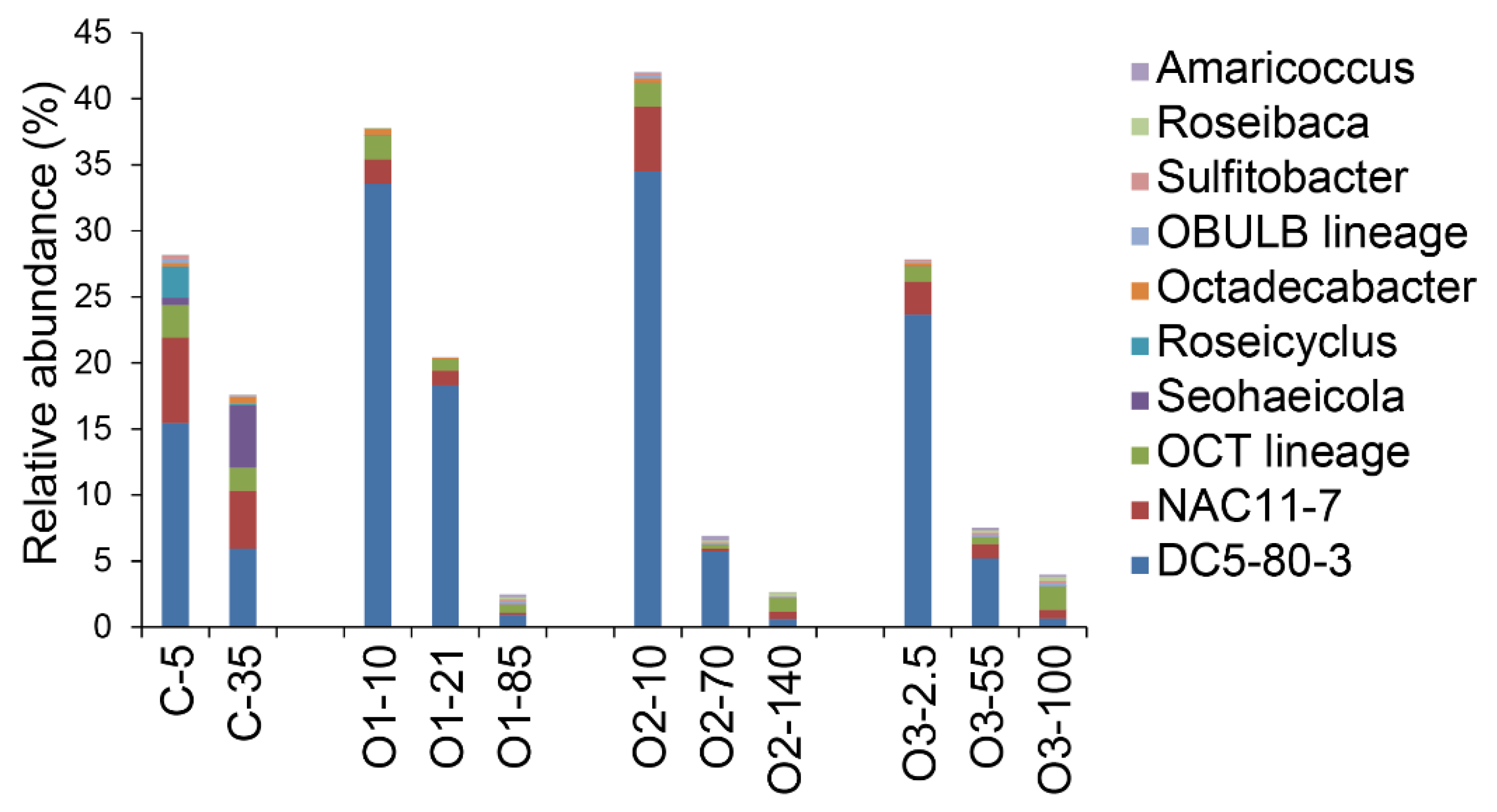
4.5. Other Abundant Groups
4.6. Influence of Environmental Factors on Bacterial Communities
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Karl, D.M. Nutrient dynamics in the deep blue sea. Trends Microbiol. 2002, 10, 410–418. [Google Scholar] [CrossRef]
- Fenchel, T. The microbial loop-25 years later. J. Exp. Mar. Biol. Ecol. 2008, 366, 99–103. [Google Scholar] [CrossRef]
- Amon, R.M.W.; Fitznar, H.P.; Benner, R. Linkages among the bioreactivity, chemical composition, and diagenetic state of marine dissolved organic matter. Limnol. Oceanogr. 2001, 46, 287–297. [Google Scholar] [CrossRef]
- Pommier, T.; Canbäck, B.; Riemann, L.; Boström, K.H.; Simu, K.; Lundberg, P.; Tunlid, A.; Hagström, Å. Global patterns of diversity and community structure in marine bacterioplankton. Mol. Ecol. 2007, 16, 867–880. [Google Scholar] [CrossRef] [PubMed]
- Giovannoni, S.J.; Rappé, M.S.; Vergin, K.L.; Adair, N.L. 16S rRNA genes reveal stratified open ocean bacterioplankton populations related to the Green Non-Sulfur bacteria. Proc. Natl. Acad. Sci. USA 1996, 93, 7979–7984. [Google Scholar] [CrossRef]
- Field, K.G.; Gordon, D.; Wright, T.; Rappé, M.; Urbach, E.; Vergin, K.; Giovannoni, S.J. Diversity and depth-specific distribution of SAR11 cluster rRNA genes from marine planktonic bacteria. Appl. Environ. Microbiol. 1997, 63, 63–70. [Google Scholar] [PubMed]
- Karner, M.B.; Delong, E.F.; Karl, D.M. Archaeal dominance in the mesopelagic zone of the Pacific Ocean. Nature 2001, 409, 507–510. [Google Scholar] [CrossRef] [PubMed]
- DeLong, E.F.; Preston, C.M.; Mincer, T.; Rich, V.; Hallam, S.J.; Frigaard, N.U.; Martinez, A.; Sullivan, M.B.; Edwards, R.; Brito, B.R.; et al. Community genomics among stratified microbial assemblages in the ocean’s interior. Science 2006, 311, 496–503. [Google Scholar] [CrossRef] [PubMed]
- Pham, V.D.; Konstantinidis, K.T.; Palden, T.; DeLong, E.F. Phylogenetic analyses of ribosomal DNA-containing bacterioplankton genome fragments from a 4000 m vertical profile in the North Pacific Subtropical Gyre. Environ. Microbiol. 2008, 10, 2313–2330. [Google Scholar] [CrossRef] [PubMed]
- Giovannoni, S.J.; Stingl, U. Molecular diversity and ecology of microbial plankton. Nature 2005, 437, 343–348. [Google Scholar] [CrossRef] [PubMed]
- Fuhrman, J.A.; Steele, J.A.; Hewson, I.; Schwalbach, M.S.; Brown, M.V.; Green, J.L.; Brown, J.H. A latitudinal diversity gradient in planktonic marine bacteria. Proc. Natl. Acad. Sci. USA 2008, 105, 7774–7778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howarth, R.W.; Marino, R. Nitrogen as the limiting nutrient for eutrophication in coastal marine ecosystems: Evolving views over three decades. Limnol. Oceanogr. 2006, 51, 364–376. [Google Scholar] [CrossRef] [Green Version]
- Nixon, S.W.; Granger, S.L.; Nowicki, B.L. An assessment of the annual mass balance of carbon, nitrogen, and phosphorus in Narragansett Bay. Biogeochemistry 1995, 31, 15–61. [Google Scholar] [CrossRef]
- Agogué, H.; Brink, M.; Dinasquet, J.; Herndl, G.J. Major gradients in putatively nitrifying and non-nitrifying Archaea in the deep North Atlantic. Nature 2008, 456, 788–792. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, A.K.; Lovejoy, C.; Galand, P.E.; Ingram, R.G. Water masses and biogeography of picoeukaryote assemblages in a cold hydrographically complex system. Limnol. Oceanogr. 2008, 53, 922–935. [Google Scholar] [CrossRef] [Green Version]
- Varela, M.M.; Van Aken, H.M.; Sintes, E.; Herndl, G.J. Latitudinal trends of Crenarchaeota and Bacteria in the meso- and bathypelagic water masses of the Eastern North Atlantic. Environ. Microbiol. 2008, 10, 110–124. [Google Scholar] [CrossRef]
- Galand, P.E.; Potvin, M.; Casamayor, E.O.; Lovejoy, C. Hydrography shapes bacterial biogeography of the deep Arctic Ocean. ISME J. 2010, 4, 564–576. [Google Scholar] [CrossRef]
- Fu, Y.; Keats, K.F.; Rivkin, R.B.; Lang, A.S. Water mass and depth determine the distribution and diversity of Rhodobacterales in an Arctic marine system. FEMS Microbiol. Ecol. 2013, 84, 564–576. [Google Scholar] [CrossRef]
- Matz, C.; Jürgens, K. Interaction of Nutrient Limitation and Protozoan Grazing Determines the Phenotypic Structure of a Bacterial Community. Microb. Ecol. 2003, 45, 384–398. [Google Scholar] [CrossRef]
- Hooper, D.U.; Chapin Iii, F.S.; Ewel, J.J.; Hector, A.; Inchausti, P.; Lavorel, S.; Lawton, J.H.; Lodge, D.M.; Loreau, M.; Naeem, S.; et al. Effects of biodiversity on ecosystem functioning: A consensus of current knowledge. Ecol. Monogr. 2005, 75, 3–35. [Google Scholar] [CrossRef]
- Pernthaler, J. Predation on prokaryotes in the water column and its ecological implications. Nat. Rev. Microbiol. 2005, 3, 537–546. [Google Scholar] [CrossRef] [PubMed]
- Giovannoni, S.J.; Britschgi, T.B.; Moyer, C.L.; Field, K.G. Genetic diversity in Sargasso Sea bacterioplankton. Nature 1990, 345, 60–63. [Google Scholar] [CrossRef] [PubMed]
- García-Martínez, J.; Rodríguez-Valera, F. Microdiversity of uncultured marine prokaryotes: The SAR11 cluster and the marine Archaea of Group I. Mol. Ecol. 2000, 9, 935–948. [Google Scholar] [CrossRef]
- Morris, R.M.; Rappe, M.S.; Connon, S.A.; Vergin, K.L.; Siebold, W.A.; Carlson, C.A.; Giovannoni, S.J. SAR11 clade dominates ocean surface bacterioplankton communities. Nature 2002, 420, 806–810. [Google Scholar] [CrossRef] [PubMed]
- Alonso, C.; Pernthaler, J. Roseobacter and SAR11 dominate microbial glucose uptake in coastal North Sea waters. Environ. Microbiol. 2006, 8, 2022–2030. [Google Scholar] [CrossRef] [PubMed]
- Brinkmeyer, R.; Rappe, M.; Gallacher, S.; Medlin, L. Development of clade—(Roseobacter and Alteromonas) and taxon-specific oligonucleotide probes to study interactions between toxic dinoflagellates and their associated bacteria. Eur. J. Phycol. 2000, 35, 315–329. [Google Scholar] [CrossRef]
- González, J.M.; Simó, R.; Massana, R.; Covert, J.S.; Casamayor, E.O.; Pedrós-Alió, C.; Moran, M.A. Bacterial community structure associated with a dimethylsulfoniopropionate-producing North Atlantic algal bloom. Appl. Environ. Microbiol. 2000, 66, 4237–4246. [Google Scholar] [CrossRef] [PubMed]
- Selje, N.; Simon, M.; Brinkhoff, T. A newly discovered Roseobacter cluster in temperate and polar oceans. Nature 2004, 427, 445–448. [Google Scholar] [CrossRef] [PubMed]
- Alderkamp, A.C.; Sintes, E.; Herndl, G.J. Abundance and activity of major groups of prokaryotic plankton in the coastal North Sea during spring and summer. Aquat. Microb. Ecol. 2006, 45, 237–246. [Google Scholar] [CrossRef]
- Sperling, M.; Giebel, H.; Rink, B.; Grayek, S.; Staneva, J.; Stanev, E.; Simon, M. Differential effects of hydrographic and biogeochemical properties on the SAR11 clade and Roseobacter RCA cluster in the North Sea. Aquat. Microb. Ecol. 2012, 67, 25–34. [Google Scholar] [CrossRef] [Green Version]
- Newton, R.J.; Griffin, L.E.; Bowles, K.M.; Meile, C.; Gifford, S.; Givens, C.E.; Howard, E.C.; King, E.; Oakley, C.A.; Reisch, C.R.; et al. Genome characteristics of a generalist marine bacterial lineage. ISME J. 2010, 4, 784–798. [Google Scholar] [CrossRef] [Green Version]
- Bouvier, T.C.; del Giorgio, P.A. Compositional changes in free-living bacterial communities along a salinity gradient in two temperate estuaries. Limnol. Oceanogr. 2002, 47, 453–470. [Google Scholar] [CrossRef]
- Hoppe, H.G.; Gocke, K.; Koppe, R.; Begler, C. Bacterial growth and primary production along a north-south transect of the Atlantic Ocean. Nature 2002, 416, 168–171. [Google Scholar] [CrossRef]
- Baldwin, A.J.; Moss, J.A.; Pakulski, J.D.; Catala, P.; Joux, F.; Jeffrey, W.H. Microbial diversity in a Pacific Ocean transect from the Arctic to Antarctic circles. Aquat. Microb. Ecol. 2005, 41, 91–102. [Google Scholar] [CrossRef]
- Taniguchi, A.; Hamasaki, K. Community structures of actively growing bacteria shift along a north-south transect in the western North Pacific. Environ. Microbiol. 2008, 10, 1007–1017. [Google Scholar] [CrossRef] [Green Version]
- Bouman, H.A.; Ulloa, O.; Scanlan, D.J.; Zwirglmaier, K.; Li, W.K.; Platt, T.; Stuart, V.; Barlow, R.; Leth, O.; Clementson, L.; et al. Oceanographic basis of the global surface distribution of Prochlorococcus ecotypes. Science 2006, 312, 918–921. [Google Scholar] [CrossRef]
- Eiler, A.; Hayakawa, D.H.; Church, M.J.; Karl, D.M.; Rappé, M.S. Dynamics of the SAR11 bacterioplankton lineage in relation to environmental conditions in the oligotrophic North Pacific subtropical gyre. Environ. Microbiol. 2009, 11, 2291–2300. [Google Scholar] [CrossRef]
- Varaljay, V.A.; Gifford, S.M.; Wilson, S.T.; Sharma, S.; Karl, D.M.; Moran, M.A. Bacterial dimethylsulfoniopropionate degradation genes in the oligotrophic North Pacific subtropical gyre. Appl. Environ. Microbiol. 2012, 78, 2775–2782. [Google Scholar] [CrossRef]
- Kirchman, D.L.; Cottrell, M.T.; Lovejoy, C. The structure of bacterial communities in the western Arctic Ocean as revealed by pyrosequencing of 16S rRNA genes. Environ. Microbiol. 2010, 12, 1132–1143. [Google Scholar] [CrossRef]
- Comeau, A.M.; Li, W.K.W.; Tremblay, J.É.; Carmack, E.C.; Lovejoy, C. Arctic ocean microbial community structure before and after the 2007 record sea ice minimum. PLoS ONE 2011, 6, e27492. [Google Scholar] [CrossRef]
- Han, D.; Kang, I.; Ha, H.K.; Kim, H.C.; Kim, O.-S.; Lee, B.Y.; Cho, J.-C.; Hur, H.-G.; Lee, Y.K. Bacterial communities of surface mixed layer in the Pacific sector of the western Arctic Ocean during sea-ice melting. PLoS ONE 2014, 9, e86887. [Google Scholar] [CrossRef]
- Bowman, J.S.; Rasmussen, S.; Blom, N.; Deming, J.W.; Rysgaard, S.; Sicheritz-Ponten, T. Microbial community structure of Arctic multiyear sea ice and surface seawater by 454 sequencing of the 16S RNA gene. ISME J. 2012, 6, 11–20. [Google Scholar] [CrossRef]
- McBean, G. Arctic Climate: Past and Present. In ACIA; Symon, C., Ed.; Cambridge University Press: Cambridge, UK, 2005; pp. 21–60. [Google Scholar]
- Dittmar, T.; Kattner, G. The biogeochemistry of the river and shelf ecosystem of the Arctic Ocean: A review. Mar. Chem. 2003, 83, 103–120. [Google Scholar] [CrossRef]
- Forest, A.; Babin, M.; Stemmann, L.; Picheral, M.; Sampei, M.; Fortier, L.; Gratton, Y.; Bélanger, S.; Devred, E.; Sahlin, J.; et al. Ecosystem function and particle flux dynamics across the Mackenzie Shelf (Beaufort Sea, Arctic Ocean): An integrative analysis of spatial variability and biophysical forcings. Biogeosciences 2013, 10, 2833–2866. [Google Scholar] [CrossRef]
- Carson, M.A.; Jasper, J.N.; Conly, P.M. Magnitude and sources of sediment input to the Mackenzie Delta, Northwest Territories, 1974–94. Arctic 1998, 51, 116–124. [Google Scholar] [CrossRef]
- Holmes, R.M.; McClelland, J.W.; Peterson, B.J.; Shiklomanov, I.A.; Shiklomanov, A.I.; Zhulidov, A.V.; Gordeev, V.V.; Bobrovitskaya, N.N. A circumpolar perspective on fluvial sediment flux to the Arctic ocean. Glob. Biogeochem. Cycles 2002, 16, 45-1–45-14. [Google Scholar] [CrossRef]
- Ortega-Retuerta, E.; Joux, F.; Jeffrey, W.H.; Ghiglione, J.F. Spatial variability of particle-attached and free-living bacterial diversity in surface waters from the Mackenzie River to the Beaufort Sea (Canadian Arctic). Biogeosciences 2013, 10, 2747–2759. [Google Scholar] [CrossRef] [Green Version]
- Vallières, C.; Retamal, L.; Ramlal, P.; Osburn, C.L.; Vincent, W.F. Bacterial production and microbial food web structure in a large arctic river and the coastal Arctic Ocean. J. Mar. Syst. 2008, 74, 756–773. [Google Scholar] [CrossRef]
- Barbeau, K.; Kujawinski, E.B.; Moffett, J.W. Remineralization and recycling of iron, thorium and organic carbon by heterotrophic marine protists in culture. Aquat. Microb. Ecol. 2001, 24, 69–81. [Google Scholar] [CrossRef]
- Strom, S.L. Microbial ecology of ocean biogeochemistry: A community perspective. Science 2008, 320, 1043–1045. [Google Scholar] [CrossRef]
- Kirchman, D.L.; Morán, X.A.G.; Ducklow, H. Microbial growth in the polar oceans—Role of temperature and potential impact of climate change. Nat. Rev. Microbiol. 2009, 7, 451. [Google Scholar] [CrossRef]
- Wassmann, P.; Duarte, C.M.; Agustí, S.; Sejr, M.K. Footprints of climate change in the Arctic marine ecosystem. Glob. Chang. Biol. 2011, 17, 1235–1249. [Google Scholar] [CrossRef]
- Matsuoka, A.; Bricaud, A.; Benner, R.; Para, J.; Sempéré, R.; Prieur, L.; Bélanger, S.; Babin, M. Tracing the transport of colored dissolved organic matter in water masses of the Southern Beaufort Sea: Relationship with hydrographic characteristics. Biogeosciences 2012, 9, 925. [Google Scholar] [CrossRef]
- Marie, D.; Brussaard, C.P.D.; Thyrhaug, R.; Bratbak, G.; Vaulot, D. Enumeration of marine viruses in culture and natural samples by flow cytometry. Appl. Environ. Microbiol. 1999, 65, 45–52. [Google Scholar]
- Li, W.K.W.; Dickie, P.M. Monitoring phytoplankton, bacterioplankton, and virioplankton in a coastal inlet (Bedford Basin) by flow cytometry. Cytometry 2001, 44, 236–246. [Google Scholar] [CrossRef]
- Hobbie, J.E.; Daley, R.J.; Jasper, S. Use of nuclepore filters for counting bacteria by fluorescence microscopy. Appl. Environ. Microbiol. 1977, 33, 1225–1228. [Google Scholar] [Green Version]
- Simon, M.; Azam, F. Protein-content and protein-synthesis rates of planktonic marine-bacteria. Mar. Ecol. Prog. Ser. 1989, 51, 201–213. [Google Scholar] [CrossRef]
- Hale, M.S.; Rivkin, R.B.; Matthews, P.; Agawin, N.S.; Li, W.K.W. Microbial response to mesoscale iron enrichment in the NE Subarctic Pacific: Bacterial dynamics. Deep Sea Res. Part II 2006, 53, 2231–2247. [Google Scholar] [CrossRef]
- Huber, J.A.; Mark Welch, D.B.; Morrison, H.G.; Huse, S.M.; Neal, P.R.; Butterfield, D.A.; Sogin, M.L. Microbial population structures in the deep marine biosphere. Science 2007, 318, 97–100. [Google Scholar] [CrossRef]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef]
- Vergin, K.L.; Beszteri, B.; Monier, A.; Cameron Thrash, J.; Temperton, B.; Treusch, A.H.; Kilpert, F.; Worden, A.Z.; Giovannoni, S.J. High-resolution SAR11 ecotype dynamics at the Bermuda Atlantic Time-series Study site by phylogenetic placement of pyrosequences. ISME J. 2013, 7, 1322–1332. [Google Scholar] [CrossRef] [Green Version]
- Bano, N.; Hollibaugh, J.T. Phylogenetic Composition of Bacterioplankton Assemblages from the Arctic Ocean. Appl. Environ. Microbiol. 2002, 68, 505. [Google Scholar] [CrossRef]
- Tatusova, T.; Ciufo, S.; Fedorov, B.; O’Neill, K.; Tolstoy, I. RefSeq microbial genomes database: New representation and annotation strategy. Nucleic Acids Res. 2014, 42, D553–D559. [Google Scholar] [CrossRef]
- Rappé, M.S.; Kemp, P.F.; Giovannoni, S.J. Phylogenetic diversity of marine coastal picoplankton 16S rRNA genes cloned from the continental shelf off Cape Hatteras, North Carolina. Limnol. Oceanogr. 1997, 42, 811–826. [Google Scholar] [CrossRef] [Green Version]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis Version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [Green Version]
- Shannon, C.E.; Weaver, W. The Mathematical Theory of Communication; University of Illinois Press: Urbana, IL, USA, 1949; 117p. [Google Scholar]
- Simpson, E.H. Measurement of Diversity. Nature 1949, 163, 688. [Google Scholar] [CrossRef]
- Pielou, E.C. An Introduction to Mathematical Ecology; Wiley-Interscience: Hoboken, NJ, USA, 1969. [Google Scholar]
- Chao, A. Nonparametric-Estimation of the Number of Classes in a Population. Scand. J. Stat. 1984, 11, 265–270. [Google Scholar]
- Yue, J.C.; Clayton, M.K. A Similarity Measure Based on Species Proportions. Commun. Stat. Theory Methods 2005, 34, 2123–2131. [Google Scholar] [CrossRef] [Green Version]
- Oksanen, J.; Blanchet, F.G.; Kindt, R.; Legendre, P.; Minchin, P.R.; O’Hara, R.; Simpson, G.L.; Solymos, P.; Stevens, M.H.H.; Wagner, H. Package ‘vegan’. Community Ecol. Package Version 2013, 2, 1–295. [Google Scholar]
- Zeng, Y.X.; Zhang, F.; He, J.F.; Lee, S.H.; Qiao, Z.Y.; Yu, Y.; Li, H.R. Bacterioplankton community structure in the Arctic waters as revealed by pyrosequencing of 16S rRNA genes. Antonie Van Leeuwenhoek 2013, 103, 1309–1319. [Google Scholar] [CrossRef]
- Spencer-Cervato, C.; Thierstein, H.R. First appearance of Globorotalia truncatulinoides: Cladogenesis and immigration. Mar. Micropaleontol. 1997, 30, 267–291. [Google Scholar] [CrossRef]
- Ortmann, A.C.; Santos, T.T.L. Spatial and temporal patterns in the Pelagibacteraceae across an estuarine gradient. FEMS Microbiol. Ecol. 2016, 92. [Google Scholar] [CrossRef]
- Agogué, H.; Lamy, D.; Neal, P.R.; Sogin, M.L.; Herndl, G.J. Water mass-specificity of bacterial communities in the North Atlantic revealed by massively parallel sequencing. Mol. Ecol. 2011, 20, 258–274. [Google Scholar] [CrossRef]
- Chow, C.E.T.; Sachdeva, R.; Cram, J.A.; Steele, J.A.; Needham, D.M.; Patel, A.; Parada, A.E.; Fuhrman, J.A. Temporal variability and coherence of euphotic zone bacterial communities over a decade in the Southern California Bight. ISME J. 2013, 7, 2259–2273. [Google Scholar] [CrossRef] [Green Version]
- Aksenov, Y.; Ivanov, V.V.; Nurser, A.J.G.; Bacon, S.; Polyakov, I.V.; Coward, A.C.; Naveira-Garabato, A.C.; Beszczynska-Moeller, A. The Arctic Circumpolar Boundary Current. J. Geophys. Res. 2011, 116. [Google Scholar] [CrossRef] [Green Version]
- Giebel, H.A.; Kalhoefer, D.; Lemke, A.; Thole, S.; Gahl-Janssen, R.; Simon, M.; Brinkhoff, T. Distribution of Roseobacter RCA and SAR11 lineages in the North Sea and characteristics of an abundant RCA isolate. ISME J. 2011, 5, 8–19. [Google Scholar] [CrossRef]
- Malmstrom, R.R.; Straza, T.R.A.; Cottrell, M.T.; Kirchman, D.L. Diversity, abundance, and biomass production of bacterial groups in the western Arctic Ocean. Aquat. Microb. Ecol. 2007, 47, 45–55. [Google Scholar] [CrossRef] [Green Version]
- Giebel, H.A.; Brinkhoff, T.; Zwisler, W.; Selje, N.; Simon, M. Distribution of Roseobacter RCA and SAR11 lineages and distinct bacterial communities from the subtropics to the Southern Ocean. Environ. Microbiol. 2009, 11, 2164–2178. [Google Scholar] [CrossRef]
- Sapp, M.; Wichels, A.; Wiltshire, K.H.; Gerdts, G. Bacterial community dynamics during the winter-spring transition in the North Sea. FEMS Microbiol. Ecol. 2007, 59, 622–637. [Google Scholar] [CrossRef]
- Giebel, H.A.; Kalhoefer, D.; Gahl-Janssen, R.; Choo, Y.J.; Lee, K.; Cho, J.C.; Tindall, B.J.; Rhiel, E.; Beardsley, C.; Aydogmus, O.O.; et al. Planktomarina temperata gen. nov., sp. nov., belonging to the globally distributed RCA cluster of the marine Roseobacter clade, isolated from the German Wadden Sea. Int. J. Syst. Evol. Microbiol. 2013, 63, 4207–4217. [Google Scholar] [CrossRef]
- Voget, S.; Wemheuer, B.; Brinkhoff, T.; Vollmers, J.; Dietrich, S.; Giebel, H.-A.; Beardsley, C.; Sardemann, C.; Bakenhus, I.; Billerbeck, S.; et al. Adaptation of an abundant Roseobacter RCA organism to pelagic systems revealed by genomic and transcriptomic analyses. ISME J. 2015, 9, 371–384. [Google Scholar] [CrossRef]
- Luo, H.; Swan, B.K.; Stepanauskas, R.; Hughes, A.L.; Moran, M.A. Evolutionary analysis of a streamlined lineage of surface ocean Roseobacters. ISME J. 2014, 8, 1428. [Google Scholar] [CrossRef]
- Luo, H.; Csűros, M.; Hughes, A.L.; Moran, M.A. Evolution of Divergent Life History Strategies in Marine Alphaproteobacteria. MBio 2013, 4. [Google Scholar] [CrossRef]
- West, N.J.; Obernosterer, I.; Zemb, O.; Lebaron, P. Major differences of bacterial diversity and activity inside and outside of a natural iron-fertilized phytoplankton bloom in the Southern Ocean. Environ. Microbiol. 2008, 10, 738–756. [Google Scholar] [CrossRef]
- Buchan, A.; González, J.M.; Moran, M.A. Overview of the marine Roseobacter lineage. Appl. Environ. Microbiol. 2005, 71, 5665–5677. [Google Scholar] [CrossRef]
- Zubkov, M.V.; Fuchs, B.M.; Archer, S.D.; Kiene, R.P.; Amann, R.; Burkill, P.H. Linking the composition of bacterioplankton to rapid turnover of dissolved dimethylsulphoniopropionate in an algal bloom in the North Sea. Environ. Microbiol. 2001, 3, 304–311. [Google Scholar] [CrossRef]
- Yao, D.; Buchan, A.; Suzuki, M.T. In Situ activity of NAC11-7 roseobacters in coastal waters off the Chesapeake Bay based on ftsZ expression. Environ. Microbiol. 2011, 13, 1032–1041. [Google Scholar] [CrossRef]
- El-Swais, H.; Dunn, K.A.; Bielawski, J.P.; Li, W.K.; Walsh, D.A. Seasonal assemblages and short-lived blooms in coastal north-west Atlantic Ocean bacterioplankton. Environ. Microbiol. 2015, 17, 3642–3661. [Google Scholar] [CrossRef]
- Sowell, S.M.; Abraham, P.E.; Shah, M.; Verberkmoes, N.C.; Smith, D.P.; Barofsky, D.F.; Giovannoni, S.J. Environmental proteomics of microbial plankton in a highly productive coastal upwelling system. ISME J. 2010. [Google Scholar] [CrossRef]
- Wilson, B.; Muller, O.; Nordmann, E.; Seuthe, L.; Bratbak, G.; Ovreas, L. Changes in Marine Prokaryote Composition with Season and Depth Over an Arctic Polar Year. Front. Mar. Sci. 2017, 4. [Google Scholar] [CrossRef] [Green Version]
- Han, D.; Ha, H.K.; Hwang, C.Y.; Lee, B.Y.; Hur, H.G.; Lee, Y.K. Bacterial communities along stratified water columns at the Chukchi Borderland in the western Arctic Ocean. Deep Sea Res. Part II 2015, 120, 52–60. [Google Scholar] [CrossRef]
- Li, Y.; Wang, Z.; Lin, X. Microbial community structure of Arctic seawater as revealed by pyrosequencing. Acta Oceanol. Sin. 2016, 35, 78–84. [Google Scholar] [CrossRef]
- Brinkhoff, T.; Giebel, H.A.; Simon, M. Diversity, ecology, and genomics of the Roseobacter clade: A short overview. Arch. Microbiol. 2008, 189, 531–539. [Google Scholar] [CrossRef]
- Giovannoni, S.J.; Tripp, H.J.; Givan, S.; Podar, M.; Vergin, K.L.; Baptista, D.; Bibbs, L.; Eads, J.; Richardson, T.H.; Noordewier, M.; et al. Genetics: Genome streamlining in a cosmopolitan oceanic bacterium. Science 2005, 309, 1242–1245. [Google Scholar] [CrossRef]
- Giovannoni, S.J. SAR11 Bacteria: The Most Abundant Plankton in the Oceans. Ann. Rev. Mar. Sci. 2017, 9, 231–255. [Google Scholar] [CrossRef]
- Zeng, Y.; Yu, Y.; Li, H.; He, J.; Lee, S.H.; Sun, K. Phylogenetic diversity of planktonic bacteria in the Chukchi Borderland region in summer. Acta Oceanol. Sin. 2013, 32, 66–74. [Google Scholar] [CrossRef]
- Brown, M.V.; Lauro, F.M.; Demaere, M.Z.; Muir, L.; Wilkins, D.; Thomas, T.; Riddle, M.J.; Fuhrman, J.A.; Andrews-Pfannkoch, C.; Hoffman, J.M.; et al. Global biogeography of SAR11 marine bacteria. Mol. Syst. Biol. 2012, 8. [Google Scholar] [CrossRef]
- Dinasquet, J.; Ortega-Retuerta, E.; Lovejoy, C.C.; Obernosterer, I. Editorial: Microbiology of the rapidly changing polar environments. Front. Mar. Sci. 2018, 5, 154. [Google Scholar] [CrossRef]
- Murphy, E.J.; Cavanagh, R.D.; Drinkwater, K.F.; Grant, S.M.; Heymans, J.J.; Hofmann, E.E.; Hunt, G.L.; Johnston, N.M. Understanding the structure and functioning of polar pelagic ecosystems to predict the impacts of change. Proc. R. Soc. B Biol. Sci. 2016, 283, 20161646. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample ID | Date | Longitude (°W) | Latitude (°N) | Depth (m) | Relative Light Intensity (%) | Water Mass | Temp. (°C) | Salinity (psu) | Sea Ice Coverage (%) |
|---|---|---|---|---|---|---|---|---|---|
| C-5 | 2009–08-30 | 137.9964 | 69.5008 | 4.7 | 50 | Coastal | 3.13 | 23.75 | 0 |
| C-35 | 2009–08-30 | 137.9964 | 69.5008 | 35 | 1 | Coastal | 0.58 | 30.40 | 0 |
| O1–10 | 2009–09-01 | 139.0255 | 71.1059 | 10 | Surface | UPML | −1.02 | 25.78 | 70 |
| O1–21 | 2009–08-31 | 139.0090 | 71.0922 | 21.8 | 50 | UPML | −0.90 | 28.18 | 70 |
| O1–85 | 2009–08-31 | 139.0090 | 71.0922 | 85 | 1 | PSW | −1.27 | 31.75 | 70 |
| O2–10 | 2009–09-09 | 136.7900 | 72.5158 | 9.9 | 50 | UPML | −1.34 | 25.26 | 90 |
| O2–70 | 2009–09-09 | 136.7900 | 72.5158 | 69.5 | 1 | LPML | −1.06 | 30.67 | 90 |
| O2–140 | 2009–09-09 | 136.7900 | 72.5158 | 139.8 | 0.1 | PSW | −1.36 | 32.46 | 90 |
| O3–2.5 | 2009–09-04 | 137.1223 | 74.5937 | 2.7 | Surface | UPML | −1.41 | 26.71 | 90 |
| O3–55 | 2009–09-04 | 137.1223 | 74.5937 | 54.6 | 1 | LPML | −1.03 | 30.56 | 90 |
| O3–100 | 2009–09-04 | 137.1223 | 74.5937 | 100 | 0.1 | PSW | −1.16 | 31.98 | 90 |
| Sample ID | Bacterial Biomass (µg C L−1) | Bacterial Abundance (103 cells mL−1) | Cellular Carbon (fg/cell) | Bacterial Production (µg C L−1 d−1) | Bacterial Growth Rate (d−1) | Chl a (µg L−1) |
|---|---|---|---|---|---|---|
| C-5 | 9.03 | 530 | 17.04 | 0.62 | 0.06 | 0.14 |
| C-35 | 9.57 | 417 | 22.95 | 1.22 | 0.15 | 0.11 |
| O1–10 | 3.63 | 200 | 17.99 | 0.11 | 0.03 | 0.18 |
| O1–21 | 3.68 | 222 | 16.56 | 0.05 | 0.01 | 0.05 |
| O1–85 | 2.08 | 124 | 16.81 | 0.03 | 0.01 | 0.12 |
| O2–10 | 3.80 | 230 | 16.53 | 0.23 | 0.06 | 0.06 |
| O2–70 | 3.65 | 281 | 13.00 | 0.09 | 0.02 | 0.15 |
| O2–140 | 1.28 | 88 | 14.58 | 0.01 | 0.01 | 0.01 |
| O3–2.5 | 3.99 | 237 | 16.85 | 0.14 | 0.03 | 0.09 |
| O3–55 | 4.00 | 236 | 16.96 | 0.06 | 0.02 | 0.19 |
| O3–100 | 1.90 | 117 | 16.22 | 0.02 | 0.01 | 0.03 |
| Number of Sequences | Number of OTUs | Shannon Index (H’) | Chao | Inverse Simpson (1/D) | |
|---|---|---|---|---|---|
| Total community | |||||
| C-5 | 181,488 | 3886 | 3.83 | 10,619.34 | 14.003 |
| C-35 | 205,484 | 2805 | 3.37 | 7242.58 | 11.881 |
| O1–10 | 158,302 | 4177 | 2.87 | 6544.76 | 5.668 |
| O1–21 | 153,607 | 2333 | 3.18 | 5465.40 | 7.192 |
| O1–85 | 185,815 | 2201 | 4.11 | 11,808.09 | 10.470 |
| O2–10 | 150,701 | 4501 | 3.11 | 6861.16 | 6.048 |
| O2–70 | 99,495 | 2334 | 3.97 | 6391.22 | 12.284 |
| O2–140 | 162,668 | 2473 | 4.30 | 13,157.34 | 11.756 |
| O3–2.5 | 48,356 | 1200 | 3.25 | 2781.98 | 7.199 |
| O3–55 | 142,258 | 3444 | 4.19 | 9256.73 | 12.481 |
| O3–100 | 125,773 | 3756 | 4.45 | 9982.94 | 13.938 |
| Rhodobacterales | |||||
| C-5 | 44,137 | 461 | 1.62 | 3143.73 | 2.786 |
| C-35 | 30,626 | 300 | 1.74 | 1860.32 | 3.968 |
| O1–10 | 53,691 | 284 | 0.54 | 1718.79 | 1.220 |
| O1–21 | 30,117 | 195 | 0.70 | 1295.25 | 1.340 |
| O1–85 | 6598 | 196 | 2.31 | 728.83 | 4.630 |
| O2–10 | 57,235 | 346 | 0.75 | 1692.90 | 1.420 |
| O2–70 | 8586 | 127 | 1.35 | 564.10 | 2.278 |
| O2–140 | 5346 | 222 | 2.57 | 1037.06 | 6.135 |
| O3–2.5 | 12,362 | 106 | 0.72 | 448.33 | 1.370 |
| O3–55 | 12,006 | 191 | 1.63 | 994.08 | 2.732 |
| O3–100 | 5295 | 202 | 2.59 | 841.40 | 6.329 |
| Pelagibacterales | |||||
| C-5 | 34,209 | 339 | 0.73 | 1340.28 | 1.399 |
| C-35 | 26,576 | 211 | 0.65 | 1106.59 | 1.330 |
| O1–10 | 43,824 | 323 | 0.93 | 1809.57 | 1.731 |
| O1–21 | 54,449 | 313 | 0.86 | 1493.67 | 1.623 |
| O1–85 | 50,809 | 415 | 0.84 | 1846.03 | 1.494 |
| O2–10 | 36,343 | 269 | 0.97 | 1266.5 | 1.814 |
| O2–70 | 26,518 | 222 | 0.79 | 914.652 | 1.514 |
| O2–140 | 37,500 | 419 | 1.04 | 2394.55 | 1.704 |
| O3–2.5 | 15,565 | 153 | 0.93 | 543.056 | 1.711 |
| O3–55 | 38,762 | 352 | 0.80 | 1346.38 | 1.468 |
| O3–100 | 28,721 | 285 | 1.00 | 1177.22 | 1.714 |
| Variable 1 | Total Community | Rhodobacterales | Pelagibacterales | |||
|---|---|---|---|---|---|---|
| F | p-Value | F | p-value | F | p-Value | |
| Water Mass | 23.93 2 | <0.001 | 109.52 | <0.001 | 8.13 | 0.011 |
| Sampling station | 0.51 | 0.686 | 0.19 | 0.900 | 3.10 | 0.099 |
| r | p-value | r | p-value | r | p-value | |
| Temperature | −0.011 | 0.975 | 0.059 | 0.862 | −0.639 | 0.034 |
| Salinity | 0.703 | 0.016 | 0.736 | 0.010 | 0.103 | 0.763 |
| Density | 0.701 | 0.016 | 0.721 | 0.012 | 0.139 | 0.684 |
| BB | −0.316 | 0.344 | −0.196 | 0.564 | −0.816 | 0.002 |
| BA | −0.267 | 0.427 | −0.258 | 0.444 | −0.794 | 0.004 |
| BP | −0.258 | 0.444 | −0.000 | 1.000 | −0.720 | 0.013 |
| BGR | −0.374 | 0.257 | −0.112 | 0.744 | −0.624 | 0.040 |
| Chl a | −0.175 | 0.606 | −0.336 | 0.312 | −0.584 | 0.059 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fu, Y.; Rivkin, R.B.; Lang, A.S. Effects of Vertical Water Mass Segregation on Bacterial Community Structure in the Beaufort Sea. Microorganisms 2019, 7, 385. https://doi.org/10.3390/microorganisms7100385
Fu Y, Rivkin RB, Lang AS. Effects of Vertical Water Mass Segregation on Bacterial Community Structure in the Beaufort Sea. Microorganisms. 2019; 7(10):385. https://doi.org/10.3390/microorganisms7100385
Chicago/Turabian StyleFu, Yunyun, Richard B. Rivkin, and Andrew S. Lang. 2019. "Effects of Vertical Water Mass Segregation on Bacterial Community Structure in the Beaufort Sea" Microorganisms 7, no. 10: 385. https://doi.org/10.3390/microorganisms7100385
APA StyleFu, Y., Rivkin, R. B., & Lang, A. S. (2019). Effects of Vertical Water Mass Segregation on Bacterial Community Structure in the Beaufort Sea. Microorganisms, 7(10), 385. https://doi.org/10.3390/microorganisms7100385