Proteomic and Transcriptional Profiles of Human Stem Cell-Derived β Cells Following Enteroviral Challenge

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells
2.2. Infection
2.3. Immunofluorescence
2.4. Perifusion
2.5. Viability Studies
2.6. Proteomics
2.7. Transcriptome (RNA-Seq)
2.8. Statistics
3. Results
3.1. Immunostaining Revealed Colocalization of Coxsackie Viral Protein and Insulin in SC-β Cells
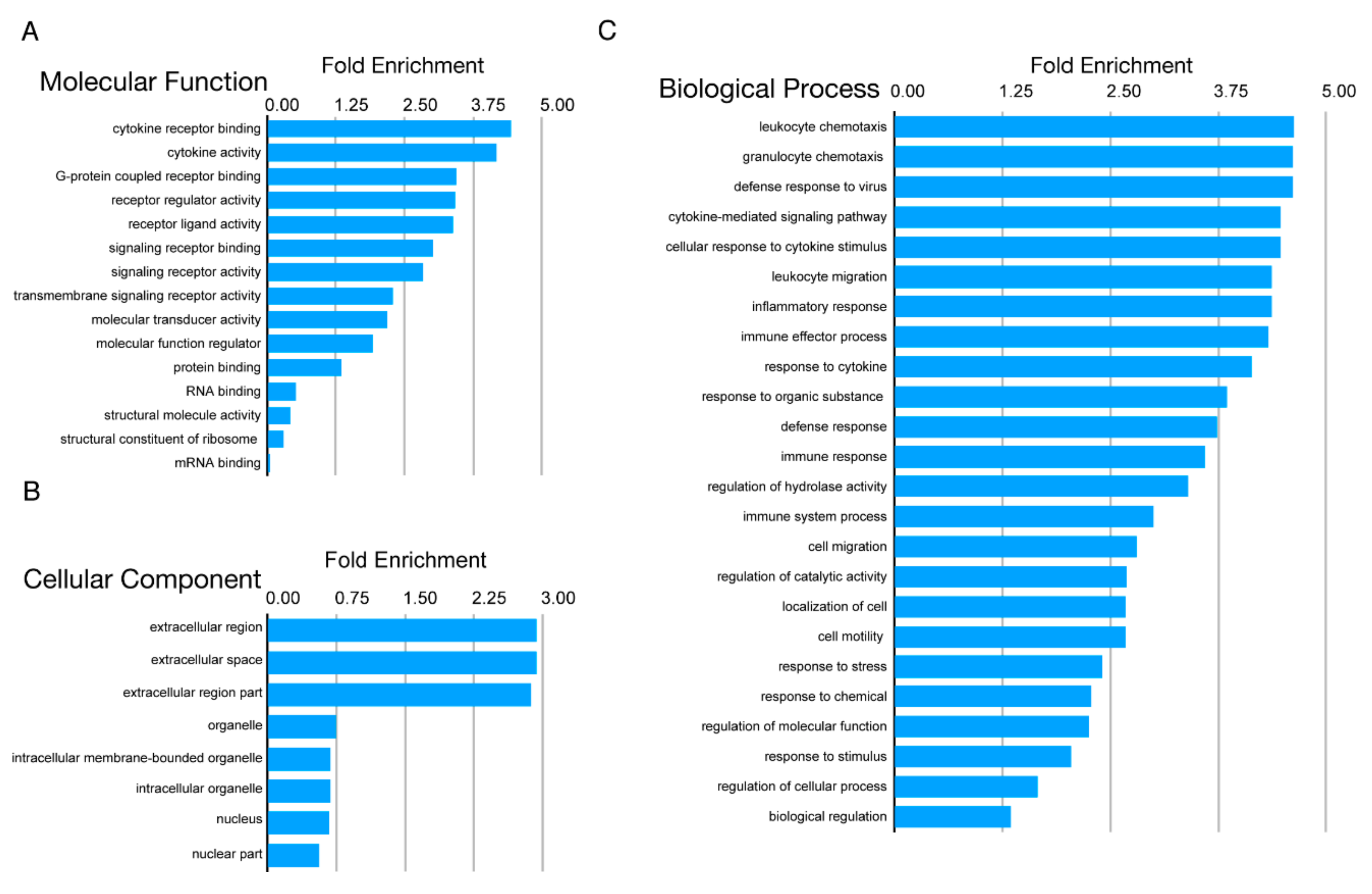
3.2. Proteomic Analysis Revealed Activation of Inflammatory Pathways, IFN, and MHC Class I in SC-β Cells over Time, with Comparative Analysis with Proteomics of Primary Human Islets
3.3. Transcriptome Analyses of CVB4-Infected SC-β Cells Showed a Temporal Increase in Viral Transcripts and Decreases in Genes Associated with the β Cell Phenotype
3.4. Perifusion Studies Showed Impaired Insulin Release by CVB4-Infected Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Craig, M.E.; Nair, S.; Stein, H.; Rawlinson, W.D. Viruses and type 1 diabetes: A new look at an old story. Pediatric Diabetes 2013, 14, 149–158. [Google Scholar] [CrossRef]
- Yeung, W.C.; Rawlinson, W.D.; Craig, M.E. Enterovirus infection and type 1 diabetes mellitus: Systematic review and meta-analysis of observational molecular studies. BMJ 2011, 342, d35. [Google Scholar] [CrossRef] [Green Version]
- Richardson, S.J.; Willcox, A.; Bone, A.J.; Foulis, A.K.; Morgan, N.G. The prevalence of enteroviral capsid protein vp1 immunostaining in pancreatic islets in human type 1 diabetes. Diabetologia 2009, 52, 1143–1151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smyth, D.J.; Cooper, J.D.; Bailey, R.; Field, S.; Burren, O.; Smink, L.J.; Guja, C.; Ionescu-Tirgoviste, C.; Widmer, B.; Dunger, D.B.; et al. A genome-wide association study of nonsynonymous SNPs identifies a type 1 diabetes locus in the interferon-induced helicase (IFIH1) region. Nat. Genet. 2006, 38, 617–619. [Google Scholar] [CrossRef] [PubMed]
- Cinek, O.; Stene, L.C.; Kramna, L.; Tapia, G.; Oikarinen, S.; Witso, E.; Rasmussen, T.; Torjesen, P.A.; Hyoty, H.; Ronningen, K.S. Enterovirus RNA in longitudinal blood samples and risk of islet autoimmunity in children with a high genetic risk of type 1 diabetes: The MIDIA study. Diabetologia 2014, 57, 2193–2200. [Google Scholar] [CrossRef] [PubMed]
- Oikarinen, S.; Martiskainen, M.; Tauriainen, S.; Huhtala, H.; Ilonen, J.; Veijola, R.; Simell, O.; Knip, M.; Hyoty, H. Enterovirus RNA in blood is linked to the development of type 1 diabetes. Diabetes 2011, 60, 276–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hodik, M.; Anagandula, M.; Fuxe, J.; Krogvold, L.; Dahl-Jorgensen, K.; Hyoty, H.; Sarmiento, L.; Frisk, G.; Consortium, P.-V. Coxsackie-adenovirus receptor expression is enhanced in pancreas from patients with type 1 diabetes. BMJ Open Diabetes Res. Care 2016, 4, e000219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vehik, K.; Lynch, K.F.; Wong, M.C.; Tian, X.; Ross, M.C.; Gibbs, R.A.; Ajami, N.J.; Petrosino, J.F.; Rewers, M.; Toppari, J.; et al. Prospective virome analyses in young children at increased genetic risk for type 1 diabetes. Nat. Med. 2019, 25, 1865–1872. [Google Scholar] [CrossRef]
- Ronnblom, L.; Eloranta, M.L. The interferon signature in autoimmune diseases. Curr. Opin. Rheumatol. 2013, 25, 248–253. [Google Scholar] [CrossRef]
- Crow, M.K.; Olferiev, M.; Kirou, K.A. Targeting of type I interferon in systemic autoimmune diseases. Transl. Res. 2015, 165, 296–305. [Google Scholar] [CrossRef] [Green Version]
- Nejentsev, S.; Walker, N.; Riches, D.; Egholm, M.; Todd, J.A. Rare variants of IFIH1, a gene implicated in antiviral responses, protect against type 1 diabetes. Science 2009, 324, 387–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nyalwidhe, J.O.; Gallagher, G.R.; Glenn, L.M.; Morris, M.A.; Vangala, P.; Jurczyk, A.; Bortell, R.; Harlan, D.M.; Wang, J.P.; Nadler, J.L. Coxsackievirus-Induced Proteomic Alterations in Primary Human Islets Provide Insights for the Etiology of Diabetes. J. Endocr. Soc. 2017, 1, 1272–1286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anagandula, M.; Richardson, S.J.; Oberste, M.S.; Sioofy-Khojine, A.B.; Hyoty, H.; Morgan, N.G.; Korsgren, O.; Frisk, G. Infection of human islets of Langerhans with two strains of Coxsackie B virus serotype 1: Assessment of virus replication, degree of cell death and induction of genes involved in the innate immunity pathway. J. Med. Virol. 2014, 86, 1402–1411. [Google Scholar] [CrossRef] [PubMed]
- Schulte, B.M.; Lanke, K.H.; Piganelli, J.D.; Kers-Rebel, E.D.; Bottino, R.; Trucco, M.; Huijbens, R.J.; Radstake, T.R.; Engelse, M.A.; de Koning, E.J.; et al. Cytokine and chemokine production by human pancreatic islets upon enterovirus infection. Diabetes 2012, 61, 2030–2036. [Google Scholar] [CrossRef] [Green Version]
- Gallagher, G.R.; Brehm, M.A.; Finberg, R.W.; Barton, B.A.; Shultz, L.D.; Greiner, D.L.; Bortell, R.; Wang, J.P. Viral infection of engrafted human islets leads to diabetes. Diabetes 2015, 64, 1358–1369. [Google Scholar] [CrossRef] [Green Version]
- Pagliuca, F.W.; Millman, J.R.; Gurtler, M.; Segel, M.; Van Dervort, A.; Ryu, J.H.; Peterson, Q.P.; Greiner, D.; Melton, D.A. Generation of functional human pancreatic beta cells in vitro. Cell 2014, 159, 428–439. [Google Scholar] [CrossRef] [Green Version]
- Kong, Y.; Ebrahimpour, P.; Liu, Y.; Yang, C.; Alonso, L.C. Pancreatic Islet Embedding for Paraffin Sections. J. Vis. Exp. 2018. [Google Scholar] [CrossRef]
- Burch, T.C.; Morris, M.A.; Campbell-Thompson, M.; Pugliese, A.; Nadler, J.L.; Nyalwidhe, J.O. Proteomic Analysis of Disease Stratified Human Pancreas Tissue Indicates Unique Signature of Type 1 Diabetes. PLoS ONE 2015, 10, e0135663. [Google Scholar] [CrossRef]
- Cox, J.; Neuhauser, N.; Michalski, A.; Scheltema, R.A.; Olsen, J.V.; Mann, M. Andromeda: A peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 2011, 10, 1794–1805. [Google Scholar] [CrossRef]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Tyanova, S.; Cox, J. Perseus: A Bioinformatics Platform for Integrative Analysis of Proteomics Data in Cancer Research. Methods Mol. Biol. 2018, 1711, 133–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemaire, K.; Schuit, F. Integrating insulin secretion and ER stress in pancreatic beta-cells. Nat. Cell Biol. 2012, 14, 979–981. [Google Scholar] [CrossRef] [PubMed]
- Raghavan, M.; Del Cid, N.; Rizvi, S.M.; Peters, L.R. MHC class I assembly: Out and about. Trends Immunol. 2008, 29, 436–443. [Google Scholar] [CrossRef] [Green Version]
- Blodgett, D.M.; Nowosielska, A.; Afik, S.; Pechhold, S.; Cura, A.J.; Kennedy, N.J.; Kim, S.; Kucukural, A.; Davis, R.J.; Kent, S.C.; et al. Novel Observations From Next-Generation RNA Sequencing of Highly Purified Human Adult and Fetal Islet Cell Subsets. Diabetes 2015, 64, 3172–3181. [Google Scholar] [CrossRef] [Green Version]
- Roivainen, M.; Rasilainen, S.; Ylipaasto, P.; Nissinen, R.; Ustinov, J.; Bouwens, L.; Eizirik, D.L.; Hovi, T.; Otonkoski, T. Mechanisms of coxsackievirus-induced damage to human pancreatic beta-cells. J. Clin. Endocrinol. Metab. 2000, 85, 432–440. [Google Scholar] [CrossRef]
- Benirschke, K.; Kibrick, S. Acute aseptic myocarditis and meningoencephalitis in the newborn child infected with coxsackle virus group B, type 3. N. Engl. J. Med. 1956, 255, 883–889. [Google Scholar] [CrossRef]
- Mena, I.; Fischer, C.; Gebhard, J.R.; Perry, C.M.; Harkins, S.; Whitton, J.L. Coxsackievirus infection of the pancreas: Evaluation of receptor expression, pathogenesis, and immunopathology. Virology 2000, 271, 276–288. [Google Scholar] [CrossRef] [Green Version]
- Op de Beeck, A.; Eizirik, D.L. Viral infections in type 1 diabetes mellitus—Why the beta cells? Nat. Rev. Endocrinol. 2016, 12, 263–273. [Google Scholar] [CrossRef] [Green Version]
- Cabrera, S.M.; Henschel, A.M.; Hessner, M.J. Innate inflammation in type 1 diabetes. Transl. Res. 2016, 167, 214–227. [Google Scholar] [CrossRef] [Green Version]
- Russell, M.A.; Redick, S.D.; Blodgett, D.M.; Richardson, S.J.; Leete, P.; Krogvold, L.; Dahl-Jorgensen, K.; Bottino, R.; Brissova, M.; Spaeth, J.M.; et al. HLA Class II Antigen Processing and Presentation Pathway Components Demonstrated by Transcriptome and Protein Analyses of Islet beta-Cells From Donors With Type 1 Diabetes. Diabetes 2019, 68, 988–1001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sherry, N.A.; Tsai, E.B.; Herold, K.C. Natural history of beta-cell function in type 1 diabetes. Diabetes 2005, 54 (Suppl. 2), S32–S39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomaidou, S.; Kracht, M.J.L.; van der Slik, A.; Laban, S.; de Koning, E.J.; Carlotti, F.; Hoeben, R.C.; Roep, B.O.; Zaldumbide, A. Beta-Cell Stress Shapes CTL Immune Recognition of Preproinsulin Signal Peptide by Post-Transcriptional Regulation of Endoplasmic Reticulum Aminopeptidase 1. Diabetes 2020. [Google Scholar] [CrossRef] [PubMed]
- Millman, J.R.; Xie, C.; Van Dervort, A.; Gurtler, M.; Pagliuca, F.W.; Melton, D.A. Generation of stem cell-derived beta-cells from patients with type 1 diabetes. Nat. Commun. 2016, 7, 11463. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 24 h | |||
| = -LOG (P-value) | Fold Difference (log2 transformed) | Protein IDs | Gene name/Protein name |
| 7.897847 | 9.588954 | P08292 | POLG_CXB4J/Genome polyprotein |
| 3.013728 | −2.054616 | Q99816 | TSG101/Tumor susceptibility gene 101 protein |
| 3.153537 | −2.260496 | P30519 | HMOX2/Heme oxygenase 2 |
| 2.616789 | −2.945653 | P43307 | SSR1/Translocon-associated protein subunit alpha |
| 48 h | |||
| = -LOG (P-value) | Fold Difference (log2 transformed) | Protein IDs | Gene name/Protein name |
| 3.789608 | 9.046398 | P08292 | POLG_CXB4J/Genome Polyprotein |
| 3.216631 | 4.741543 | P20591 | MX1/Interferon-induced GTP-binding protein Mx1 |
| 3.350592 | 4.65571 | P12955 | PEPD/Xaa-Pro dipeptidase |
| 3.742026 | 3.205037 | Q04941 | PLP2/Proteolipid protein 2 |
| 5.440948 | 3.004789 | P62072 | TIMM10/Mitochondrial import inner membrane translocase subunit Tim10 |
| 6.367597 | 2.369012 | P07996 | THBS1/Thrombospondin-1 |
| 4.888743 | 2.081825 | P80162 | CXCL6/C-X-C motif chemokine 6 |
| 1.863447 | 1.912146 | Q6KCM7 | SLC25A25/Calcium-binding mitochondrial carrier protein SCaMC-2 |
| 3.944867 | 1.822556 | P10145 | CXCL8/Interleukin-8 |
| 2.789387 | 1.807034 | O95786 | DDX58/Probable ATP-dependent RNA helicase DDX58 |
| 3.484639 | 1.529275 | P09914 | IFIT1/Interferon-induced protein with tetratricopeptide repeats 1 |
| 2.11946 | 1.525606 | P30480 | HLA-B/HLA class I histocompatibility antigen |
| 5.962474 | −1.66812 | P22676 | CALB2/Calretinin |
| 4.316548 | −1.70801 | P67809 | YBX1/Nuclease-sensitive element-binding protein 1 |
| 3.94315 | −1.72381 | Q13310 | PABPC4/Polyadenylate-binding protein 4 |
| 2.155246 | −1.80032 | P30519 | HMOX2/Heme oxygenase 2 |
| 2.691352 | −1.98576 | Q9UMX0 | UBQLN1/Ubiquilin-1 |
| 1.847708 | −2.11509 | Q9UHD9 | UBQLN2/Ubiquilin-2 |
| 3.702343 | −2.17211 | Q9Y2W2 | WBP11/WW domain-binding protein 11 |
| 3.326408 | −2.22052 | Q9NX14 | NDUFB11/NADH dehydrogenase [ubiquinone] 1 beta subcomplex subunit 11 |
| 3.449024 | −2.28579 | P50402 | EMD/Emerin |
| 2.690032 | −2.39827 | Q9UNH7 | SNX6/Sorting nexin-6;Sorting nexin-6, N-terminally processed |
| 3.795319 | −2.47833 | Q9H3P7 | ACBD3/Golgi resident protein GCP60 |
| 2.292801 | −3.22337 | P43307 | SSR1/Translocon-associated protein subunit alpha |
| Upstream Regulator | Molecule Type | Predicted Activation State | Activation z-Score | p-Value of Overlap | Target Molecules in Dataset |
|---|---|---|---|---|---|
| IFN-α | group | Activated | 2.369 | 3.04E-11 | APOL2, EIF2AK2, GBP1, IFIT1, IFIT2, IFIT3, ISG15, PML |
| TLR7 | transmembrane receptor | Activated | 2.433 | 5.28E-09 | ICAM1, IFIT1, IFIT3, ISG15, MX1, STAT1 |
| PRL | cytokine | Activated | 2.236 | 1.88E-07 | EIF2AK2, IFIT1, IFIT3, ISG15, SAMHD1 |
| JAK1 | kinase | Activated | 2 | 2.32E-08 | HLA-A, IFIT2, MX1, STAT1 |
| IL27 | cytokine | Activated | 2.414 | 5.44E-10 | B2M, HLA-A, HLA-B, ICAM1, STAT1, TAP1 |
| IFNL1 | cytokine | Activated | 3.113 | 4.56E-18 | EIF2AK2, GBP1, IFIT1, IFIT2, IFIT3, ISG15, MX1, PML, STAT1, UBE2L6 |
| TNF | cytokine | Activated | 2.706 | 5.82E-08 | GBP1, HLA-B, ICAM1, IFIT3, PML, STAT1, TAP1, TAPBP, TYMP |
| TGM2 | enzyme | Activated | 2.646 | 4.99E-08 | HLA-B, IFIT1, IFIT2, IFIT3, SAMHD1, STAT1, TAP1 |
| TLR9 | transmembrane receptor | Activated | 2.236 | 2.15E-08 | IFIT1, IFIT3, ISG15, MX1, STAT1 |
| IFNB1 | cytokine | Activated | 2.623 | 1.35E-13 | IFIT1, IFIT2, IFIT3, ISG15, MX1, PML, STAT1 |
| IFNG | cytokine | Activated | 3.096 | 4.58E-17 | EIF2AK2, GBP1, HLA-A, HLA-B, ICAM1, IFIT1, IFIT3, INS, ISG15, MX1, PML, STAT1, TAP1, TYMP |
| IFNA2 | cytokine | Activated | 3.148 | 1.26E-16 | EIF2AK2, GBP1, IFIT1, IFIT2, IFIT3, ISG15, MX1, PML, STAT1, UBE2L6 |
| EIF2AK2* | kinase | Activated | 2.412 | 5.44E-10 | EIF2AK2, IFIT1, ISG15, SAMHD1, STAT1, UBE2L6 |
| EBI3 | cytokine | Activated | 2.449 | 9.50E-13 | B2M, HLA-A, HLA-B, ICAM1, STAT1, TAP1 |
| IL1RN | cytokine | Inhibited | -2.433 | 2.16E-09 | GBP1, ICAM1, IFIT3, INS, MX1, PML |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nyalwidhe, J.O.; Jurczyk, A.; Satish, B.; Redick, S.; Qaisar, N.; Trombly, M.I.; Vangala, P.; Racicot, R.; Bortell, R.; Harlan, D.M.; et al. Proteomic and Transcriptional Profiles of Human Stem Cell-Derived β Cells Following Enteroviral Challenge. Microorganisms 2020, 8, 295. https://doi.org/10.3390/microorganisms8020295
Nyalwidhe JO, Jurczyk A, Satish B, Redick S, Qaisar N, Trombly MI, Vangala P, Racicot R, Bortell R, Harlan DM, et al. Proteomic and Transcriptional Profiles of Human Stem Cell-Derived β Cells Following Enteroviral Challenge. Microorganisms. 2020; 8(2):295. https://doi.org/10.3390/microorganisms8020295
Chicago/Turabian StyleNyalwidhe, Julius O., Agata Jurczyk, Basanthi Satish, Sambra Redick, Natasha Qaisar, Melanie I. Trombly, Pranitha Vangala, Riccardo Racicot, Rita Bortell, David M. Harlan, and et al. 2020. "Proteomic and Transcriptional Profiles of Human Stem Cell-Derived β Cells Following Enteroviral Challenge" Microorganisms 8, no. 2: 295. https://doi.org/10.3390/microorganisms8020295