Abstract
The Egyptian mongoose (Herpestes ichneumon) is a medium-size carnivore that, in Europe, is restricted to Iberia. The bio-ecology of this species remains to be elucidated in several dimensions, including gut microbiota that is nowadays recognized as a fundamental component of mammals. In this work, we investigated the gut microbiota of this herpestid by single-molecule real-time sequencing of twenty paired male (n = 10) and female (n = 10) intestinal samples. This culture-independent approach enabled microbial profiling based on 16S rDNA and investigation of taxonomical and functional features. The core gut microbiome of the adult subpopulation was dominated by Firmicutes, Fusobacteria, Actinobacteria, and Proteobacteria. Eight genera were uniquely found in adults and five in non-adults. When comparing gut bacterial communities across sex, four genera were exclusive of females and six uniquely found in males. Despite these compositional distinctions, alpha- and beta-diversity analyses showed no statistically significant differences across sex or between adult and non-adult specimens. However, when function was inferred, males presented a significantly higher abundance of amino acid and citrate cycle metabolic pathways, compared to the significant overrepresentation in females of galactose metabolic pathways. Additionally, adults exhibited a significantly higher abundance of cationic antimicrobial peptide resistance pathways, while non-adults bared a significant overrepresentation of two-component systems associated with antibiotic synthesis, flagellin and biofilm production, and chemotaxis control. This study adds new insights into the mongoose bio-ecology palette, highlighting taxonomical and functional microbiome dissimilarities across sex and age classes, possibly related to primary production resources and life-history traits that impact on behavior and diet.
1. Introduction
The bioecology of each mammal species is a conjugation of different domains, such as geographic range and habitat, diet, genetics, morpho-physiology, social behavior and gut microbiota, the latter of which has been progressively acknowledged as a fundamental component of mammal biology [1]. The Egyptian mongoose (Herpestes ichneumon (Linnaeus, 1758)) is a carnivore species from the Herpestidae family with opportunistic feeding behavior and whose diet in the Mediterranean includes wild rabbit, other small mammals like rodents, reptiles, amphibians, birds, crayfish, eggs, and even carrion [2]. This species is mostly present in the African continent, but also in the Mediterranean Middle East, Turkey, and the Iberian Peninsula (Portugal and Spain) [3]. The historical process underlying Egyptian mongoose colonization of Iberia is an issue under debate. While Gaubert et al. (2011) support that mongooses reached Iberia through the Strait of Gibraltar during the Middle to Late Pleistocene sea-level fluctuations [4], more recently, Detry et al. (2018) suggested that this species might have been introduced by the Romans during their establishment in Hispania [5]. In Portugal, the species distribution in the early 20th century was restricted to south of the Tagus River [6], but it has gradually, and remarkably, expanded into central and north-eastern regions [7]. The drivers for this geographic expansion are subject to speculation but land-use changes in shrub-dominated ecosystems, deforestation, the transformation of agricultural practices, and climate change [8] seem to have jointly contributed to this phenomenon. The Egyptian mongoose has a home-range of about 3 km², inhabiting locals with scrub vegetation in coastal, lacustrine, and riparian habitats, avoiding humid forests and extreme deserts. In the Iberian Peninsula, it is found in Mediterranean maqui. Listed as of Least Concern, the species is widespread, common, and present in many protected areas. Ecological features of mongooses such as morpho- and stress-physiology, diet, body condition, or reproduction, have been unraveled in recent years [2], driven by the opportunity to explore a large array of specimen samples in Portugal, where it is a game species under the Portuguese hunting law [9].
In Portugal, both sexual and regional dimorphism in body size has been reported, attributed to different feeding behaviors across sex and regions, resulting in larger and heavier male adults in the south [10]. H. ichneumon exhibit variability in social organization, ranging from solitary individuals to groups, which show cooperative tendencies, particularly in areas with abundant food resources. The exclusive home-range use of males in high-density populations suggests the existence of a polygynous mating system, which is accomplished by the spatial distribution of females, in combination with the absence of paternal care behavior [11]. This species microbiota has been investigated through culture-dependent methods in two separate approaches: first, a preliminary study based on the limited bacteriological screening of 53 specimens [12], and the latter focused on the microbial characterization of the gut of 20 males and females using a broad range, systematic culturomics-like strategy [13]. This study enabled the isolation and characterization of a large array of aerobic and anaerobic bacterial microbiota, sporobiota, and mycobiota [13]. However, a deeper insight into comprehensive gut communities can only be accomplished by a culture-independent approach that complementarily allows the characterization of non-viable or viable but non-culturable bacteria. Third generation technologies have been revolutionizing genomic sequencing. Pacific Biosciences Single-Molecule Real-Time (PacBio SMRT) is one of these, producing an average read length of 10 kb to 15 kb (up to a maximum of 80 kb), with a throughput per run of 0.5 Gb to 1 Gb [14], which allows a more accurate taxonomical identification compared to previous sequencing techniques that generate much smaller reads.
In this work, we thus set out to explore the gut microbiota of Egyptian mongooses sampled in South Portugal using a phylogenetic marker gene sequencing approach based on the 16S rRNA gene. The aims of this study were to: (1) characterize the gut bacterial microbiota of the Egyptian mongoose population; (2) investigate sex and age class-related taxonomic and functional differences; (3) identify statistically significant associations between microbiota and biotic and/or abiotic factors.
2. Materials and Methods
2.1. Egyptian Mongoose Specimens
Egyptian mongoose carcasses (ten male and ten female) obtained from legal predator density control actions were opportunistically used for this work. These carcasses were donated by hunters for scientific purposes and, after death, were frozen at −20 °C until necropsy. No animals were sacrificed for this study. The twenty animals under analysis were selected from a wider array of available mongooses based on several biological factors, namely sex, age class, geographic location, land-use and stomach content at the time of death. They were harvested from the Baixo Alentejo region, south of the Tagus River, from a landscape predominated by agroforestry and agriculture. The selected animals had the same stomach content at death, mostly composed of mammal and egg items [2]. Age class distribution was 16 adults and four non-adults (two subadults and two juveniles).
Mongooses were subjected to necropsy and specimen collection by pathologists at the necropsy facilities of the National Institute for Agrarian and Veterinary Research (INIAV, IP), the National Reference Laboratory for Animal Health. No signs of putrefaction or disease were detected. The abdominal cavity of each specimen was opened and the intestines isolated. Solid intestinal content (colon) was collected from each animal using a sterile feces collection tube and immediately processed for further analysis.
2.2. DNA Extraction, Quantification, Sequencing and Reads Processing
DNA was extracted from 500 mg of feces from each mongoose using the NZYSoil gDNA isolation kit (NZYTech, Lisbon, Portugal) following the manufacturer’s instructions. DNA was quantified using a Qubit fluorometer (Qiagen, Venlo, Netherlands), following the manufacturer’s instructions. The full-length 16S rRNA gene was amplified from 2.5 ng/µL of total DNA from each intestinal sample, using the universal primers 27F (5’-AGAGTTTGATCCTGGCTCAG-3′) and BS-R1407 (5′-GACGGGCGGTGWGTRC-3′), resulting in the amplification of variable regions (V) V1 to V8, originating an expected size amplicon of 1381 bp [15]. These amplicons were commercially sequenced on the PacBio SMRT RS-II platform (Pacific Biosciences, CA, USA; commercially available at Eurofins Genomics, Germany). The more recent PacBio P6-C4 chemistry was used.
Data were preprocessed using the PacBio SMRT Analysis Portal (Pacific Biosciences, CA, USA), generating single-molecule circular consensus sequences (CCS). The resulting CCS were analyzed using the EzBioCloud platform [16] (Seoul, Republic of Korea; accessed on January 2019). The PKSSU4.0 taxonomy database of prokaryotic 16S rRNA gene sequences was used. The uploaded CCS were trimmed of primers and quality filtered under several criteria, namely sequence length out of the expected range (<80 bp or >2000 bp), sequences with an average Q value lower than 25, sequences not predicted as being from the 16S rRNA gene by the Hidden Markov Model, and sequences found to be singletons when clustering using a cutoff of 97% similarity by the UCLUST method [17]. After, the pool of CCS reads was denoised using the Discrete Universal Denoiser (DUDE)-Seq software [18] and non-redundant reads were extracted. Next, the resulting reads were taxonomically assigned using USEARCH [17]. The following similarity cutoff values were used for taxonomical identification: species (≥97%), genus (between 96.9% and 94.5%), family (between 94.4% and 86.5%), order (between 86.4% and 82%), class (between 81.9% and 78.5%), and phylum (between 78.4% and 75%) [19]. Following, the non-assigned sequences were subjected to the UCHIME program [20] for the detection of chimerical sequences.
The valid reads (all reads that passed the previous filters) were then used for Operational Taxonomic Unit (OTU) picking, using an “open-reference” method with the following steps: (1) species-level identification clustering using the taxonomic assigned data; (2) OTU clustering using the UCLUST tool; and (3) a conjugation of the clusters obtained by the two previous steps. Singletons were omitted from further analysis. Also, the Good’s coverage of library was calculated to assess the representativity of the obtained reads when compared with the actual population [21].
2.3. Estimation of Alpha-Diversity Indices
Using OTU information, several diversity indices were estimated to measure bacterial species richness (Abundance-based Coverage Estimator (ACE) [22], Chao1 [23], Jackknife [24], and number of OTU) and evenness (Shannon [25], Simpson [25], and non-parametric Shannon (NPShannon) [26]), using the EzBioCloud platform. Besides these estimators, rarefaction and rank abundance curves were also plotted [27,28].
2.4. Comparison between Subpopulations Using Beta-Diversity Indices
The microbial communities at the genus level of each individual host were grouped according to host biological features, namely host sex (male and female) and age (adult and non-adult). Next, the subpopulations were compared using both ordination analysis and hierarchical clustering. First, a distance matrix was calculated using four possible metrics: UniFrac [29], Generalized UniFrac [30], Bray-Curtis [31], and Jensen-Shannon [32]. Then, the resulting matrix was used to compute a principal coordinate analysis (PCoA) or cluster using the Unweighted Pair Group Method with Arithmetic Mean (UPGMA) algorithm, originating a dendrogram. The beta-diversity analysis was also performed using the EzBioCloud platform.
2.5. Taxonomic and Functional Biomarker Discovery
To assess the taxonomic biomarkers specifically found in each host subpopulation, multiple comparisons between the different taxa were performed using a Kruskal-Wallis H test (α = 0.05) coupled with a Benjamini-Hochberg correction test. To infer and predict the differential functional profile of the host subpopulations, the Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt) algorithm was used, using an up-to-date taxonomic database of 16S rRNA genes, species genomes, and the Kyoto Encyclopedia of Genes and Genomes (KEGG) information as the input. This algorithm predicts which gene families are present in the sample using 16S rRNA data and estimates the overall metagenome composition and function [33]. The obtained functional profile is compared between host subpopulations using a linear discriminant analysis effect size (LEfSe) (α = 0.05) [34]. Both analyses were performed using the EzBioCloud platform.
2.6. Abiotic and Biotic Data Integration
For microbiota and bio-environmental data integration, we performed a Principal Component Analysis (PCA) using available information for the 20 Egyptian mongoose specimens [2,10]. The abiotic variables used were related to land-use, climatic data, road net, river net, and population data. The biotic variables used were related to stomach content at the time of death and different body measurements. Both biotic and abiotic data available were coupled with the gut microbiota genera detected and a PCA was calculated, enabling the prediction of the most influential biotic and abiotic variables to the gut microbiota composition of the Egyptian mongoose. Data integration was performed using the R software (Version 3.6.1, Vienna, Austria).
2.7. Data Analysis
Considering alpha-diversity indices, results from indices estimations were displayed as means of values of the individuals belonging to each subpopulation with respective standard deviation. When comparing two conditions, the Wilcoxon rank-sum test (α = 0.05) was performed. Considering beta-diversity, comparison of microbial communities between subpopulations of hosts were evaluated using beta set-significance analysis, namely a permutational multivariate analysis of variance (PERMANOVA) test. Both statistical analyses were performed using the EzBioCloud platform.
3. Results
3.1. Analysis of the Sequencing Data
A total of 46,508 reads across all samples were generated through SMRT sequencing. The trimming based on quality detected 1091 low-quality amplicons. Next, the clustering and taxonomic assignment detected seven non-target amplicons. Also, the chimera search detected 4187 chimerical amplicons. So, the total number of valid reads was 41,223 (88.6%). The average length of these reads was approximately 1416.6 (± 10.8) nucleotides, with an average minimum length of 1320 (± 10.53) nucleotides and an average maximum length of 1478 (± 26.86) nucleotides. A total of 37,565 (91.1%) sequences were identified at a species level. Together, this work accomplished a Good’s coverage of library of 99.18% (±0.46). For more information regarding individual sample data, see Supplementary Table S1.
3.2. Bacterial Composition of the Egyptian Mongoose Gut on a Population and Individual Level
The assignment of consensus taxonomy resulted in the identification of 11 phyla being represented across the intestinal samples of the Egyptian mongoose population. On a populational level, all intestinal samples were dominated by Firmicutes (86%), followed by Actinobacteria (6%), Fusobacteria (3%), Proteobacteria (3%), and Bacteroidetes (1%) (Figure 1a). Besides these phyla, some rare phyla were also detected (0.04%), namely Chloroflexi, Cyanobacteria, Planctomycetes, Saccharibacteria, TM6, and Verrucomicrobia.
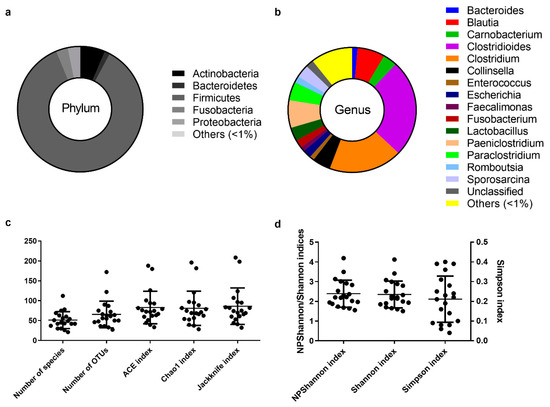
Figure 1.
The core intestinal bacterial microbiota of the Egyptian mongoose population. (a) Relative abundance on a phylum and (b) genus level. (c) Species richness (number of species, number of OTUs, ACE, Chao1, and Jackknife indices) and (d) species evenness (NPShannon, Shannon, and Simpson indices).
The initial genus-level analysis focused on OTUs with an abundance greater than 1%. Genus level OTU classification resulted in the identification of 16 genera present in the gastrointestinal tract of Egyptian mongooses, with a relative abundance greater than 1% in any given sample (Figure 1b). Clostridioides (25%) and Clostridium (19%) were the most abundant genera, representing almost half of the bacterial population (44%). At a lower, but still high, abundance, Blautia (7%) and Paniclostridium (7%) genera were identified. Besides these genera, Collinsella (5%), Paraclostridium (5%), Carnobacterium (4%), Lactobacillus (3%), Sporosarcina (3%), Escherichia (2%), Fusobacterium (2%), Romboutsia (2%), Bacteroides (1%), Enterococcus (1%), and Faecalimonas (1%) were also detected. A total of 10% of reads were assigned to genera present in <1% relative abundance and only a total of 2% of the reads were not assigned to a genus.
On an individual level, Egyptian mongoose specimens exhibited similar microbial profiles, with Firmicutes prevailing (Figure 2a). However, some notorious individual differences could be highlighted in relation to the overall population, namely mongoose individual HI467 that holds a higher abundance of Actinobacteria and mongoose HI509 that presents a higher abundance of Fusobacteria. Regarding genera compositions (Figure 2b), a higher dissimilarity among individuals could be detected, with a more pronounced difference in mongoose HI674 which possessed a higher abundance of Sporosarcina sp. (66%); HI508 with a higher abundance of Carnobacteria sp. (61%); HI501 with a higher abundance of Lactobacillus sp. (47%); HI516 with a higher abundance of Romboutsia sp. (26%); and HI466 with a higher abundance of Paraclostridium sp. (25%). Mongoose HI460 shows a higher abundance of unclassified reads (28%) at the genus level.
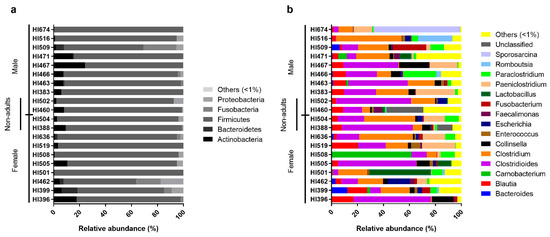
Figure 2.
Relative abundance of the individual intestinal bacterial microbiota of the Egyptian mongoose on a phylum (a) and genus (b) level.
Comparative analysis of OTUs across male and female intestinal samples evidenced that the majority of OTUs were shared across the individuals (Figure 3). Of the 174 different genera identified in the gastrointestinal tract of Egyptian mongoose individuals (including OTUs with <1% relative abundance), 19 were present in >1% relative abundance in at least one subpopulation (male or female); nine were found to be shared between sexes, four were found to be unique to the female hosts (Bacteroides, Carnobacterium, Cetobacterium, and Enterococcus), and six unique to the male hosts (Coprococcus, Faecalimonas, Romboutsia, Slackia, Sporosarcina, and Staphylococcus) (Figure 3b). Apart from these dissimilarities, similar proportions of phyla and genera were observed, with Bacteroidetes being uniquely found in the gastrointestinal tract of females (Figure 3a). However, these differences were statistically non-significant.
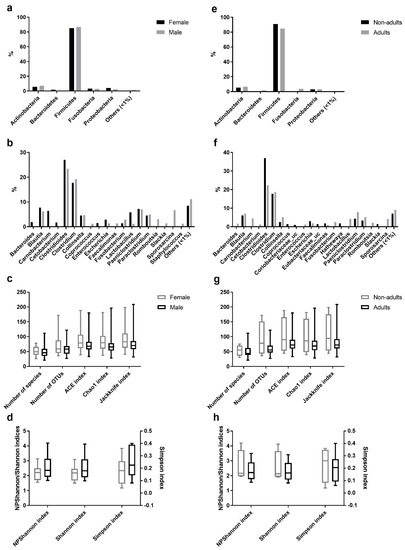
Figure 3.
Comparison of the intestinal bacterial microbiota of the Egyptian mongoose between sex and age class. (a) Relative abundance on a phylum and (b) genus level of female and male individuals. (c) Species richness (number of species, number of OTUs, ACE, Chao1, and Jackknife indices) and (d) species evenness (NPShannon, Shannon, and Simpson indices) of female and male individuals. (e) Relative abundance on a phylum and (f) genus level of adult and non-adult individuals. (g) Species richness (number of species, number of OTUs, ACE, Chao1, and Jackknife indices) and (h) species evenness (NPShannon, Shannon, and Simpson indices) of adult and non-adult individuals.
A comparative analysis of OTUs identified in the adult and non-adult intestinal samples found that the majority of OTUs were shared across individuals (Figure 3). Of the 174 different genera identified in the gastrointestinal tract of Egyptian mongoose individuals (including OTUs with <1% relative abundance), 21 were present in >1% relative abundance in at least one subpopulation (adult or non-adult); eight were shared between age classes; eight were unique to the adult hosts (Bacteroides, Carnobacterium, Cetobacterium, Enterococcus, Fusobacterium, Lactobacillus, Romboutsia, and Sporosarcina), and five unique to the non-adult hosts (Coprococcus, Coriobacteriaceae_uc, Eubacterium_uc, Hathewaya, and Slackia) (Figure 3f). Besides, similar proportions of phyla and genera were observed. However, Bacteroidetes and Fusobacteria were uniquely found in the gastrointestinal tract of adults (Figure 3e). Furthermore, these differences were statistically non-significant.
3.3. Estimation of Alpha Diversity Indices
Among the species richness indices, ACE, Chao1, Jackknife, and the number of OTUs were estimated. ACE and Chao1 are only sensitive indicators to rare OTUs [22,23], and Jackknife is sensitive to abundant OTUs [24], with higher values indicating higher diversity. Besides the difference in sensitivity, all species richness indicators showed similar values for all fecal samples (Figure 1c) and no significant differences across sex (Figure 3c) and age (Figure 3g) were found, suggesting low variability across this Egyptian mongoose population sample.
Among the species evenness indices, Shannon, Simpson, and NPShannon, were estimated. The Shannon and Simpson indicators are the most commonly used in microbial community studies, with the first displaying values higher than zero, and the maximum value corresponding to an equal distribution among all species present [25]. The second displays the probability of two randomly selected sequences belonging to the same species and uses this as a proxy to define even populations to the ones showing lower values [25]. NPShanon is a non-parametric estimation of the Shannon index and takes into consideration undetectable species and species of unknown abundance in a given sample [26]. Besides individual differences in considered variables, all species evenness indicators showed similar values across all fecal samples (Figure 1d). No significant differences across sex (Figure 3d) and age (Figure 3h) were found, suggesting, once again, limited variability across this Egyptian mongoose subpopulation.
Besides these estimators, rarefaction and rank abundance curves were also plotted. The rarefaction curve is the correlation between the number of reads and the number of OTUs and the steeper the slope, the higher the diversity [27]. Rarefaction curves show a good depth of sequence coverage, with curves beginning to level off after 1500 reads (Supplementary Figure S1). The rank abundance curve is the correlation between the rank of OTUs and the relative abundance of OTUs at each rank—the steeper the slope, the lower the diversity [28]. Rank abundance curves obtained from the OTU list evidence low species evenness across all fecal samples, indicated by the relatively steep curve, highlighting a greater abundance of high-ranking species comparing to low-ranking ones (Supplementary Figure S2).
3.4. Comparison between Subpopulations Using Beta-Diversity Analysis
The beta-diversity analysis helps to assess the relationship of microbial communities of different subpopulations. This can be performed using different metrics to calculate the dissimilarity/distance matrix, such as UniFrac, Generalized UniFrac, Bray-Curtis, and Jensen-Shannon. UniFrac is a distance measure obtained from the comparison between microbial communities based on the phylogenetic analysis of their OTU [29]. Generalized UniFrac distance unifies the weighted and unweighted UniFrac, correcting their limitations by decreasing the importance given to either abundant or rare OTUs, respectively [30]. The Bray-Curtis dissimilarity measure is obtained by comparing the counts of each OTU in different communities [31]. The Jensen-Shannon distance is the square root of the Jensen–Shannon divergence and is based on the comparison of the probability distribution of two microbial communities [32]. These dissimilarity/distance matrices can be used in an ordination analysis (such as Principal Coordination Analysis (PCoA)) or clustering in a hierarchical analysis.
Besides numerical differences in the PCoA obtained from the different metrics, samples clustered in a similar way, with samples clustering throughout the plot and with no clear differentiation between host sex (Supplementary Figure S3) or host age-class (Supplementary Figure S4), suggesting a comparable phylogenetic diversity across all fecal samples on a bacterial genus level. However, the fecal samples from non-adult individuals seem to be plotted in close proximity, suggesting a similar phylogenetic diversity. The hierarchical analysis using the UPGMA agglomeration metric shows similar dendrograms between UniFrac and Generalized UniFrac (Supplementary Figures S3c,f and S4c,f), and between Bray-Curtis and Jensen-Shannon (Supplementary Figures S3i,l and S4i,l), but they were different between them. The observed clusters do not group intestinal samples according to host sex or age class, reinforcing the idea of a comparable phylogenetic diversity. PERMANOVA results show no significant difference between bacterial gut microbiota of male and female hosts and between non-adult and adult hosts (Supplementary Table S2).
3.5. Taxonomic Biomarker Discovery
To assess the taxonomic biomarkers of male, female, adults, and non-adults, a Kruskal-Wallis H test was performed. Regarding sex (Supplementary Table S3), only two genera and three species were significantly more abundant (p-value = 0.03) in male mongooses when compared to female mongooses, and one species was significantly more abundant (p-value = 0.03) in female mongooses when compared to male. Regarding age class (Supplementary Table S4), one class (p-value = 0.034), two orders (p-value = 0.034 and 0.048), six families (p-value = 0.003 to 0.054), 24 genera (p-value = 0.004 to 0.051), and 53 species (p-value = 0.004 to 0.055) were significantly more abundant in non-adult mongooses when compared with adults.
3.6. Functional Biomarker Discovery
To predict the differential functional profile of female, male, adults, and non-adults, a LEfSe analysis was performed. LEfSe identifies units that are highly associated with a group in a dataset, using a non-parametric statistical test and estimating a size effect score for each differentially abundant feature using linear discriminant analysis [34]. The functional analysis can be based on three KEGG categories: orthology, module, or pathway [35]. Regarding sex (Supplementary Table S5), 23 orthologs (p-value = 0.007 to 0.051), two modules (p-value = 0.029 and 0.035), and three pathways (p-value = 0.037 to 0.054) were significantly different in abundance, with seven orthologs and one pathway being significantly more abundant in samples of female individuals, and 16 orthologs, two modules, and two pathways being significantly more abundant in samples of male individuals. Regarding age class (Supplementary Table S6), 19 orthologs (p-value = 0.006 to 0.049), one module (p-value = 0.038), and one pathway (p-value = 0.048) were significantly different in abundance, with four orthologs and one module being significantly more abundant in samples of adult individuals, and 15 orthologs and one pathway being significantly more abundant in samples of non-adult individuals.
3.7. Abiotic and Biotic Data Integration
For microbiota and bio-environmental data integration, we performed a Principal Component Analysis integrating 35 variables, including 19 biotic factors, with the microbiota data. Reptile and mammal stomach contents and all biometric factors tested (body weight (BW), snout-tail length (STL), tail length (TL), head and body length (HBL), right hind leg length (RHLL), right hind foot length (RHFL), shoulder height (SH), neck perimeter (NP), head diameter (HD), head width (HW), spleen weight (SW), kidney weight (KW), solid fat index (SFI), and perivisceral fat index (PFI)) were more influential on the bacterial microbiota in the Egyptian mongoose gut (Figure 4a,b). Additionally, several abiotic factors were also tested (n = 16), with land-use, temperature, rainfall, river net, and road net being the more influential on the gut microbiota of the Egyptian mongoose (Figure 4c,d).
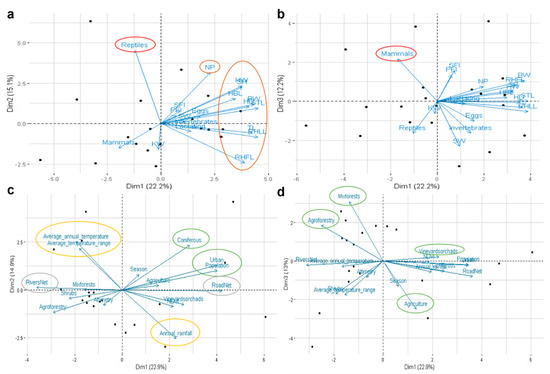
Figure 4.
Interaction between bacterial gut microbiota and biotic and abiotic factors of Egyptian mongoose. Principal Component Analysis using: (a,b) biological variables related to stomach content at the time of death and different body measurements; (c,d) environmental variables related to land-use, and climatic, topographic, and population data. The more influential factors are delimited using different colors—red: stomach content; brown: biometric data; green: land-use; yellow: climatic data; grey: topographic data. BW: body weight; STL: snout-tail length; TL: tail length; HBL: head and body length; RHLL: right hind leg length; RHFL: right hind foot length; SH: shoulder height; NP: neck perimeter; HD: head diameter; HW: head width; SW: spleen weight; KW: kidney weight; SFI: solid fat index; PFI: perivisceral fat index.
4. Discussion
Gut microbiota is nowadays an interdisciplinary and central research topic due to its recognized importance in shaping mammalian biology. In this study, we generated an extended sequence library of the gut microbiota of the Egyptian mongoose, highlighting sex and age class-related differences on taxonomic and functional levels. To accomplish this level of information on the gut microbiome of this carnivore species, a culture-independent approach was used to complement the first insights generated by previous culturomic-based studies [12,13]. The use of PacBio SMRT technology enabled the production of single long reads with the full extension of the 16S rRNA gene sequence, leading to a more reliable taxonomic identification of bacterial communities with high resolution, sensitivity, and accuracy. This phylogenetic marker gene strategy reinforced the notion that the gut microbiota of the Egyptian mongoose adult population is remarkably dominated by Firmicutes (Blautia, Clostridioides, Clostridium, and Lactobacillus), followed, in markedly lower proportions (in decreasing order), by Actinobacteria (Collinsella), Fusobacteria (Fusobacterium), and Proteobacteria (Escherichia).
Firmicutes are one of the most abundant bacterial phyla in the mammalian gastrointestinal tract, assuring protein degradation, the preservation of gut homeostasis and host immunity development [36]. Within Firmicutes, Clostridia members represented 66% of the bacterial gut microbiota of the Egyptian mongoose. Their ability to breakdown carbohydrates and proteins and to promote nutrient absorption [37] place these proteolytic bacteria as central within mammal microbiota [38] under the presence of high-protein content diet [37,39]. Detection of these and other bacterial phyla in our study is consistent with the carnivorous diet of the Egyptian mongoose [10,40]. The Bacilli members, like Lactobacillus spp., which are also reported in the gut of vertebrates [41], possessing prebiotic and probiotic activities [42], were also found in the mongoose gut.
Interestingly, the Actinobacteria phylum members that are usually a minor fraction of the gut microbiota of mammals [36] were the second most represented phyla in this study. Collinsella, previously correlated with human lipid metabolism [43], was the most prevalent genus within that phylum and is probably related to a high-fat diet, reinforcing, once again, the consistency of our findings with the carnivorous dietary patterns of the Egyptian mongoose.
Also in agreement was the detection of the Fusobacteria phylum, particularly Fusobacterium spp. that ferment carbohydrates and amino acids to produce a variety of organic acids, such as acetic acid, formic acid, butyric acid [36] and short-chain fatty acids, which account for host energy sources and regulate the metabolism of glucose, cholesterol, and fatty acids [44].
Proteobacteria members, particularly Enterobacteriaceae such as Escherichia spp., were also found. These are common commensals in mammals, with extremely diverse metabolism that includes the ability to break down and ferment complex sugars and produce vitamins [36]. They have been reported as predominant in other Carnivora members, such as grizzly bears and giant pandas [37,45,46]. A high ratio of Proteobacteria/Bacteroidetes (calculated ratio of 3) was evident in the Egyptian mongoose samples surveyed. This finding has been previously related with a carnivorous or scavenger diet, namely in carnivores like cheetah, Tasmanian devil, spotted hyena, and polar bear [47], and also with a very efficient harvest of energy [48].
A previous study by our group using culture-dependent methods followed by 16sRNA gene sequencing of selected isolates was performed upon the same 20 Egyptian mongoose specimens surveyed in this work [13]. The detection of the phyla Firmicutes (67%), Proteobacteria (32%), and Actinobacteria (1%) was registered, enclosing twenty genera. Strikingly, the Delftia, Ralstonia, Rummelibacillus, Stenotrophomonas, Pantoea, Solibacillus, and Robinsoniella genera were exclusively found when using a culture-dependent approach. The disparities between the data generated by both methods can be caused by the length of the 16S rRNA gene sequence used to identify the bacterial isolates in the former study, which sometimes were of shorter length, possibly leading to taxonomic misidentification, allied with the strong bias that can be associated with culture-dependent methods performed on limited cultivability conditions (limited number of growth media, oxygenation, temperature, among others). However, culture-independent approaches have a minimum sequence concentration threshold, which could explain the lack of detection of poorly represented OTUs. This limitation can be overcome by culture-dependent and taxonomical enrichment approaches [49]. Discrepancies between culture-dependent and culture-independent methodologies are frequently reported by others [50,51,52,53].
Regarding sex-related differences, the male-specific bacterial groups detected here were the Faecalimonas genus that is usually found in the mammalian gut [54,55], the Romboutsia genus that is normally found in humans and rats [56,57], and the Sporosarcina genus typically found in birds [58]. Female-specific taxa comprised Carnobacterium, Enterococcus, and Cetobacterium genera, together with Bacteroidetes. The first genus was previously detected in food (fish, meat, and some dairy products) and natural environments (sediments and water) [59]; however, to our knowledge, Carnobacterium spp. has only been identified in the gastrointestinal microbiota of the Egyptian mongoose both by sequence-dependent and -independent methods [12,13]. Detection of Enterococci is in agreement with other works on wild animals conducted in Portugal, particularly carnivores [13,60,61,62]. The Cetobacterium genus is commonly found in the intestines of freshwater fish species [63,64,65], humans [66], and dog feces [67]. This genus has the ability to ferment peptides and carbohydrates and to produce vitamin B12 that can be absorbed by the host [65,66]. Bacteroidetes are cosmopolitan bacteria, being one of the most frequently found members in mammalian gastrointestinal microbiota [36]. These bacteria have the ability to degrade proteins and carbohydrates. The fermentation of these compounds releases short-chain fatty acids [36,68]. Also, they can interact with the immune system of the host, activating T-cells and protecting the gastrointestinal tract from pathogenic bacteria [69], although some sporadic reports of opportunistic infections are also available [70].
In our previous culture-dependent study [13], the microbial load of intestinal samples seeded in a rich medium under anaerobiosis was higher in females than in males, as indicated by aerobic/anaerobic vegetative and sporobiota communities. Also, Paeniclostridium spp., Pantoea spp., Sporosarcina spp., Solibacillus spp., and Stenotrophomonas spp. isolation was restricted to female individuals, while Paenibacillus spp., Propionibacterium spp., Robinsoniella spp. and Staphylococcus spp. were only isolated from males.
Regarding the taxonomic biomarker discovery analysis, we detected a significantly higher abundance of Kocuria spp., Hathewaya spp., and Clostridium haemolytium in male samples. The Kocuria genus is a typical initial gut colonizer and is normally found in the intestine, but more typically present on other mucosae of mammals [71]. Clostridium haemolytium is the causative agent of bacillary hemoglobinuria, mostly occurring in cattle [72]. Female samples showed a significantly higher abundance of Clostridium mediterraneense, a recently discovered species isolated from the human gut in France [73]. Besides these compositional differences, the beta-diversity analysis showed no statistically significant differences across sex. Compositional sex differences in gut microbiota could be the result of dietary, behavioral and/or host physiology distinctions. In fact, previous studies exploring biometric, diet and splenic data of Egyptian mongooses from the same biogeographic region found dietary and immune system differences across sex [2,74]. Other studies in primates have also shown sex-specific bacterial microbiota frameworks [75,76,77].
Regarding age-related differences, several main bacterial groups were absent from non-adults, while the Coprococcus genus was specifically associated with this age class, as was Slackia spp. previously found in gut samples from porcupine, beaver, coyote, and Arctic wolf [78]. Besides the latter, other members of Coriobacteriaceae are common mammalian symbionts, responsible for the conversion of bile salts and steroids and activation of polyphenols [79]. Moreover, Eubacterium and Hathewaya genera were also confined to non-adult specimens. The presence of these species can indicate an increased digestive and absorptive capacity, promoting an increase in weight and size, which is essential for the normal development of juveniles and an increase in the immune system capacity when reaching the adult period due to an increase of vitamin production and the interaction of bacterial-epithelial cells in the gastrointestinal tract.
Reinforcing these notions, the taxonomic biomarker discovery analysis detected a significantly higher abundance of Tissierellia and Clostridia classes, Erysipelotrichales order, Eubacteriaceae, Cellullomonadaceae, Rikenellaceae, and Peptoniphilaceae families in non-adult samples. Tissierellia members have been previously detected in the gut microbiota of Canadian mink [80] and humans [81,82]. Erysipelotrichales members were associated with lipidemic profiles of human hosts, probably associated with high-fat content diets [83], together with Rikenellaceae members [84]. Eubacteriaceae bacteria have been reported as major members of the gut microbiota of Forest Musk deer [85]. Cellulomonadaceae are considered probiotic species that can convert cellulose into other metabolites [86]. Peptoniphilaceae were reported in the gut microbiota of children [81] and women [87]. Besides compositional differences, the beta-diversity analysis also showed no statistically significant differences across age classes of the Egyptian mongoose. In contrast, results from the culture-dependent methodology [13] evidenced similarities between adults and juveniles, with sub-adults clustering separately. This common framework for adults and cubs was attributed to the social behavior of the species which relies on the protection and feeding of the cubs, scent marking and social latrines [11]. This higher proximity and interaction between adults and juveniles can increase diet similarity and fecal-oral transmission of microbiota [11]. This phenomenon was previously reported in mice, birds, and humans [55].
For the first time, the differential functional profile of the Egyptian mongoose population was evaluated. Male Egyptian mongooses showed a significantly higher abundance of catabolic pathways of valine, leucine, and isoleucine amino acids. This degradation is usually performed by members of the Clostridia class [88], such as Coprococcus, Faecalimonas, and Romboutsia genera, which are more abundant in male gut microbiota. The male microbiota also had a significantly higher abundance of tryptophan metabolism pathways. This metabolic route is usually conducted by Peptostreptococcus spp., Lactobacillus spp., and Clostridium spp. [89]. Male individuals did have an overrepresentation of closely related members of Peptostreptococcus (i.e., Romboutsia). In contrast, the female microbiota showed an overrepresentation of closely related members of Lactobacillus (i.e., Carnobacterium and Enterococcus). Male hosts also revealed a significantly higher abundance of citrate cycle modules in their microbiota. The citrate cycle includes several amino acid metabolites, such as valine, leucine, isoleucine, and tryptophan, all of which have differentially functioning synthesis in the bacterial community of the male gut. Female mongooses exhibited a significantly higher abundance of galactose metabolic pathways, normally performed by Bacteroides (in particular, Bacteroides vulgaris [90]), which is usually overrepresented in female hosts. These findings suggest that the bacterial gut microbiome of the Egyptian mongoose is modulated by sex-specific strategies to produce energy.
In adult Egyptian mongooses, cationic antimicrobial peptide (CAMP) resistance genes were significantly overrepresented, specifically the dltABCD operon of Gram-positive bacteria. CAMP are short peptides secreted by immune and epithelial cells in response to bacterial products, such as lipopolysaccharide (LPS) or other inflammatory signals [91]. The resistance to CAMP enables bacterial virulence and resistance to innate immunity mechanisms, increasing immune evasion [91]. This finding may indicate the presence of opportunistic pathogenic bacteria in the bacterial community of the adult gut, even though no signs of disease could be perceived during necropsy. Also, no high percentage of opportunistic pathogenic Gram-positive bacteria could be found. In non-adult Egyptian mongooses, two-component systems associated with antibiotic synthesis, flagellin production, chemotaxis control, and biofilm formation genes were significantly overrepresented. All these processes are related to bacterial virulence and pathogenesis, which may indicate an overrepresentation of opportunistic bacteria in the gut of juvenile Egyptian mongooses, possibly enabled by an immature immune system, even though signs of disease were not perceived.
Abiotic and biotic factors such as land-use, climatic and topological data, feeding, and biometric data were found to exert an influence in the bacterial gut of the Egyptian mongoose. The effect exerted by abiotic factors on mammalian microbiota, such as alterations in native habitat, has been reported by others [92]. Shifts in land-use and topography that cause shifts in food availability, quality, or composition have been described to impact the gut microbiome [93,94,95]. Temperature and rainfall can affect bacterial microbiota by directly altering environmental microbial communities and indirectly by inducing changes in primary production and host physiology [96,97,98]. Biotic factors such as host biometry can modify bacterial abundance and diversity, being associated with host health [95]. Other intrinsic host-associated factors, such as diet, and extrinsic features, such as land-use and climatic changes, can both lead to gut microbiota adaptations as a result of the Egyptian mongoose expanding its range across environmental gradients [8].
5. Conclusions
This study represents the first description of the Egyptian mongoose gastrointestinal microbiota using culture-independent methods, improving our knowledge on the bioecology of this species. It leads to a better understanding of an until now poorly characterized carnivore, together with a sex and age class comparative analyses, that help to assess the indirect effects exerted by host behavior, diet, reproduction, and other biological characteristics on gut microbiota composition and function. Altogether, our results also reinforce the need to use a combination of both culture-dependent and culture-independent strategies when the aim is to capture the maximum biodiversity across complex samples. Finally, our efforts emphasize the value of microbiome studies to fully comprehend mammalian species ecology in light of behavior, diet, and geographic features.
Supplementary Materials
The following are available online at https://www.mdpi.com/2076-2607/8/3/392/s1, Table S1: Data of read analysis for the 20 intestinal samples of Egyptian mongoose specimens, Table S2: PERMANOVA test results of the microbial community of Egyptian mongoose comparison between female and male and between non-adult and adult, Table S3: Taxonomic biomarkers discovery of female and male Egyptian mongoose fecal bacterial microbiota using a Kruskal-Wallis H test, Table S4: Taxonomic biomarkers discovery of non-adult and adult Egyptian mongoose fecal bacterial microbiota using a Kruskal-Wallis H test, Table S5: Functional biomarkers discovery of female and male Egyptian mongoose fecal bacterial microbiota applying a LEfSe analysis, Table S6: Functional biomarkers discovery of non-adult and adult Egyptian mongoose fecal bacterial microbiota applying a LEfSe analysis, Figure S1: Rarefaction curves for the twenty different fecal samples of Egyptian mongoose measured individuals using the observed species metric, Figure S2: Rank abundance curves for the twenty different fecal samples of Egyptian mongoose individuals using the observed species metric, Figure S3: Beta-diversity analysis to compare the fecal bacterial microbiota, on a genus level, of male and female Egyptian mongoose, Figure S4: Beta-diversity analysis to compare the fecal bacterial microbiota, on a genus level, of adult and non-adult Egyptian mongoose.
Author Contributions
Conceptualization, M.V.C.; Formal analysis, A.C.P.; Funding acquisition, M.V.C.; Investigation, A.C.P. and M.V.C.; Resources, C.F. and M.V.C.; Writing—original draft, A.C.P. and M.V.C.; Writing—review and editing, V.B. and M.V.C. All authors have read and agreed to the published version of the manuscript.
Funding
This work was partially funded by Fundação para a Ciência e a Tecnologia (FCT/MEC), Portugal, through strategic funding to cE3c (UID/BIA/00329/2020), BioISI (UID/Multi/04046/2020) and CESAM (UID/AMB/50017/2019) Research Units and a PhD fellowship to VB (SFRH/BD/51540/2011).
Acknowledgments
We are grateful to Madalena Monteiro, Paulo Carvalho, Paula Mendonça (INIAV, IP) and Tânia Barros (CESAM) for collaboration in necropsies in the scope of another research work. Jacinto Amaro (FENCAÇA) and other hunting federations/associations, as well as hunters and land owners, are gratefully appreciated for collaboration in animal corpse collection and donation for scientific purposes.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Jones, K.E.; Safi, K. Ecology and evolution of mammalian biodiversity. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2011, 366, 2451–2461. [Google Scholar] [CrossRef] [PubMed]
- Bandeira, V.; Virgós, E.; Carvalho, J.; Barros, T.; Cunha, M.; Fonseca, C. Diet footprint of Egyptian mongoose along ecological gradients: Effects of primary productivity and life history traits. Mamm. Biol. 2017, 88, 16–25. [Google Scholar] [CrossRef]
- Kingdon, J. The kingdon field guide to african mammals. J. Mammal 1999, 80, 692–693. [Google Scholar]
- Gaubert, P.; López-Bao, J.V.; Machordom, A.; Morales, A.; Palomares, F.; Tània, B.; Veron, G. Comparative phylogeography of two african carnivorans presumably introduced into europe: Disentangling natural versus human-mediated dispersal across the strait of gibraltar. Blackwell Publ. 2011, 38, 341–358. [Google Scholar] [CrossRef]
- Detry, C.; Bicho, N.; Fernandes, H.; Fernandes, C. The emirate of Córdoba (756–929 ad) and the introduction of the Egyptian mongoose (Herpestes ichneumon) in Iberia: The remains from Muge, Portugal. J. Archaeol. Sci. 2011, 38, 3518–3523. [Google Scholar] [CrossRef]
- Borralho, R.; Rego, F.; Palomares, F.; Hora, A. The distribution of the Egyptian mongoose Herpestes ichneumon (L.) in Portugal. Mammal Rev. 1996, 26, 1–8. [Google Scholar] [CrossRef]
- Balmori, A.; Carbonell, R. Expansion and distribution of the Egyptian mongoose (Herpestes ichneumon) in the Iberian Peninsula. Galemys 2012, 24, 83–85. [Google Scholar] [CrossRef]
- Barros, T.; Carvalho, J.; Pereira, M.J.R.; Ferreira, J.P.; Fonseca, C. Following the trail: Factors underlying the sudden expansion of the Egyptian mongoose (Herpestes ichneumon) in Portugal. PLoS ONE 2015, 10, e0133768. [Google Scholar] [CrossRef]
- Do Linh San, E.; Maddock, A.H.; Gaubert, P.; Palomares, F. Herpestes ichneumon. The IUCN red list of threatened species 2016. IUCN Red List Threat. Species 2016. [Google Scholar] [CrossRef]
- Bandeira, V.; Virgós, E.; Barros, T.; Cunha, M.V.; Fonseca, C. Geographic variation and sexual dimorphism in body size of the Egyptian mongoose, Herpestes ichneumon in the western limit of its European distribution. Zoologischer Anzeiger J. Comp. Zool. 2016, 264, 1–10. [Google Scholar] [CrossRef]
- Schneider, T.C.; Kappeler, P.M. Social systems and life-history characteristics of mongooses. Biol. Rev. 2014, 89, 173–198. [Google Scholar] [CrossRef]
- Cunha, M.V.; Albuquerque, T.; Themudo, P.; Fonseca, C.; Bandeira, V.; Rosalino, L.M. Implications of Egyptian mongoose microbiome in the management of Mediterranean carnivores. Ecohealth, 3 February 2020; under review. [Google Scholar]
- Pereira, A.C.; Bandeira, V.; Fonseca, C.; Cunha, M.V. Understanding the cross-talk between microbiota, host life-history, and the environment using Egyptian mongoose (Herpestes ichneumon) as a model. (manuscript under preparation for submission).
- Van Dijk, E.L.; Jaszczyszyn, Y.; Naquin, D.; Thermes, C. The third revolution in sequencing technology. Trends Genet. 2018, 34, 666–681. [Google Scholar] [CrossRef] [PubMed]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glöckner, F.O. Evaluation of general 16s ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013, 41, e1. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.-H.; Ha, S.-M.; Kwon, S.; Lim, J.; Kim, Y.; Seo, H.; Chun, J. Introducing ezbiocloud: A taxonomically united database of 16s rRNA gene sequences and whole-genome assemblies. Int. J. Syst. Evol. Microbiol. 2017, 67, 1613–1617. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. Search and clustering orders of magnitude faster than blast. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef]
- Lee, B.; Moon, T.; Yoon, S.; Weissman, T. Dude-seq: Fast, flexible, and robust denoising for targeted amplicon sequencing. PLoS ONE 2017, 12, e0181463. [Google Scholar] [CrossRef]
- Yarza, P.; Yilmaz, P.; Pruesse, E.; Glockner, F.O.; Ludwig, W.; Schleifer, K.H.; Whitman, W.B.; Euzeby, J.; Amann, R.; Rossello-Mora, R. Uniting the classification of cultured and uncultured bacteria and archaea using 16s rRNA gene sequences. Nat. Rev. Microbiol. 2014, 12, 635–645. [Google Scholar] [CrossRef]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. Uchime improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef]
- Good, I.J. The population frequencies of species and the estimation of population parameters. Biometrika 1953, 40, 237–264. [Google Scholar] [CrossRef]
- Chao, A.; Lee, S.-M. Estimating the number of classes via sample coverage. J. Am. Stat. Assoc. 1992, 87, 210–217. [Google Scholar] [CrossRef]
- Chao, A. Nonparametric estimation of the number of classes in a population. Scand. J. Stat. 1984, 265–270. [Google Scholar]
- Burnham, K.P.; Overton, W.S. Robust estimation of population size when capture probabilities vary among animals. Ecology 1979, 60, 927–936. [Google Scholar] [CrossRef]
- Magurran, A.E. Measuring Biological Diversity; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2013. [Google Scholar]
- Chao, A.; Shen, T.-J. Nonparametric prediction in species sampling. J. Agric. Biol. Environ. Stat. 2004, 9, 253–269. [Google Scholar] [CrossRef]
- Heck, K.L.; van Belle, G.; Simberloff, D. Explicit calculation of the rarefaction diversity measurement and the determination of sufficient sample size. Ecology 1975, 56, 1459–1461. [Google Scholar] [CrossRef]
- Whittaker, R.H. Dominance and diversity in land plant communities: Numerical relations of species express the importance of competition in community function and evolution. Sci. N. Y. 1965, 147, 250–260. [Google Scholar] [CrossRef] [PubMed]
- Lozupone, C.; Knight, R. Unifrac: A new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol. 2005, 71, 8228–8235. [Google Scholar] [CrossRef]
- Chen, J.; Bittinger, K.; Charlson, E.S.; Hoffmann, C.; Lewis, J.; Wu, G.D.; Collman, R.G.; Bushman, F.D.; Li, H. Associating microbiome composition with environmental covariates using generalized unifrac distances. Bioinformatics 2012, 28, 2106–2113. [Google Scholar] [CrossRef]
- Bray, J.R.; Curtis, J.T. An ordination of the upland forest communities of southern Wisconsin. Ecol. Monogr. 1957, 27, 325–349. [Google Scholar] [CrossRef]
- Endres, D.M.; Schindelin, J.E. A new metric for probability distributions. IEEE Trans. Inf. Theory 2003, 49, 1858–1860. [Google Scholar] [CrossRef]
- Langille, M.G.I.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Vega Thurber, R.L.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16s rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Sato, Y.; Furumichi, M.; Morishima, K.; Tanabe, M. New approach for understanding genome variations in KEGG. Nucleic Acids Res. 2019, 47, D590–D595. [Google Scholar] [CrossRef] [PubMed]
- Colston, T.J.; Jackson, C.R. Microbiome evolution along divergent branches of the vertebrate tree of life: What is known and unknown. Mol. Ecol. 2016, 25, 3776–3800. [Google Scholar] [CrossRef] [PubMed]
- Schwab, C.; Cristescu, B.; Northrup, J.M.; Stenhouse, G.B.; Gänzle, M. Diet and environment shape fecal bacterial microbiota composition and enteric pathogen load of grizzly bears. PLoS ONE 2011, 6, e27905. [Google Scholar] [CrossRef] [PubMed]
- McFarland, L.V.; Mulligan, M.E.; Kwok, R.Y.; Stamm, W.E. Nosocomial acquisition of clostridium difficile infection. N. Engl. J. Med. 1989, 320, 204–210. [Google Scholar] [CrossRef] [PubMed]
- Glad, T.; Bernhardsen, P.; Nielsen, K.M.; Brusetti, L.; Andersen, M.; Aars, J.; Sundset, M.A. Bacterial diversity in faeces from polar bear (Ursus maritimus) in arctic Svalbard. BMC Microbiol. 2010, 10, 10. [Google Scholar] [CrossRef]
- Rosalino, L.M.; Santos, M.J.; Pereira, I.; Santos-Reis, M. Sex-driven differences in Egyptian mongoose’s (Herpestes ichneumon) diet in its Northwestern European range. Eur. J. Wildl. Res. 2009, 55, 293. [Google Scholar] [CrossRef]
- Maurice, C.F.; Cl Knowles, S.; Ladau, J.; Pollard, K.S.; Fenton, A.; Pedersen, A.B.; Turnbaugh, P.J. Marked seasonal variation in the wild mouse gut microbiota. ISME J. 2015, 9, 2423–2434. [Google Scholar] [CrossRef]
- Walter, J. Ecological role of lactobacilli in the gastrointestinal tract: Implications for fundamental and biomedical research. Appl. Environ. Microbiol. 2008, 74, 4985–4996. [Google Scholar] [CrossRef]
- Lahti, L.; Salonen, A.; Kekkonen, R.A.; Salojärvi, J.; Jalanka-Tuovinen, J.; Palva, A.; Orešič, M.; de Vos, W.M. Associations between the human intestinal microbiota, Lactobacillus rhamnosus GG and serum lipids indicated by integrated analysis of high-throughput profiling data. PeerJ 2013, 1, e32. [Google Scholar] [CrossRef] [PubMed]
- Den Besten, G.; van Eunen, K.; Groen, A.K.; Venema, K.; Reijngoud, D.-J.; Bakker, B.M. The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J. Lipid Res. 2013, 54, 2325–2340. [Google Scholar] [CrossRef] [PubMed]
- Schwab, C.; Ganzle, M. Comparative analysis of fecal microbiota and intestinal microbial metabolic activity in captive polar bears. Can. J. Microbiol. 2011, 57, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Xue, Z.; Zhang, W.; Wang, L.; Hou, R.; Zhang, M.; Fei, L.; Zhang, X.; Huang, H.; Bridgewater, L.C.; Jiang, Y.; et al. The bamboo-eating giant panda harbors a carnivore-like gut microbiota, with excessive seasonal variations. mBio 2015, 6, e00022-15. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Fox, S.; Pemberton, D.; Hogg, C.; Papenfuss, A.T.; Belov, K. The tasmanian devil microbiome—Implications for conservation and management. Microbiome 2015, 3, 76. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Ley, R.E.; Mahowald, M.A.; Magrini, V.; Mardis, E.R.; Gordon, J.I. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 2006, 444, 1027–1031. [Google Scholar] [CrossRef]
- Hugon, P.; Lagier, J.-C.; Robert, C.; Lepolard, C.; Papazian, L.; Musso, D.; Vialettes, B.; Raoult, D. Molecular studies neglect apparently gram-negative populations in the human gut microbiota. J. Clin. Microbiol. 2013, 51, 3286–3293. [Google Scholar] [CrossRef]
- Hugon, P.; Lagier, J.C.; Colson, P.; Bittar, F.; Raoult, D. Repertoire of human gut microbes. Microb. Pathog. 2017, 106, 103–112. [Google Scholar] [CrossRef]
- Lagier, J.-C.; Hugon, P.; Khelaifia, S.; Fournier, P.-E.; La Scola, B.; Raoult, D. The rebirth of culture in microbiology through the example of culturomics to study human gut microbiota. Clin. Microbiol. Rev. 2015, 28, 237–264. [Google Scholar] [CrossRef]
- Lagier, J.C.; Armougom, F.; Million, M.; Hugon, P.; Pagnier, I.; Robert, C.; Bittar, F.; Fournous, G.; Gimenez, G.; Maraninchi, M.; et al. Microbial culturomics: Paradigm shift in the human gut microbiome study. Clin. Microbiol. Infect. Off. Publ. Eur. Soc. Clin. Microbiol. Infect. Dis. 2012, 18, 1185–1193. [Google Scholar] [CrossRef]
- Lagier, J.C.; Million, M.; Hugon, P.; Armougom, F.; Raoult, D. Human gut microbiota: Repertoire and variations. Front. Cell. Infect. Microbiol. 2012, 2, 136. [Google Scholar] [CrossRef] [PubMed]
- Ley, R.E.; Hamady, M.; Lozupone, C.; Turnbaugh, P.J.; Ramey, R.R.; Bircher, J.S.; Schlegel, M.L.; Tucker, T.A.; Schrenzel, M.D.; Knight, R.; et al. Evolution of mammals and their gut microbes. Sci. N.Y. 2008, 320, 1647–1651. [Google Scholar] [CrossRef] [PubMed]
- Ley, R.E.; Lozupone, C.A.; Hamady, M.; Knight, R.; Gordon, J.I. Worlds within worlds: Evolution of the vertebrate gut microbiota. Nat. Rev. Microbiol. 2008, 6, 776–788. [Google Scholar] [CrossRef] [PubMed]
- Gerritsen, J.; Fuentes, S.; Grievink, W.; van Niftrik, L.; Tindall, B.J.; Timmerman, H.M.; Rijkers, G.T.; Smidt, H. Characterization of Romboutsia ilealis gen. nov., sp. nov., isolated from the gastro-intestinal tract of a rat, and proposal for the reclassification of five closely related members of the genus Clostridium into the genera Romboutsia gen. nov., Intestinibacter gen. nov., Terrisporobacter gen. nov. and Asaccharospora gen. nov. Int. J. Syst. Evol. Microbiol. 2014, 64, 1600–1616. [Google Scholar]
- Ricaboni, D.; Mailhe, M.; Khelaifia, S.; Raoult, D.; Million, M. Romboutsia timonensis, a new species isolated from human gut. New Microbes New Infect. 2016, 12, 6–7. [Google Scholar] [CrossRef]
- Yang, L.; Liu, S.; Ding, J.; Dai, R.; He, C.; Xu, K.; Honaker, C.F.; Zhang, Y.; Siegel, P.; Meng, H. Gut microbiota co-microevolution with selection for host humoral immunity. Front. Microbiol. 2017, 8, 1243. [Google Scholar] [CrossRef]
- Leisner, J.J.; Laursen, B.G.; Prévost, H.; Drider, D.; Dalgaard, P. Carnobacterium: Positive and negative effects in the environment and in foods. FEMS Microbiol. Rev. 2007, 31, 592–613. [Google Scholar] [CrossRef]
- Sousa, M.; Goncalves, A.; Silva, N.; Serra, R.; Alcaide, E.; Zorrilla, I.; Torres, C.; Canica, M.; Igrejas, G.; Poeta, P. Acquired antibiotic resistance among wild animals: The case of Iberian Lynx (Lynx pardinus). Vet. Q. 2014, 34, 105–112. [Google Scholar] [CrossRef]
- Poeta, P.; Costa, D.; Saenz, Y.; Klibi, N.; Ruiz-Larrea, F.; Rodrigues, J.; Torres, C. Characterization of antibiotic resistance genes and virulence factors in faecal enterococci of wild animals in Portugal. J. Vet. Med. B Infect. Dis. Vet. Public Health 2005, 52, 396–402. [Google Scholar] [CrossRef]
- Lebreton, F.; Willems, R.J.L.; Gilmore, M.S. Enterococcus diversity, origins in nature, and gut colonization. In Enterococci: From Commensals to Leading Causes of Drug Resistant Infection; Gilmore, M.S., Clewell, D.B., Ike, Y., Shankar, N., Eds.; Massachusetts Eye and Ear Infirmary: Boston, MA, USA, 2014. [Google Scholar]
- Seedorf, H.; Griffin, N.W.; Ridaura, V.K.; Reyes, A.; Cheng, J.; Rey, F.E.; Smith, M.I.; Simon, G.M.; Scheffrahn, R.H.; Woebken, D.; et al. Bacteria from diverse habitats colonize and compete in the mouse gut. Cell 2014, 159, 253–266. [Google Scholar] [CrossRef]
- Nakashima, K.; Kimura, S.; Ogawa, Y.; Watanabe, S.; Soma, S.; Kaneko, T.; Yamada, L.; Sawada, H.; Tung, C.-H.; Lu, T.-M.; et al. Chitin-based barrier immunity and its loss predated mucus-colonization by indigenous gut microbiota. Nat. Commun. 2018, 9, 3402. [Google Scholar] [CrossRef]
- Tsuchiya, C.; Sakata, T.; Sugita, H. Novel ecological niche of Cetobacterium somerae, an anaerobic bacterium in the intestinal tracts of freshwater fish. Lett. Appl. Microbiol. 2008, 46, 43–48. [Google Scholar] [CrossRef]
- Finegold, S.M.; Vaisanen, M.L.; Molitoris, D.R.; Tomzynski, T.J.; Song, Y.; Liu, C.; Collins, M.D.; Lawson, P.A. Cetobacterium somerae sp. nov. from human feces and emended description of the genus Cetobacterium. Syst. Appl. Microbiol. 2003, 26, 177–181. [Google Scholar] [CrossRef] [PubMed]
- Hand, D.; Wallis, C.; Colyer, A.; Penn, C.W. Pyrosequencing the canine faecal microbiota: Breadth and depth of biodiversity. PLoS ONE 2013, 8, e53115. [Google Scholar] [CrossRef] [PubMed]
- Keijser, B.J.; Zaura, E.; Huse, S.M.; van der Vossen, J.M.; Schuren, F.H.; Montijn, R.C.; ten Cate, J.M.; Crielaard, W. Pyrosequencing analysis of the oral microflora of healthy adults. J. Dent. Res. 2008, 87, 1016–1020. [Google Scholar] [CrossRef] [PubMed]
- Mazmanian, L. Has the microbiota played a critical role in the evolution of the adaptive immune system? Sci. N.Y. 2010, 330, 1768–1773. [Google Scholar]
- Wexler, H.M. Bacteroides: The good, the bad, and the nitty-gritty. Clin. Microbiol. Rev. 2007, 20, 593–621. [Google Scholar] [CrossRef]
- Alipour, M.J.; Jalanka, J.; Pessa-Morikawa, T.; Kokkonen, T.; Satokari, R.; Hynönen, U.; Iivanainen, A.; Niku, M. The composition of the perinatal intestinal microbiota in cattle. Sci. Rep. 2018, 8, 10437. [Google Scholar] [CrossRef]
- Navarro, M.; Dutra, F.; Uzal, F.A. Bacillary Hemoglobinuria. In Clostridial Diseases of Animals, 1st ed.; Uzal, F.A., Songer, W.G., Prescott, J.F., Eds.; Willey & Blackwell: Ames, IA, USA; Hoboken, NJ, USA, 2016; pp. 265–274. [Google Scholar]
- Ndongo, S.; Cassir, N.; Delerce, J.; Raoult, D.; La Scola, B. ‘Clostridium mediterraneense,’ a new bacterial species isolated from the human gut. New Microbes New Infect. 2017, 20, 25–26. [Google Scholar] [CrossRef]
- Bandeira, V.; Virgós, E.; Azevedo, A.; Carvalho, J.; Cunha, M.V.; Fonseca, C. Sex and season explain spleen weight variation in the Egyptian mongoose. Curr. Zool. 2018, 65, 11–20. [Google Scholar] [CrossRef]
- Aivelo, T.; Laakkonen, J.; Jernvall, J. Population- and individual-level dynamics of the intestinal microbiota of a small primate. Appl. Environ. Microbiol. 2016, 82, 3537–3545. [Google Scholar] [CrossRef] [PubMed]
- Degnan, P.H.; Pusey, A.E.; Lonsdorf, E.V.; Goodall, J.; Wroblewski, E.E.; Wilson, M.L.; Rudicell, R.S.; Hahn, B.H.; Ochman, H. Factors associated with the diversification of the gut microbial communities within chimpanzees from Gombe national park. Proc. Natl. Acad. Sci. USA 2012, 109, 13034–13039. [Google Scholar] [CrossRef] [PubMed]
- Amato, K.R.; Leigh, S.R.; Kent, A.; Mackie, R.I.; Yeoman, C.J.; Stumpf, R.M.; Wilson, B.A.; Nelson, K.E.; White, B.A.; Garber, P.A. The role of gut microbes in satisfying the nutritional demands of adult and juvenile wild, black howler monkeys (Alouatta pigra). Am. J. Phys. Anthropol. 2014, 155, 652–664. [Google Scholar] [CrossRef] [PubMed]
- Finlayson-Trick, E.C.L.; Getz, L.J.; Slaine, P.D.; Thornbury, M.; Lamoureux, E.; Cook, J.; Langille, M.G.I.; Murray, L.E.; McCormick, C.; Rohde, J.R.; et al. Taxonomic differences of gut microbiomes drive cellulolytic enzymatic potential within hind-gut fermenting mammals. PLoS ONE 2017, 12, e0189404. [Google Scholar] [CrossRef]
- Clavel, T.; Lepage, P.; Charrier, C. The Family Coriobacteriaceae; Springer Nature: Basel, Switzerland, 2014; Volume 49, pp. 201–238. [Google Scholar]
- Compo, N.R.; Gomez, D.E.; Tapscott, B.; Weese, J.S.; Turner, P.V. Fecal bacterial microbiota of Canadian commercial mink (Neovison vison): Yearly, life stage, and seasonal comparisons. PLoS ONE 2018, 13, e0207111. [Google Scholar] [CrossRef]
- Tidjani Alou, M.; Million, M.; Traore, S.I.; Mouelhi, D.; Khelaifia, S.; Bachar, D.; Caputo, A.; Delerce, J.; Brah, S.; Alhousseini, D.; et al. Gut bacteria missing in severe acute malnutrition, can we identify potential probiotics by culturomics? Front. Microbiol. 2017, 8, 899. [Google Scholar] [CrossRef]
- Goodrich, J.K.; Waters, J.L.; Poole, A.C.; Sutter, J.L.; Koren, O.; Blekhman, R.; Beaumont, M.; Van Treuren, W.; Knight, R.; Bell, J.T.; et al. Human genetics shape the gut microbiome. Cell 2014, 159, 789–799. [Google Scholar] [CrossRef]
- Kaakoush, N.O. Insights into the role of Erysipelotrichaceae in the human host. Front. Cell. Infect. Microbiol. 2015, 5, 84. [Google Scholar] [CrossRef]
- Clarke, S.F.; Murphy, E.F.; O’Sullivan, O.; Ross, R.P.; O’Toole, P.W.; Shanahan, F.; Cotter, P.D. Targeting the microbiota to address diet-induced obesity: A time dependent challenge. PLoS ONE 2013, 8, e65790. [Google Scholar] [CrossRef]
- Hu, X.; Liu, G.; Li, Y.; Wei, Y.; Lin, S.; Liu, S.; Zheng, Y.; Hu, D. High-throughput analysis reveals seasonal variation of the gut microbiota composition within forest musk deer (Moschus berezovskii). Front. Microbiol. 2018, 9, 1674. [Google Scholar] [CrossRef]
- Zhao, G.; Zhou, L.; Dong, Y.; Cheng, Y.; Song, Y. The gut microbiome of hooded cranes (Grus monacha) wintering at Shengjin lake, China. Microbiol. Open 2017, 6, e00447. [Google Scholar] [CrossRef] [PubMed]
- Moon, J.-Y.; Zolnik, C.P.; Wang, Z.; Qiu, Y.; Usyk, M.; Wang, T.; Kizer, J.R.; Landay, A.L.; Kurland, I.J.; Anastos, K.; et al. Gut microbiota and plasma metabolites associated with diabetes in women with, or at high risk for, HIV infection. EBioMedicine 2018, 37, 392–400. [Google Scholar] [CrossRef] [PubMed]
- Neis, E.P.; Dejong, C.H.; Rensen, S.S. The role of microbial amino acid metabolism in host metabolism. Nutrients 2015, 7, 2930–2946. [Google Scholar] [CrossRef] [PubMed]
- Agus, A.; Planchais, J.; Sokol, H. Gut microbiota regulation of tryptophan metabolism in health and disease. Cell Host Microbe 2018, 23, 716–724. [Google Scholar] [CrossRef]
- Hobbs, M.E.; Williams, H.J.; Hillerich, B.; Almo, S.C.; Raushel, F.M. l-galactose metabolism in Bacteroides vulgatus from the human gut microbiota. Biochemistry 2014, 53, 4661–4670. [Google Scholar] [CrossRef]
- Brown, K.L.; Hancock, R.E. Cationic host defense (antimicrobial) peptides. Curr. Opin. Immunol. 2006, 18, 24–30. [Google Scholar] [CrossRef]
- Trevelline, B.K.; Fontaine, S.S.; Hartup, B.K.; Kohl, K.D. Conservation biology needs a microbial renaissance: A call for the consideration of host-associated microbiota in wildlife management practices. Proc. R. Soc. B Biol. Sci. 2019, 286, 20182448. [Google Scholar] [CrossRef]
- Amato, K.R.; Yeoman, C.J.; Kent, A.; Righini, N.; Carbonero, F.; Estrada, A.; Rex Gaskins, H.; Stumpf, R.M.; Yildirim, S.; Torralba, M.; et al. Habitat degradation impacts black howler monkey (Alouatta pigra) gastrointestinal microbiomes. ISME J. 2013, 7, 1344. [Google Scholar] [CrossRef]
- Barelli, C.; Albanese, D.; Donati, C.; Pindo, M.; Dallago, C.; Rovero, F.; Cavalieri, D.; Tuohy, K.M.; Hauffe, H.C.; de Filippo, C. Habitat fragmentation is associated to gut microbiota diversity of an endangered primate: Implications for conservation. Sci. Rep. 2015, 5, 14862. [Google Scholar] [CrossRef]
- Menke, S.; Meier, M.; Mfune, J.K.E.; Melzheimer, J.; Wachter, B.; Sommer, S. Effects of host traits and land-use changes on the gut microbiota of the Namibian black-backed jackal (Canis mesomelas). FEMS Microbiol. Ecol. 2017, 93. [Google Scholar] [CrossRef]
- Kikuchi, Y.; Tada, A.; Musolin, D.L.; Hari, N.; Hosokawa, T.; Fujisaki, K.; Fukatsu, T. Collapse of insect gut symbiosis under simulated climate change. mBio 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Bestion, E.; Jacob, S.; Zinger, L.; Di Gesu, L.; Richard, M.; White, J.; Cote, J. Climate warming reduces gut microbiota diversity in a vertebrate ectotherm. Nat. Ecol. Amp. Evol. 2017, 1, 0161. [Google Scholar] [CrossRef] [PubMed]
- Fontaine, S.S.; Novarro, A.J.; Kohl, K.D. Environmental temperature alters the digestive performance and gut microbiota of a terrestrial amphibian. J. Exp. Biol. 2018, 221. [Google Scholar] [CrossRef] [PubMed]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).