
Deciphering Estrus Expression in Gilts: The Role of Alternative Polyadenylation and LincRNAs in Reproductive Transcriptomics
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Sequencing of Samples
2.3. LincRNAs Identification
2.4. Analysis of Differential Expression and Function Enrichment Analysis
2.5. Analysis of Alternative Polyadenylation in the 3′-UTR
2.6. Analysis of 3′-UTR Sequences and Functional Elements
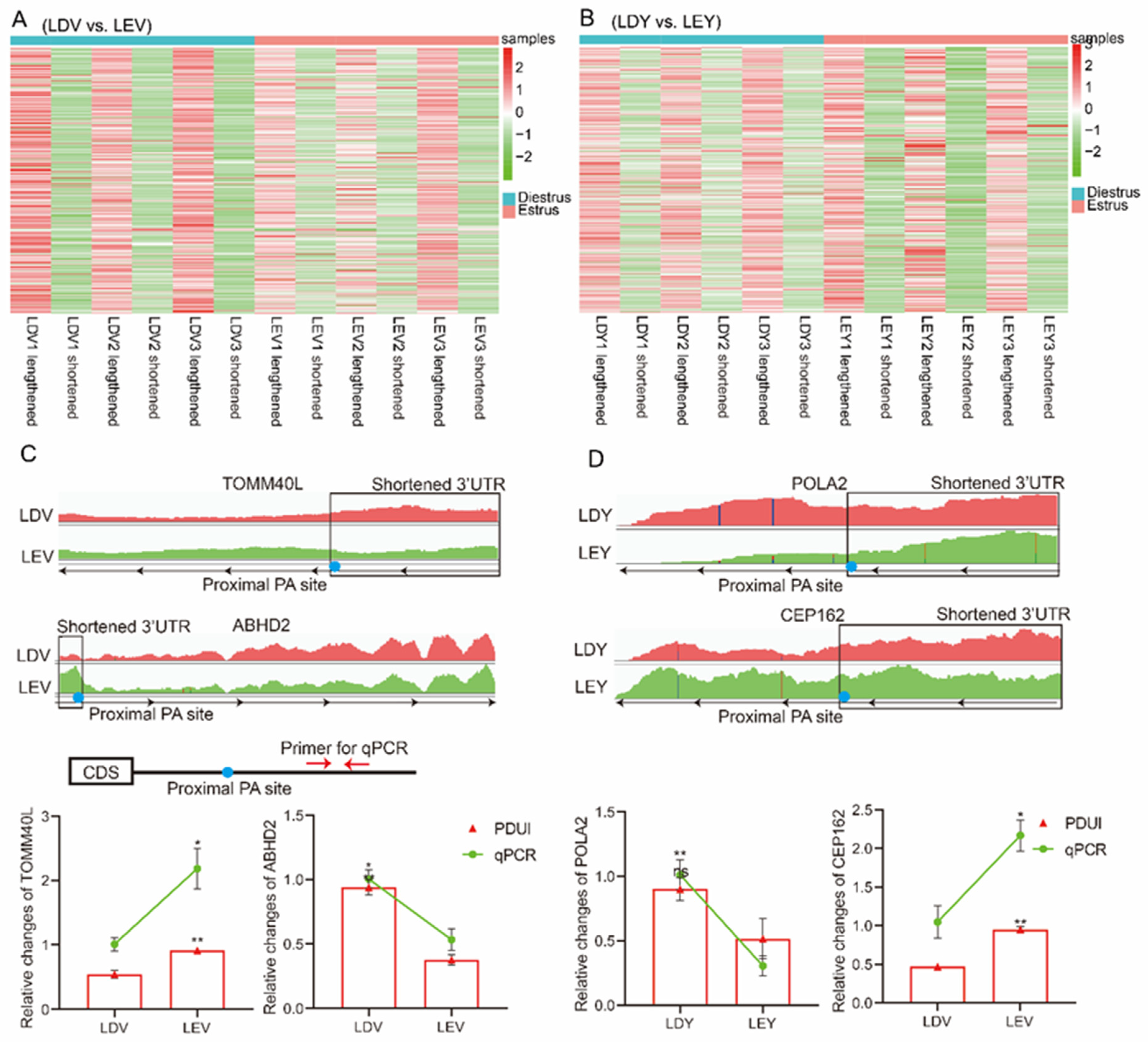
2.7. RT-qPCR Verification
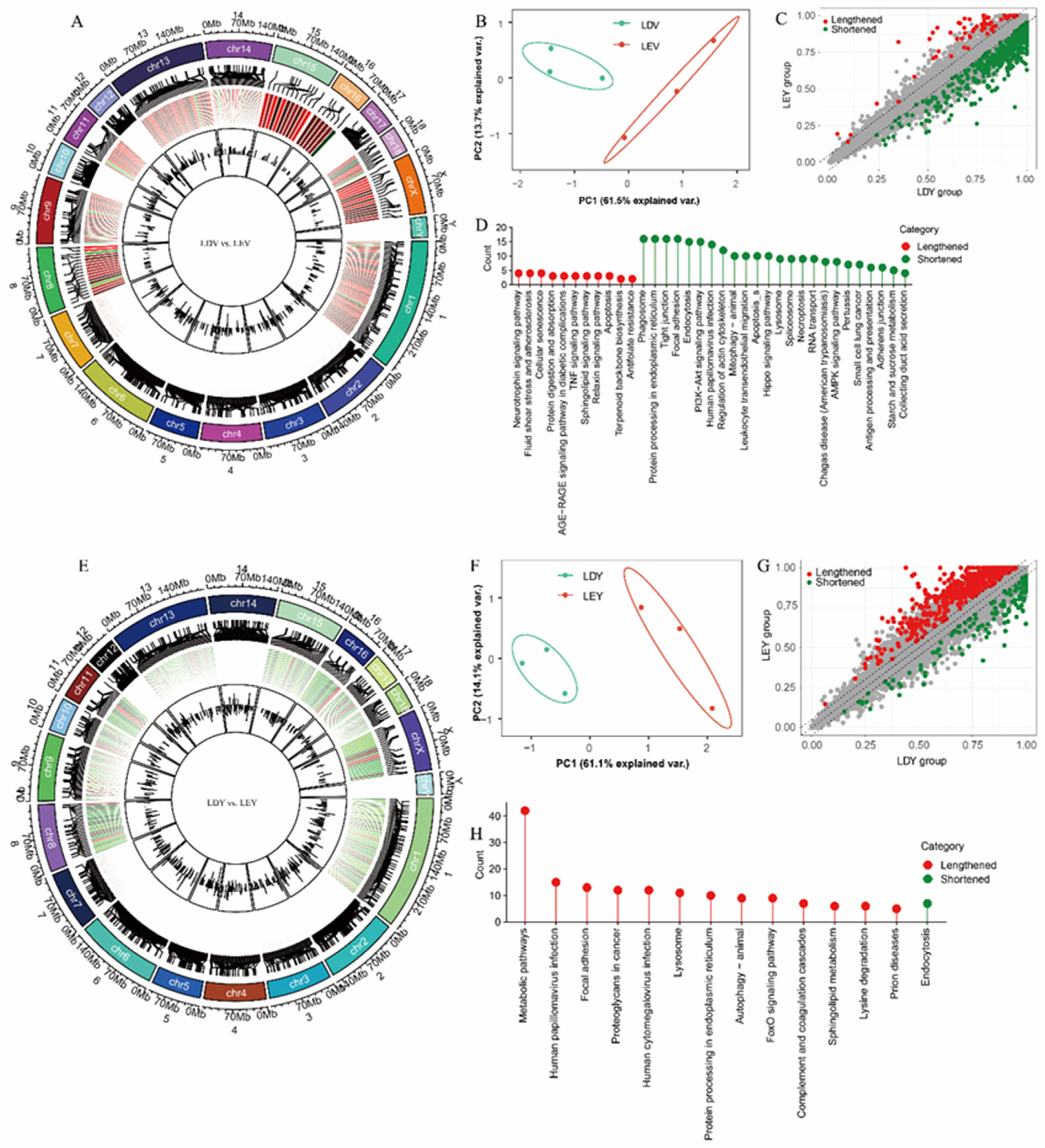
3. Results
3.1. Analysis of Differentially Expressed Genes in the Vulva and Vagina
3.2. Analysis of Differentially Expressed lincRNAs in the Vulva and Vagina
3.3. Identification and Analysis of APA Sites in the Estrus Cycles
3.4. Visualization and Validation of 3′-UTR Expression Abundance
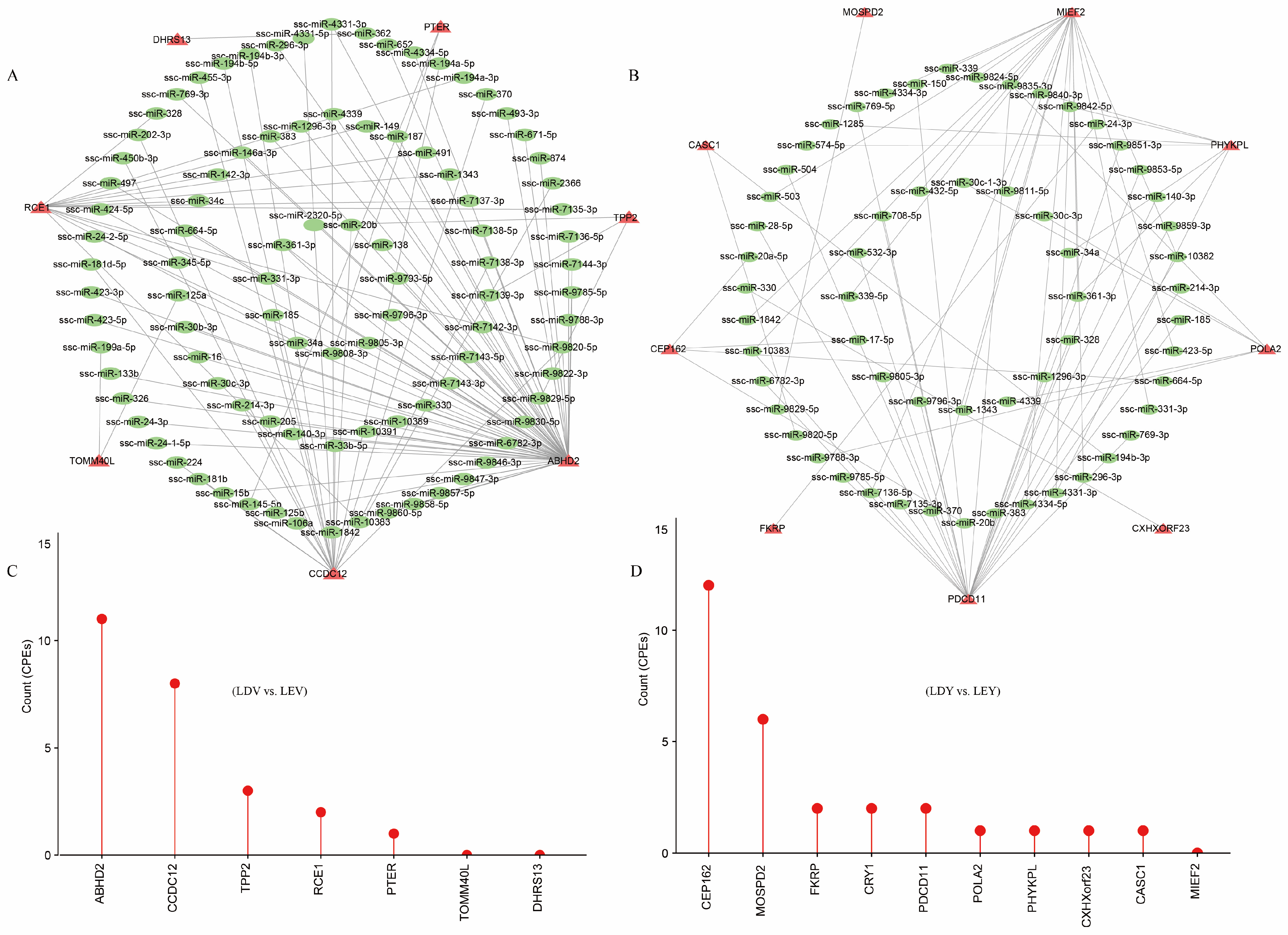
3.5. Regulatory Elements in Lengthened 3′-UTR
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Xue, H.X.; Chen, J.X.; Ding, Q.A.; Sun, Y.W.; Shen, M.X.; Liu, L.S.; Chen, X.D.; Zhou, J.Y. Automatic detection of sow posture and estrus based on convolutional neural network. Front. Phys. 2022, 10, 1037129. [Google Scholar] [CrossRef]
- Niu, X.; Huang, Y.L.; Lu, H.; Li, S.; Huang, S.H.; Ran, X.Q.; Wang, J.F. CircRNAs in Xiang pig ovaries among diestrus and estrus stages. Porc. Health Manag. 2022, 8, 29. [Google Scholar] [CrossRef]
- Yang, S.B.; Zhou, X.L.; Pei, Y.; Wang, H.; He, K.; Zhao, A.Y. Identification of Differentially Expressed Genes in Porcine Ovaries at Proestrus and Estrus Stages Using RNA-Seq Technique. Biomed. Res. Int. 2018, 2018, 9150723. [Google Scholar] [CrossRef]
- Verhoeven, S.; Chantziaras, I.; Bernaerdt, E.; Loicq, M.; Verhoeven, L.; Maes, D. The evaluation of an artificial intelligence system for estrus detection in sows. Porc. Health Manag. 2023, 9, 9. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.T.; Ran, X.Q.; Mao, N.; Zhang, F.P.; Niu, X.; Ruan, Y.Q.; Yi, F.L.; Li, S.; Wang, J.F. Analysis of alternative splicing events by RNA sequencing in the ovaries of Xiang pig at estrous and diestrous. Theriogenology 2018, 119, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Shade, K.A.; Stewart, K.R.; Johnson, J.S. Characterizing body temperature and movement differences at the onset of estrus in replacement gilts. J. Anim. Sci. 2016, 94, 194. [Google Scholar] [CrossRef]
- Liu, M.; Xu, Q.; Zhao, J.; Guo, Y.; Zhang, C.; Chao, X.; Cheng, M.; Schinckel, A.P.; Zhou, B. Comprehensive Transcriptome Analysis of Follicles from Two Stages of the Estrus Cycle of Two Breeds Reveals the Roles of Long Intergenic Non-Coding RNAs in Gilts. Biology 2022, 11, 716. [Google Scholar] [CrossRef] [PubMed]
- Glencorse, D.; Grupen, C.G.; Bathgate, R. Vaginal and vestibular electrical resistance as an alternative marker for optimum timing of artificial insemination with liquid-stored and frozen-thawed spermatozoa in sows. Sci. Rep. 2023, 13, 12103. [Google Scholar] [CrossRef] [PubMed]
- Yilma, T.; Sobiraj, A. Relationships Between Intra-Vaginal Electrical Impedance, Estrus Behavior, Plasma Levels of Ovarian Steroids and the Pre-Ovulatory Luteinizing Hormone Surge in the Prediction of the Optimal Insemination Time in Normal Cycling Pigs. Indian J. Anim. Res. 2012, 46, 114–120. [Google Scholar]
- Cao, J.; Kuyumcu-Martinez, M.N. Alternative polyadenylation regulation in cardiac development and cardiovascular disease. Cardiovasc. Res. 2023, 119, 1324–1335. [Google Scholar] [CrossRef]
- Derti, A.; Garrett-Engele, P.; MacIsaac, K.D.; Stevens, R.C.; Sriram, S.; Chen, R.H.; Rohl, C.A.; Johnson, J.M.; Babak, T. A quantitative atlas of polyadenylation in five mammals. Genome Res. 2012, 22, 1173–1183. [Google Scholar] [CrossRef]
- Ji, Z.; Luo, W.T.; Li, W.C.; Hoque, M.; Pan, Z.H.; Zhao, Y.; Tian, B. Transcriptional activity regulates alternative cleavage and polyadenylation. Mol. Syst. Biol. 2011, 7, 534. [Google Scholar] [CrossRef]
- Wang, Y.; Wu, Z.W.; Mou, Q.; Chen, L.; Fang, T.; Zhang, Y.Q.; Yin, Z.J.; Du, Z.Q.; Yang, C.X. Global 3’-UTRome of porcine immature Sertoli cells altered by acute heat stress. Theriogenology 2023, 196, 79–87. [Google Scholar] [CrossRef]
- Stotts, M.J.; Zhang, Y.Z.; Zhang, S.W.; Michal, J.J.; Velez, J.; Hans, B.; Maquivar, M.; Jiang, Z.H. Alternative polyadenylation events in epithelial cells sense endometritis progression in dairy cows. J. Integr. Agric. 2023, 22, 1820–1832. [Google Scholar] [CrossRef]
- Ren, F.G.; Zhang, N.; Zhang, L.; Miller, E.; Pu, J.J. Alternative Polyadenylation: A new frontier in post transcriptional regulation. Biomark. Res. 2020, 8, 67. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.C.; Zheng, D.H.; Zhang, Q.; Guvenek, A.; Cheng, H.; Tian, B. Alternative 3’ UTRs play a widespread role in translation-independent mRNA association with the endoplasmic reticulum. Cell Rep. 2021, 36, 109407. [Google Scholar] [CrossRef]
- Masamha, C.P.; Wagner, E.J. The contribution of alternative polyadenylation to the cancer phenotype. Carcinogenesis 2018, 39, 2–10. [Google Scholar] [CrossRef]
- Berkovits, B.D.; Mayr, C. Alternative 3’ UTRs act as scaffolds to regulate membrane protein localization. Nature 2015, 522, 363–367. [Google Scholar] [CrossRef] [PubMed]
- Mayr, C.; Bartel, D.P. Widespread Shortening of 3’ UTRs by Alternative Cleavage and Polyadenylation Activates Oncogenes in Cancer Cells. Cell 2009, 138, 673–684. [Google Scholar] [CrossRef]
- Rane, S.; Sayed, D.; Abdellatif, M. MicroRNA with a MacroFunction. Cell Cycle 2007, 6, 1850–1855. [Google Scholar] [CrossRef]
- Wang, R.J.; Nambiar, R.; Zheng, D.H.; Tian, B. PolyA_DB 3 catalogs cleavage and polyadenylation sites identified by deep sequencing in multiple genomes. Nucleic Acids Res. 2018, 46, D315–D319. [Google Scholar] [CrossRef] [PubMed]
- Xia, Z.; Donehower, L.A.; Cooper, T.A.; Neilson, J.R.; Wheeler, D.A.; Wagner, E.J.; Li, W. Dynamic analyses of alternative polyadenylation from RNA-seq reveal a 3’-UTR landscape across seven tumour types. Nat. Commun. 2014, 5, 5274. [Google Scholar] [CrossRef]
- Mittleman, B.E.; Pott, S.; Warland, S.; Zeng, T.; Mu, Z.P.; Kaur, M.; Gilad, Y.; Li, Y. Alternative polyadenylation mediates genetic regulation of gene expression. Elife 2020, 9, e57492. [Google Scholar] [CrossRef]
- Lv, W.; Jiang, W.; Luo, H.M.; Tong, Q.; Niu, X.Y.; Liu, X.; Miao, Y.; Wang, J.N.; Guo, Y.W.; Li, J.A.; et al. Long noncoding RNA lncMREF promotes myogenic differentiation and muscle regeneration by interacting with the Smarca5/p300 complex. Nucleic Acids Res. 2022, 50, 10733–10755. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Ji, J.Z.; Cui, H.D.; Zhao, Q.; Ding, C.M.; Xu, C. Functional evaluation of LTR-derived lncRNAs in porcine oocytes and zygotes with RNA-seq and small RNA-seq. Front. Genet. 2022, 13, 1023041. [Google Scholar] [CrossRef]
- Ransohoff, J.D.; Wei, Y.N.; Khavari, P.A. The functions and unique features of long intergenic non-coding RNA. Nat. Rev. Mol. Cell Biol. 2018, 19, 143–157. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.P.; Gao, R.F.; Chen, X.M.; Geng, Y.Q.; Yin, X.; Peng, C.; Mu, X.Y.; Su, Y.; Zhang, Y.; Li, F.F.; et al. lincRNA RP24-315D19.10 promotes endometrial decidualization upregulation of hnRNPA2B1. Bba-Mol. Basis Dis. 2023, 1869, 166762. [Google Scholar] [CrossRef]
- Guttman, M.; Donaghey, J.; Carey, B.W.; Garber, M.; Grenier, J.K.; Munson, G.; Young, G.; Lucas, A.B.; Ach, R.; Bruhn, L.; et al. lincRNAs act in the circuitry controlling pluripotency and differentiation. Nature 2011, 477, 295–300. [Google Scholar] [CrossRef]
- Keniry, A.; Oxley, D.; Monnier, P.; Kyba, M.; Dandolo, L.; Smits, G.; Reik, W. The H19 lincRNA is a developmental reservoir of miR-675 that suppresses growth and Igf1r. Nat. Cell Biol. 2012, 14, 659–665. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, L.; Qiu, Q.; Zhou, Q.; Ding, J.; Lu, Y.; Liu, P. Alternative polyadenylation: Methods, mechanism, function, and role in cancer. J. Exp. Clin. Cancer Res. 2021, 40, 51. [Google Scholar] [CrossRef]
- Liu, M.; Zhang, C.; Chen, J.; Xu, Q.; Liu, S.; Chao, X.; Yang, H.; Wang, T.; Muhammad, A.; Schinckel, A.P.; et al. Characterization and analysis of transcriptomes of multiple tissues from estrus and diestrus in pigs. Int. J. Biol. Macromol. 2024, 256, 128324. [Google Scholar] [CrossRef]
- Chu, Q.; Zhou, B.; Xu, F.; Chen, R.; Shen, C.; Liang, T.; Li, Y.; Schinckel, A.P. Genome-wide differential mRNA expression profiles in follicles of two breeds and at two stages of estrus cycle of gilts. Sci. Rep. 2017, 7, 5052. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, Y.; Shi, C.; Huang, Z.; Zhang, Y.; Li, S.; Li, Y.; Ye, J.; Yu, C.; Li, Z.; et al. SOAPnuke: A MapReduce acceleration-supported software for integrated quality control and preprocessing of high-throughput sequencing data. Gigascience 2018, 7, gix120. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Li, H. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Bao, Z.; Yang, Z.; Huang, Z.; Zhou, Y.; Cui, Q.; Dong, D. LncRNADisease 2.0: An updated database of long non-coding RNA-associated diseases. Nucleic Acids Res. 2019, 47, D1034–D1037. [Google Scholar] [CrossRef] [PubMed]
- Bu, D.C.; Luo, H.T.; Huo, P.P.; Wang, Z.H.; Zhang, S.; He, Z.H.; Wu, Y.; Zhao, L.H.; Liu, J.J.; Guo, J.C.; et al. KOBAS-i: Intelligent prioritization and exploratory visualization of biological functions for gene enrichment analysis. Nucleic Acids Res. 2021, 49, W317–W325. [Google Scholar] [CrossRef] [PubMed]
- Thorvaldsdottir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef] [PubMed]
- Pique, M.; Lopez, J.M.; Foissac, S.; Guigo, R.; Mendez, R. A combinatorial code for CPE-mediated translational control. Cell 2008, 132, 434–448. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, V.; Bell, G.W.; Nam, J.W.; Bartel, D.P. Predicting effective microRNA target sites in mammalian mRNAs. eLife 2015, 4, e05005. [Google Scholar] [CrossRef]
- Long, X.; Burke, K.A.; Bigsby, R.M.; Nephew, K.P. Effects of the xenoestrogen bisphenol A on expression of vascular endothelial growth factor (VEGF) in the rat. Exp. Biol. Med. 2001, 226, 477–483. [Google Scholar] [CrossRef] [PubMed]
- Ni, Y.F.; Wu, F.; Chen, Q.Q.; Cai, J.F.; Hu, J.P.; Shen, J.C.; Zhang, J.Z. Long noncoding RNA and mRNA profiling of hypothalamic-pituitary-mammary gland axis in lactating sows under heat stress. Genomics 2020, 112, 3668–3676. [Google Scholar] [CrossRef] [PubMed]
- Li, M.X.; Liu, Y.; Xie, S.; Ma, L.P.; Zhao, Z.C.; Gong, H.B.; Sun, Y.S.; Huang, T. Transcriptome analysis reveals that long noncoding RNAs contribute to developmental differences between medium-sized ovarian follicles of Meishan and Duroc sows. Sci. Rep. 2021, 11, 22510. [Google Scholar] [CrossRef] [PubMed]
- Tian, B.; Hu, J.; Zhang, H.B.; Lutz, C.S. A large-scale analysis of mRNA polyadenylation of human and mouse genes. Nucleic Acids Res. 2005, 33, 201–212. [Google Scholar] [CrossRef] [PubMed]
- Di Giammartino, D.C.; Nishida, K.; Manley, J.L. Mechanisms and Consequences of Alternative Polyadenylation. Mol. Cell 2011, 43, 853–866. [Google Scholar] [CrossRef] [PubMed]
- Devany, E.; Park, J.Y.; Murphy, M.R.; Zakusilo, G.; Baquero, J.; Zhang, X.K.; Hoque, M.; Tian, B.; Kleiman, F.E. Intronic cleavage and polyadenylation regulates gene expression during DNA damage response through U1 snRNA. Cell Discov. 2016, 2, 16013. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.T.; Zhan, D.D.; Qu, S.; Jiang, S.; Gan, W.H.; Qin, W.S.; Zheng, C.X.; Cheng, F.; Lu, Y.H.; Liu, M.W.; et al. Transcriptomics-proteomics Integration reveals alternative polyadenylation driving inflammation-related protein translation in patients with diabetic nephropathy. J. Transl. Med. 2023, 21, 86. [Google Scholar] [CrossRef]
- Guo, S.Y.; Lin, S.B. mRNA alternative polyadenylation (APA) in regulation of gene expression and diseases. Genes Dis. 2023, 10, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.L.; Ji, G.L.; Fu, H.J.; Lin, Q.M.; Ye, C.T.; Ye, W.B.; Su, Y.R.; Wu, X.H. A survey on identification and quantification of alternative polyadenylation sites from RNA-seq data. Brief. Bioinform. 2020, 21, 1261–1276. [Google Scholar] [CrossRef]
- Yan, J.; Marr, T.G. Computational analysis of 3’-ends of ESTs shows four classes of alternative polyadenylation in human, mouse, and rat. Genome Res. 2005, 15, 369–375. [Google Scholar] [CrossRef]
- MacDonald, C.C. Tissue-specific mechanisms of alternative polyadenylation: Testis, brain, and beyond (2018 update). Wiley Interdiscip. Rev. RNA 2019, 10, e1526. [Google Scholar] [CrossRef] [PubMed]
- Imada, E.L.; Wilks, C.; Langmead, B.; Marchionni, L. REPAC: Analysis of alternative polyadenylation from RNA-sequencing data. Genome Biol. 2023, 24, 22. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.J.; Cai, T.; Liu, C.; Zhang, X.; Fu, X.D. Sequential Polyadenylation to Enable Alternative mRNA3’ End Formation. Mol. Cells 2023, 46, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Soede, N.M.; Langendijk, P.; Kemp, B. Reproductive cycles in pigs. Anim. Reprod. Sci. 2011, 124, 251–258. [Google Scholar] [CrossRef]
- Li, X.; Su, J.; Lei, Z.H.; Zhao, Y.Y.; Jin, M.M.; Fang, R.; Zheng, L.C.; Jiao, Y. Gonadotropin-inhibitory hormone (GnIH) and its receptor in the female pig: cDNA cloning, expression in tissues and expression pattern in the reproductive axis during the estrous cycle. Peptides 2012, 36, 176–185. [Google Scholar] [CrossRef]
- Gade, S.; Bennewitz, J.; Kirchner, K.; Looft, H.; Knap, P.W.; Thaller, G.; Kalm, E. A note on genetic parameters for estrus symptoms in sows. Appl. Anim. Behav. Sci. 2008, 109, 406–409. [Google Scholar] [CrossRef]
- Tian, B.; Manley, J.L. Alternative polyadenylation of mRNA precursors. Nat. Rev. Mol. Cell Bio 2017, 18, 18–30. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, M.; Chen, J.; Zhang, C.; Liu, S.; Chao, X.; Yang, H.; Muhammad, A.; Zhou, B.; Ao, W.; Schinckel, A.P. Deciphering Estrus Expression in Gilts: The Role of Alternative Polyadenylation and LincRNAs in Reproductive Transcriptomics. Animals 2024, 14, 791. https://doi.org/10.3390/ani14050791
Liu M, Chen J, Zhang C, Liu S, Chao X, Yang H, Muhammad A, Zhou B, Ao W, Schinckel AP. Deciphering Estrus Expression in Gilts: The Role of Alternative Polyadenylation and LincRNAs in Reproductive Transcriptomics. Animals. 2024; 14(5):791. https://doi.org/10.3390/ani14050791
Chicago/Turabian StyleLiu, Mingzheng, Jiahao Chen, Chunlei Zhang, Shuhan Liu, Xiaohuan Chao, Huan Yang, Asim Muhammad, Bo Zhou, Weiping Ao, and Allan P. Schinckel. 2024. "Deciphering Estrus Expression in Gilts: The Role of Alternative Polyadenylation and LincRNAs in Reproductive Transcriptomics" Animals 14, no. 5: 791. https://doi.org/10.3390/ani14050791