
Alveolar Hyperoxia and Exacerbation of Lung Injury in Critically Ill SARS-CoV-2 Pneumonia
 , , , ,
, , , ,
Abstract
:1. COVID-19
2. COVID-19, Oxygen Therapy, and Alveolar Hyperoxia
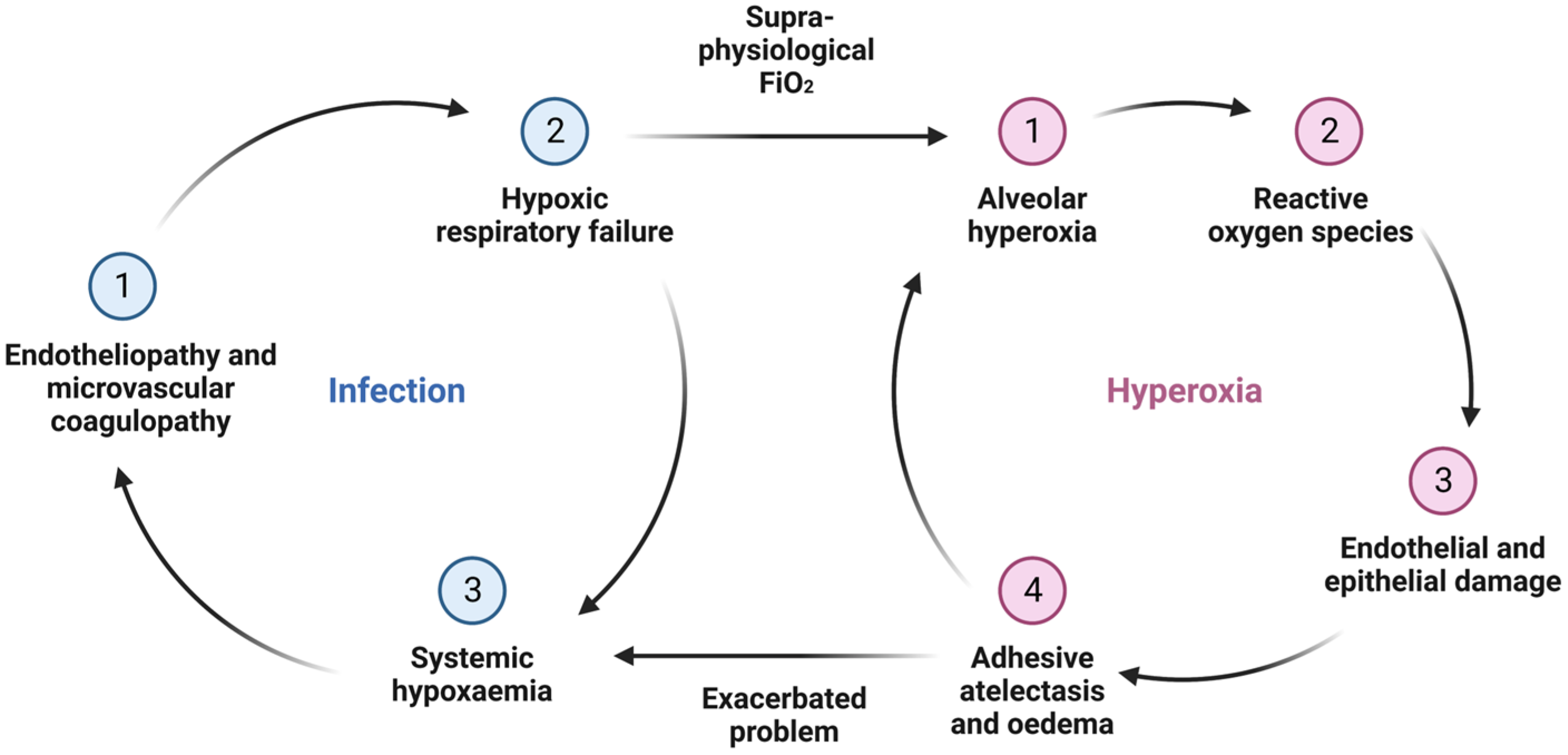
3. Consequence of Alveolar Hyperoxia and Systemic Hyperoxaemia
3.1. Ubiquitous Use of Oxygen May Be a Problem—Pre COVID-19 Studies
3.2. Oxygen Toxicity Mechanisms
3.3. Alveolar Hyperoxia Induced Surfactant Damage
3.4. Alveolar Hyperoxia and the Expression of ACE2 Receptors
3.5. Alveolar Hyperoxia Induced Pulmonary Vascular Changes
3.6. Hyperoxia Induced Immune Dysfunction and Secondary Bacterial Infections
4. SARS-CoV-2 Pathogenesis Suggest Significant Endotheliopathy, Coagulopathy, and Microangiopathy
5. How to Minimise Alveolar Hyperoxia?
5.1. Permissive Hypoxaemia
5.2. Adjunctive Measures to Optimise Oxygenation
5.2.1. Mechanical Ventilation
5.2.2. Pulmonary Vasodilators
5.2.3. Prone Positioning
5.2.4. Extracorporeal Membrane Oxygenation
5.3. Improving the Burden of Microangiopathy and Microthrombosis—Potential Targets
5.4. Improving Oxidative Stress and Inflammation—Exploratory Targets
6. Steroids for COVID-19 Patients Requiring Oxygen Therapy
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- WHO Coronavirus (COVID-19) Dashboard. Available online: https://covid19.who.int (accessed on 21 September 2023).
- Wingert, A.; Pillay, J.; Gates, M.; Guitard, S.; Rahman, S.; Beck, A.; Vandermeer, B.; Hartling, L. Risk factors for severity of COVID-19: A rapid review to inform vaccine prioritisation in Canada. BMJ Open 2021, 11, e044684. [Google Scholar] [CrossRef] [PubMed]
- Roberts, C.M.; Levi, M.; McKee, M.; Schilling, R.; Lim, W.S.; Grocott, M.P.W. COVID-19: A complex multisystem disorder. Br. J. Anaesth. 2020, 125, 238–242. [Google Scholar] [CrossRef] [PubMed]
- Wunsch, H. Mechanical Ventilation in COVID-19: Interpreting the Current Epidemiology. Am. J. Respir. Crit. Care Med. 2020, 202, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Osuchowski, M.F.; Winkler, M.S.; Skirecki, T.; Cajander, S.; Shankar-Hari, M.; Lachmann, G.; Monneret, G.; Venet, F.; Bauer, M.; Brunkhorst, F.M.; et al. The COVID-19 puzzle: Deciphering pathophysiology and phenotypes of a new disease entity. Lancet Respir. Med. 2021, 9, 622–642. [Google Scholar] [CrossRef]
- Nicolai, L.; Leunig, A.; Brambs, S.; Kaiser, R.; Weinberger, T.; Weigand, M.; Muenchhoff, M.; Hellmuth, J.C.; Ledderose, S.; Schulz, H.; et al. Immunothrombotic Dysregulation in COVID-19 Pneumonia Is Associated with Respiratory Failure and Coagulopathy. Circulation 2020, 142, 1176–1189. [Google Scholar] [CrossRef]
- Zhou, F.; Yu, T.; Du, R.; Fan, G.; Liu, Y.; Liu, Z.; Xiang, J.; Wang, Y.; Song, B.; Gu, X.; et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: A retrospective cohort study. Lancet 2020, 395, 1054–1062. [Google Scholar] [CrossRef]
- Li, L.; Li, R.; Wu, Z.; Yang, X.; Zhao, M.; Liu, J.; Chen, D. Therapeutic strategies for critically ill patients with COVID-19. Ann. Intensive Care 2020, 10, 45. [Google Scholar] [CrossRef]
- Zampieri, F.G.; Ferreira, J.C. Defining Optimal Respiratory Support for Patients with COVID-19. JAMA 2022, 327, 531–533. [Google Scholar] [CrossRef]
- Grasselli, G.; Zangrillo, A.; Zanella, A.; Antonelli, M.; Cabrini, L.; Castelli, A.; Cereda, D.; Coluccello, A.; Foti, G.; Fumagalli, R.; et al. Baseline Characteristics and Outcomes of 1591 Patients Infected With SARS-CoV-2 Admitted to ICUs of the Lombardy Region, Italy. JAMA 2020, 323, 1574–1581. [Google Scholar] [CrossRef]
- ICNARC. ICNARC Report on COVID-19 in Critical Care: England, Wales and Northern Ireland; Intensive Care National Audit & Research Centre: London, UK, June 2021; Available online: https://www.icnarc.org/Our-Audit/Audits/Cmp/Reports (accessed on 24 September 2023).
- Mach, W.J.; Thimmesch, A.R.; Pierce, J.T.; Pierce, J.D. Consequences of hyperoxia and the toxicity of oxygen in the lung. Nurs. Res. Pract. 2011, 2011, 260482. [Google Scholar] [CrossRef]
- Myti, D.; Gunjak, M.; Casado, F.; Khaghani Raziabad, S.; Nardiello, C.; Vadasz, I.; Herold, S.; Pryhuber, G.; Seeger, W.; Morty, R.E. Elevated FiO(2) increases SARS-CoV-2 co-receptor expression in respiratory tract epithelium. Am. J. Physiol. Lung Cell Mol. Physiol. 2020, 319, L670–L674. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, M.; Verleden, S.E.; Kuehnel, M.; Haverich, A.; Welte, T.; Laenger, F.; Vanstapel, A.; Werlein, C.; Stark, H.; Tzankov, A.; et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in Covid-19. N. Engl. J. Med. 2020, 383, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.; Eastwood, G.M.; Goodwin, M.D.; Noe, G.D.; Smith, P.E.; Glassford, N.; Schneider, A.G.; Bellomo, R. Atelectasis and mechanical ventilation mode during conservative oxygen therapy: A before-and-after study. J. Crit. Care 2015, 30, 1232–1237. [Google Scholar] [CrossRef] [PubMed]
- Pagano, A.; Barazzone-Argiroffo, C. Alveolar cell death in hyperoxia-induced lung injury. Ann. N. Y. Acad. Sci. 2003, 1010, 405–416. [Google Scholar] [CrossRef]
- Beyer, A.M.; Norwood Toro, L.E.; Hughes, W.E.; Young, M.; Clough, A.V.; Gao, F.; Medhora, M.; Audi, S.H.; Jacobs, E.R. Autophagy, TERT, and mitochondrial dysfunction in hyperoxia. Am. J. Physiol. Heart Circ. Physiol. 2021, 321, H985–H1003. [Google Scholar] [CrossRef]
- Elsoukkary, S.S.; Mostyka, M.; Dillard, A.; Berman, D.R.; Ma, L.X.; Chadburn, A.; Yantiss, R.K.; Jessurun, J.; Seshan, S.V.; Borczuk, A.C.; et al. Autopsy Findings in 32 Patients with COVID-19: A Single-Institution Experience. Pathobiology 2021, 88, 56–68. [Google Scholar] [CrossRef]
- Dias-Freitas, F.; Metelo-Coimbra, C.; Roncon-Albuquerque, R., Jr. Molecular mechanisms underlying hyperoxia acute lung injury. Respir. Med. 2016, 119, 23–28. [Google Scholar] [CrossRef]
- Ni, W.; Yang, X.; Yang, D.; Bao, J.; Li, R.; Xiao, Y.; Hou, C.; Wang, H.; Liu, J.; Yang, D.; et al. Role of angiotensin-converting enzyme 2 (ACE2) in COVID-19. Crit. Care 2020, 24, 422. [Google Scholar] [CrossRef]
- Kapur, N.; Nixon, G.; Robinson, P.; Massie, J.; Prentice, B.; Wilson, A.; Schilling, S.; Twiss, J.; Fitzgerald, D.A. Respiratory management of infants with chronic neonatal lung disease beyond the NICU: A position statement from the Thoracic Society of Australia and New Zealand. Respirology 2020, 25, 880–888. [Google Scholar] [CrossRef]
- O’Driscoll, B.R.; Howard, L.S.; Earis, J.; Mak, V.; British Thoracic Society Emergency Oxygen Guideline Group. BTS guideline for oxygen use in adults in healthcare and emergency settings. Thorax 2017, 72 (Suppl. S1), ii1–ii90. [Google Scholar] [CrossRef]
- Dushianthan, A.; Cumpstey, A.F.; Ferrari, M.; Thomas, W.; Moonesinghe, R.S.; Summers, C.; Montgomery, H.; Grocott, M.P. Intensive care physicians’ perceptions of the diagnosis & management of patients with acute hypoxic respiratory failure associated with COVID-19: A UK based survey. J. Intensive Care Soc. 2022, 23, 285–292. [Google Scholar] [PubMed]
- Cumpstey, A.F.; Oldman, A.H.; Martin, D.S.; Smith, A.; Grocott, M.P.W. Oxygen Targets During Mechanical Ventilation in the ICU: A Systematic Review and Meta-Analysis. Crit. Care Explor. 2022, 4, e0652. [Google Scholar] [CrossRef] [PubMed]
- Klitgaard, T.L.; Schjørring, O.L.; Nielsen, F.M.; Meyhoff, C.S.; Perner, A.; Wetterslev, J.; Rasmussen, B.S.; Barbateskovic, M. Higher versus lower fractions of inspired oxygen or targets of arterial oxygenation for adults admitted to the intensive care unit. Cochrane Database Syst. Rev. 2023, 9, CD012631. [Google Scholar] [PubMed]
- Protocol and Statistical Analysis Plan for the Mega Randomised Registry Trial Research Program Comparing Conservative versus Liberal Oxygenation Targets in Adults Receiving Unplanned Invasive Mechanical Ventilation in the ICU (Mega-ROX)|CCR. 7 July 2022. Available online: https://ccr.cicm.org.au/journal-editions/2022/june/24/original-articles/article-8 (accessed on 21 September 2023).
- UK-ROX. Intensive Care Unit Randomised Trial Comparing Two Approaches to OXygen Therapy. 7 July 2022. Available online: https://www.icnarc.org/Our-Research/Studies/Uk-Rox/About (accessed on 21 September 2023).
- Mega-ROX. In ANZICS [Internet]. 8 July 2022. Available online: https://www.anzics.com.au/current-active-endorsed-research/mega-rox/ (accessed on 24 September 2023).
- Rasmussen, B.S.; Klitgaard, T.L.; Perner, A.; Brand, B.A.; Hildebrandt, T.; Siegemund, M.; Hollinger, A.; Aagaard, S.R.; Bestle, M.H.; Marcussen, K.V.; et al. Oxygenation targets in ICU patients with COVID-19: A post hoc subgroup analysis of the HOT-ICU trial. Acta Anaesthesiol. Scand. 2022, 66, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Clark, J.M.; Lambertsen, C.J. Pulmonary oxygen toxicity: A review. Pharmacol. Rev. 1971, 23, 37–133. [Google Scholar]
- Smith, J.L. The pathological effects due to increase of oxygen tension in the air breathed. J. Physiol. 1899, 24, 19–35. [Google Scholar] [CrossRef]
- Kallet, R.H.; Matthay, M.A. Hyperoxic acute lung injury. Respir. Care 2013, 58, 123–141. [Google Scholar] [CrossRef]
- Whitehead, G.S.; Burch, L.H.; Berman, K.G.; Piantadosi, C.A.; Schwartz, D.A. Genetic basis of murine responses to hyperoxia-induced lung injury. Immunogenetics 2006, 58, 793–804. [Google Scholar] [CrossRef]
- Bhandari, V.; Elias, J.A. Cytokines in tolerance to hyperoxia-induced injury in the developing and adult lung. Free. Radic. Biol. Med. 2006, 41, 4–18. [Google Scholar] [CrossRef]
- Coalson, J.J.; King, R.J.; Winter, V.T.; Prihoda, T.J.; Anzueto, A.R.; Peters, J.I.; Johanson, W.G., Jr. O2− and pneumonia-induced lung injury. I. Pathological and morphometric studies. J. Appl. Physiol. 1989, 67, 346–356. [Google Scholar] [CrossRef]
- Fracica, P.J.; Caminiti, S.P.; Piantadosi, C.A.; Duhaylongsod, F.G.; Crapo, J.D.; Young, S.L. Natural surfactant and hyperoxic lung injury in primates. II. Morphometric analyses. J. Appl. Physiol. 1994, 76, 1002–1010. [Google Scholar] [CrossRef] [PubMed]
- Comroe, J.H.; Dripps, R.D.; Dumke, P.R.; Deming, M. Oxygen toxicity: The effect of inhalation of high concentrations of oxygen for twenty-four hours on normal men at sea level and at a simulated altitude of 18,000 feet. J. Am. Med. Assoc. 1945, 128, 710–717. [Google Scholar] [CrossRef]
- Caldwell, P.R.; Lee, W.L., Jr.; Schildkraut, H.S.; Archibald, E.R. Changes in lung volume, diffusing capacity, and blood gases in men breathing oxygen. J. Appl. Physiol. 1966, 21, 1477–1483. [Google Scholar] [CrossRef] [PubMed]
- Sackner, M.A.; Landa, J.; Hirsch, J.; Zapata, A. Pulmonary effects of oxygen breathing. A 6-hour study in normal men. Ann. Intern. Med. 1975, 82, 40–43. [Google Scholar] [CrossRef] [PubMed]
- Davis, W.B.; Rennard, S.I.; Bitterman, P.B.; Crystal, R.G. Pulmonary oxygen toxicity. Early reversible changes in human alveolar structures induced by hyperoxia. N. Engl. J. Med. 1983, 309, 878–883. [Google Scholar] [CrossRef] [PubMed]
- Tateda, K.; Deng, J.C.; Moore, T.A.; Newstead, M.W.; Paine, R., 3rd; Kobayashi, N.; Yamaguchi, K.; Standiford, T.J. Hyperoxia mediates acute lung injury and increased lethality in murine Legionella pneumonia: The role of apoptosis. J. Immunol. 2003, 170, 4209–4216. [Google Scholar] [CrossRef]
- Chu, D.K.; Kim, L.H.; Young, P.J.; Zamiri, N.; Almenawer, S.A.; Jaeschke, R.; Szczeklik, W.; Schunemann, H.J.; Neary, J.D.; Alhazzani, W. Mortality and morbidity in acutely ill adults treated with liberal versus conservative oxygen therapy (IOTA): A systematic review and meta-analysis. Lancet 2018, 391, 1693–1705. [Google Scholar] [CrossRef]
- Schjorring, O.L.; Klitgaard, T.L.; Perner, A.; Wetterslev, J.; Lange, T.; Siegemund, M.; Backlund, M.; Keus, F.; Laake, J.H.; Morgan, M.; et al. Lower or Higher Oxygenation Targets for Acute Hypoxemic Respiratory Failure. N. Engl. J. Med. 2021, 384, 1301–1311. [Google Scholar] [CrossRef]
- Barrot, L.; Asfar, P.; Mauny, F.; Winiszewski, H.; Montini, F.; Badie, J.; Quenot, J.P.; Pili-Floury, S.; Bouhemad, B.; Louis, G.; et al. Liberal or Conservative Oxygen Therapy for Acute Respiratory Distress Syndrome. N. Engl. J. Med. 2020, 382, 999–1008. [Google Scholar] [CrossRef]
- Rowe, L.A.; Degtyareva, N.; Doetsch, P.W. DNA damage-induced reactive oxygen species (ROS) stress response in Saccharomyces cerevisiae. Free Radic. Biol. Med. 2008, 45, 1167–1177. [Google Scholar] [CrossRef]
- Khomich, O.A.; Kochetkov, S.N.; Bartosch, B.; Ivanov, A.V. Redox Biology of Respiratory Viral Infections. Viruses 2018, 10, 392. [Google Scholar] [CrossRef] [PubMed]
- Ighodaro, O.M.; Akinloye, O.A. First line defence antioxidants-superoxide dismutase (SOD), catalase (CAT) and glutathione peroxidase (GPX): Their fundamental role in the entire antioxidant defence grid. Alex. J. Med. 2018, 54, 287–293. [Google Scholar] [CrossRef]
- Traber, M.G.; Stevens, J.F. Vitamins C and E: Beneficial effects from a mechanistic perspective. Free Radic. Biol. Med. 2011, 51, 1000–1013. [Google Scholar] [CrossRef] [PubMed]
- Muhammad, Y.; Kani, Y.A.; Iliya, S.; Muhammad, J.B.; Binji, A.; El-Fulaty Ahmad, A.; Kabir, M.B.; Umar Bindawa, K.; Ahmed, A. Deficiency of antioxidants and increased oxidative stress in COVID-19 patients: A cross-sectional comparative study in Jigawa, Northwestern Nigeria. SAGE Open Med. 2021, 9, 2050312121991246. [Google Scholar] [CrossRef]
- Chiscano-Camon, L.; Ruiz-Rodriguez, J.C.; Ruiz-Sanmartin, A.; Roca, O.; Ferrer, R. Vitamin C levels in patients with SARS-CoV-2-associated acute respiratory distress syndrome. Crit. Care 2020, 24, 522. [Google Scholar] [CrossRef]
- Pincemail, J.; Cavalier, E.; Charlier, C.; Cheramy-Bien, J.P.; Brevers, E.; Courtois, A.; Fadeur, M.; Meziane, S.; Goff, C.L.; Misset, B.; et al. Oxidative Stress Status in COVID-19 Patients Hospitalized in Intensive Care Unit for Severe Pneumonia. A Pilot Study. Antioxidants 2021, 10, 257. [Google Scholar] [CrossRef]
- Sena, C.M.; Leandro, A.; Azul, L.; Seica, R.; Perry, G. Vascular Oxidative Stress: Impact and Therapeutic Approaches. Front. Physiol. 2018, 9, 1668. [Google Scholar] [CrossRef]
- Porzionato, A.; Sfriso, M.M.; Mazzatenta, A.; Macchi, V.; De Caro, R.; Di Giulio, C. Effects of hyperoxic exposure on signal transduction pathways in the lung. Respir. Physiol. Neurobiol. 2015, 209, 106–114. [Google Scholar] [CrossRef]
- Walsh, C.M.; Luhrs, K.A.; Arechiga, A.F. The “fuzzy logic” of the death-inducing signaling complex in lymphocytes. J. Clin. Immunol. 2003, 23, 333–353. [Google Scholar] [CrossRef]
- Cho, H.Y.; Jedlicka, A.E.; Reddy, S.P.; Kensler, T.W.; Yamamoto, M.; Zhang, L.Y.; Kleeberger, S.R. Role of NRF2 in protection against hyperoxic lung injury in mice. Am. J. Respir. Cell Mol. Biol. 2002, 26, 175–182. [Google Scholar] [CrossRef]
- Cho, H.Y.; Kleeberger, S.R. Nrf2 protects against airway disorders. Toxicol. Appl. Pharmacol. 2010, 244, 43–56. [Google Scholar] [CrossRef] [PubMed]
- Dushianthan, A.; Goss, V.; Cusack, R.; Grocott, M.P.; Postle, A.D. Phospholipid composition and kinetics in different endobronchial fractions from healthy volunteers. BMC Pulm. Med. 2014, 14, 10. [Google Scholar] [CrossRef] [PubMed]
- Dushianthan, A.; Goss, V.; Cusack, R.; Grocott, M.P.; Postle, A.D. Altered molecular specificity of surfactant phosphatidycholine synthesis in patients with acute respiratory distress syndrome. Respir. Res. 2014, 15, 128. [Google Scholar] [CrossRef] [PubMed]
- Andersson, S.; Kheiter, A.; Merritt, T.A. Oxidative inactivation of surfactants. Lung 1999, 177, 179–189. [Google Scholar] [CrossRef]
- Rodriguez-Capote, K.; Manzanares, D.; Haines, T.; Possmayer, F. Reactive oxygen species inactivation of surfactant involves structural and functional alterations to surfactant proteins SP-B and SP-C. Biophys. J. 2006, 90, 2808–2821. [Google Scholar] [CrossRef]
- Tolle, A.; Kolleck, I.; Schlame, M.; Wauer, R.; Stevens, P.A.; Rustow, B. Effect of hyperoxia on the composition of the alveolar surfactant and the turnover of surfactant phospholipids, cholesterol, plasmalogens and vitamin E. Biochim. Biophys. Acta 1997, 1346, 198–204. [Google Scholar] [CrossRef]
- Pace, P.W.; Yao, L.J.; Wilson, J.X.; Possmayer, F.; Veldhuizen, R.A.; Lewis, J.F. The effects of hyperoxia exposure on lung function and pulmonary surfactant in a rat model of acute lung injury. Exp. Lung Res. 2009, 35, 380–398. [Google Scholar] [CrossRef]
- Putman, E.; van Golde, L.M.; Haagsman, H.P. Toxic oxidant species and their impact on the pulmonary surfactant system. Lung 1997, 175, 75–103. [Google Scholar] [CrossRef]
- Calkovska, A.; Kolomaznik, M.; Calkovsky, V. Alveolar type II cells and pulmonary surfactant in COVID-19 era. Physiol. Res. 2021, 70 (Suppl. S2), S195–S208. [Google Scholar] [CrossRef]
- Kennedy, K.A.; Snyder, J.M.; Stenzel, W.; Saito, K.; Warshaw, J.B. Vitamin E alters alveolar type II cell phospholipid synthesis in oxygen and air. Exp. Lung Res. 1990, 16, 607–615. [Google Scholar] [CrossRef]
- Polak, M.J.; Knight, M.E.; Andresen, T.L.; DeSena, C. Effects of hyperoxia and beta-adrenergic stimulation on pulmonary surfactant in neonatal rabbits. Exp. Lung Res. 1992, 18, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Peng, L.Q.; Zhao, A.L. Hyperoxia induces the apoptosis of alveolar epithelial cells and changes of pulmonary surfactant proteins. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 492–497. [Google Scholar] [PubMed]
- Smallwood, C.D.; Boloori-Zadeh, P.; Silva, M.R.; Gouldstone, A. High Oxygen Concentrations Adversely Affect the Performance of Pulmonary Surfactant. Respir. Care 2017, 62, 1085–1090. [Google Scholar] [CrossRef] [PubMed]
- Meng, H.; Sun, Y.; Lu, J.; Fu, S.; Meng, Z.; Scott, M.; Li, Q. Exogenous surfactant may improve oxygenation but not mortality in adult patients with acute lung injury/acute respiratory distress syndrome: A meta-analysis of 9 clinical trials. J. Cardiothorac. Vasc. Anesth. 2012, 26, 849–856. [Google Scholar] [CrossRef]
- Postle, A.D.; Clark, H.W.; Fink, J.; Madsen, J.; Koster, G.; Panchal, M.; Djukanovic, R.; Brealey, D.; Grocott, M.P.W.; Dushianthan, A. Rapid Phospholipid Turnover after Surfactant Nebulization in Severe COVID-19 Infection: A Randomized Clinical Trial. Am. J. Respir. Crit. Care Med. 2022, 205, 471–473. [Google Scholar] [CrossRef]
- Engstrom, P.C.; Holm, B.A.; Matalon, S. Surfactant replacement attenuates the increase in alveolar permeability in hyperoxia. J. Appl. Physiol. 1989, 67, 688–693. [Google Scholar] [CrossRef]
- Matalon, S.; Holm, B.A.; Notter, R.H. Mitigation of pulmonary hyperoxic injury by administration of exogenous surfactant. J. Appl. Physiol. 1987, 62, 756–761. [Google Scholar] [CrossRef]
- Bezerra, F.S.; Ramos, C.O.; Castro, T.F.; Araujo, N.; de Souza, A.B.F.; Bandeira, A.C.B.; Costa, G.P.; Cartelle, C.T.; Talvani, A.; Cangussu, S.D.; et al. Exogenous surfactant prevents hyperoxia-induced lung injury in adult mice. Intensive Care Med. Exp. 2019, 7, 19. [Google Scholar] [CrossRef]
- Dani, C.; Corsini, I.; Longini, M.; Burchielli, S.; Dichiara, G.; Cantile, C.; Buonocore, G. Natural surfactant combined with superoxide dismutase and catalase decreases oxidative lung injury in the preterm lamb. Pediatr. Pulmonol. 2014, 49, 898–904. [Google Scholar] [CrossRef]
- Ghio, A.J.; Fracica, P.J.; Young, S.L.; Piantadosi, C.A. Synthetic surfactant scavenges oxidants and protects against hyperoxic lung injury. J. Appl. Physiol. 1994, 77, 1217–1223. [Google Scholar] [CrossRef]
- Srivastava, S.; Garg, I.; Bansal, A.; Kumar, B. SARS-CoV-2 infection: Physiological and environmental gift factors at high altitude. Virusdisease 2020, 31, 450–452. [Google Scholar] [CrossRef] [PubMed]
- Pun, M.; Turner, R.; Strapazzon, G.; Brugger, H.; Swenson, E.R. Lower Incidence of COVID-19 at High Altitude: Facts and Confounders. High Alt. Med. Biol. 2020, 21, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Zhu, L.; Cai, J.; Lei, F.; Qin, J.J.; Xie, J.; Liu, Y.M.; Zhao, Y.C.; Huang, X.; Lin, L.; et al. Association of Inpatient Use of Angiotensin-Converting Enzyme Inhibitors and Angiotensin II Receptor Blockers with Mortality Among Patients with Hypertension Hospitalized With COVID-19. Circ. Res. 2020, 126, 1671–1681. [Google Scholar] [CrossRef] [PubMed]
- Jia, H.P.; Look, D.C.; Shi, L.; Hickey, M.; Pewe, L.; Netland, J.; Farzan, M.; Wohlford-Lenane, C.; Perlman, S.; McCray, P.B., Jr. ACE2 receptor expression and severe acute respiratory syndrome coronavirus infection depend on differentiation of human airway epithelia. J. Virol. 2005, 79, 14614–14621. [Google Scholar] [CrossRef]
- Jia, H.P.; Look, D.C.; Tan, P.; Shi, L.; Hickey, M.; Gakhar, L.; Chappell, M.C.; Wohlford-Lenane, C.; McCray, P.B., Jr. Ectodomain shedding of angiotensin converting enzyme 2 in human airway epithelia. Am. J. Physiol. Lung Cell Mol. Physiol. 2009, 297, L84–L96. [Google Scholar] [CrossRef]
- Li, X.; Molina-Molina, M.; Abdul-Hafez, A.; Uhal, V.; Xaubet, A.; Uhal, B.D. Angiotensin converting enzyme-2 is protective but downregulated in human and experimental lung fibrosis. Am. J. Physiol. Lung Cell Mol. Physiol. 2008, 295, L178–L185. [Google Scholar] [CrossRef]
- Imai, Y.; Kuba, K.; Rao, S.; Huan, Y.; Guo, F.; Guan, B.; Yang, P.; Sarao, R.; Wada, T.; Leong-Poi, H.; et al. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature 2005, 436, 112–116. [Google Scholar] [CrossRef]
- Fang, Y.; Gao, F.; Liu, Z. Angiotensin-converting enzyme 2 attenuates inflammatory response and oxidative stress in hyperoxic lung injury by regulating NF-kappaB and Nrf2 pathways. QJM 2019, 112, 914–924. [Google Scholar] [CrossRef]
- Oarhe, C.I.; Dang, V.; Dang, M.; Nguyen, H.; Gopallawa, I.; Gewolb, I.H.; Uhal, B.D. Hyperoxia downregulates angiotensin-converting enzyme-2 in human fetal lung fibroblasts. Pediatr. Res. 2015, 77, 656–662. [Google Scholar] [CrossRef]
- Writing Committee for the REMAP-CAP Investigators; Lawler, P.R.; Derde, L.P.G.; van de Veerdonk, F.L.; McVerry, B.J.; Huang, D.T.; Berry, L.R.; Lorenzi, E.; van Kimmenade, R.; Gommans, F.; et al. Effect of Angiotensin-Converting Enzyme Inhibitor and Angiotensin Receptor Blocker Initiation on Organ Support-Free Days in Patients Hospitalized With COVID-19: A Randomized Clinical Trial. JAMA 2023, 329, 1183–1196. [Google Scholar]
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.S.; Mehra, M.R.; Schuepbach, R.A.; Ruschitzka, F.; Moch, H. Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020, 395, 1417–1418. [Google Scholar] [CrossRef] [PubMed]
- Six, S.; Jaffal, K.; Ledoux, G.; Jaillette, E.; Wallet, F.; Nseir, S. Hyperoxemia as a risk factor for ventilator-associated pneumonia. Crit. Care 2016, 20, 195. [Google Scholar] [CrossRef] [PubMed]
- Maes, M.; Higginson, E.; Pereira-Dias, J.; Curran, M.D.; Parmar, S.; Khokhar, F.; Cuchet-Lourenco, D.; Lux, J.; Sharma-Hajela, S.; Ravenhill, B.; et al. Ventilator-associated pneumonia in critically ill patients with COVID-19. Crit. Care 2021, 25, 25. [Google Scholar] [CrossRef] [PubMed]
- Rawson, T.M.; Moore, L.S.P.; Zhu, N.; Ranganathan, N.; Skolimowska, K.; Gilchrist, M.; Satta, G.; Cooke, G.; Holmes, A. Bacterial and Fungal Coinfection in Individuals with Coronavirus: A Rapid Review to Support COVID-19 Antimicrobial Prescribing. Clin. Infect. Dis. 2020, 71, 2459–2468. [Google Scholar] [CrossRef]
- O’Reilly, P.J.; Hickman-Davis, J.M.; Davis, I.C.; Matalon, S. Hyperoxia impairs antibacterial function of macrophages through effects on actin. Am. J. Respir. Cell Mol. Biol. 2003, 28, 443–450. [Google Scholar] [CrossRef]
- Gauthier, A.G.; Wu, J.; Lin, M.; Sitapara, R.; Kulkarni, A.; Thakur, G.A.; Schmidt, E.E.; Perron, J.C.; Ashby, C.R., Jr.; Mantell, L.L. The Positive Allosteric Modulation of alpha7-Nicotinic Cholinergic Receptors by GAT107 Increases Bacterial Lung Clearance in Hyperoxic Mice by Decreasing Oxidative Stress in Macrophages. Antioxidants 2021, 10, 135. [Google Scholar] [CrossRef]
- Baleeiro, C.E.; Wilcoxen, S.E.; Morris, S.B.; Standiford, T.J.; Paine, R., 3rd. Sublethal hyperoxia impairs pulmonary innate immunity. J. Immunol. 2003, 171, 955–963. [Google Scholar] [CrossRef]
- Ashley, S.L.; Sjoding, M.W.; Popova, A.P.; Cui, T.X.; Hoostal, M.J.; Schmidt, T.M.; Branton, W.R.; Dieterle, M.G.; Falkowski, N.R.; Baker, J.M.; et al. Lung and gut microbiota are altered by hyperoxia and contribute to oxygen-induced lung injury in mice. Sci. Transl. Med. 2020, 12, eaau9959. [Google Scholar] [CrossRef]
- Xing, Z.; Li, Y.; Liu, G.; He, Y.; Tao, Y.; Chen, M. Hyperoxia provokes gut dysbiosis in rats. Crit. Care 2020, 24, 517. [Google Scholar] [CrossRef]
- Santamarina, M.G.; Boisier, D.; Contreras, R.; Baque, M.; Volpacchio, M.; Beddings, I. COVID-19: A hypothesis regarding the ventilation-perfusion mismatch. Crit. Care 2020, 24, 395. [Google Scholar] [CrossRef]
- Feener, E.P.; Northrup, J.M.; Aiello, L.P.; King, G.L. Angiotensin II induces plasminogen activator inhibitor-1 and -2 expression in vascular endothelial and smooth muscle cells. J. Clin. Invest. 1995, 95, 1353–1362. [Google Scholar] [CrossRef]
- Senchenkova, E.Y.; Russell, J.; Kurmaeva, E.; Ostanin, D.; Granger, D.N. Role of T lymphocytes in angiotensin II-mediated microvascular thrombosis. Hypertension 2011, 58, 959–965. [Google Scholar] [CrossRef] [PubMed]
- Nguyen Dinh Cat, A.; Montezano, A.C.; Burger, D.; Touyz, R.M. Angiotensin II, NADPH oxidase, and redox signaling in the vasculature. Antioxid. Redox Signal 2013, 19, 1110–1120. [Google Scholar] [CrossRef] [PubMed]
- Namsolleck, P.; Moll, G.N. Does activation of the protective Renin-Angiotensin System have therapeutic potential in COVID-19? Mol. Med. 2020, 26, 80. [Google Scholar] [CrossRef] [PubMed]
- Heurich, A.; Hofmann-Winkler, H.; Gierer, S.; Liepold, T.; Jahn, O.; Pohlmann, S. TMPRSS2 and ADAM17 cleave ACE2 differentially and only proteolysis by TMPRSS2 augments entry driven by the severe acute respiratory syndrome coronavirus spike protein. J. Virol. 2014, 88, 1293–1307. [Google Scholar] [CrossRef] [PubMed]
- Ramos, S.G.; Rattis, B.; Ottaviani, G.; Celes, M.R.N.; Dias, E.P. ACE2 Down-Regulation May Act as a Transient Molecular Disease Causing RAAS Dysregulation and Tissue Damage in the Microcirculatory Environment Among COVID-19 Patients. Am. J. Pathol. 2021, 191, 1154–1164. [Google Scholar] [CrossRef]
- Le Berre, A.; Boeken, T.; Caramella, C.; Afonso, D.; Nhy, C.; Saccenti, L.; Tardivel, A.M.; Gerber, S.; Frison Roche, A.; Emmerich, J.; et al. Dual-energy CT angiography reveals high prevalence of perfusion defects unrelated to pulmonary embolism in COVID-19 lesions. Insights Imaging 2021, 12, 24. [Google Scholar] [CrossRef]
- Lazzaroni, M.G.; Piantoni, S.; Masneri, S.; Garrafa, E.; Martini, G.; Tincani, A.; Andreoli, L.; Franceschini, F. Coagulation dysfunction in COVID-19: The interplay between inflammation, viral infection and the coagulation system. Blood Rev. 2021, 46, 100745. [Google Scholar] [CrossRef]
- Varikasuvu, S.R.; Varshney, S.; Dutt, N.; Munikumar, M.; Asfahan, S.; Kulkarni, P.P.; Gupta, P. D-dimer, disease severity, and deaths (3D-study) in patients with COVID-19: A systematic review and meta-analysis of 100 studies. Sci. Rep. 2021, 11, 21888. [Google Scholar] [CrossRef]
- Zhao, R.; Su, Z.; Komissarov, A.A.; Liu, S.L.; Yi, G.; Idell, S.; Matthay, M.A.; Ji, H.L. Associations of D-Dimer on Admission and Clinical Features of COVID-19 Patients: A Systematic Review, Meta-Analysis, and Meta-Regression. Front. Immunol. 2021, 12, 691249. [Google Scholar] [CrossRef]
- Pranata, R.; Lim, M.A.; Yonas, E.; Huang, I.; Nasution, S.A.; Setiati, S.; Alwi, I.; Kuswardhani, R.A.T. Thrombocytopenia as a prognostic marker in COVID-19 patients: Diagnostic test accuracy meta-analysis. Epidemiol. Infect. 2021, 149, e40. [Google Scholar] [CrossRef] [PubMed]
- Mancini, I.; Baronciani, L.; Artoni, A.; Colpani, P.; Biganzoli, M.; Cozzi, G.; Novembrino, C.; Boscolo Anzoletti, M.; De Zan, V.; Pagliari, M.T.; et al. The ADAMTS13-von Willebrand factor axis in COVID-19 patients. J. Thromb. Haemost. 2021, 19, 513–521. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, J.M.; Barouqa, M.; Krause, G.J.; Gonzalez-Lugo, J.D.; Rahman, S.; Gil, M.R. Low ADAMTS13 Activity Correlates with Increased Mortality in COVID-19 Patients. Am. J. Clin. Pathol. 2021, 5, e89–e103. [Google Scholar]
- Kanoore Edul, V.S.; Caminos Eguillor, J.F.; Ferrara, G.; Estenssoro, E.; Siles, D.S.P.; Cesio, C.E.; Dubin, A. Microcirculation alterations in severe COVID-19 pneumonia. J. Crit. Care 2021, 61, 73–75. [Google Scholar] [CrossRef]
- Magro, C.M.; Mulvey, J.; Kubiak, J.; Mikhail, S.; Suster, D.; Crowson, A.N.; Laurence, J.; Nuovo, G. Severe COVID-19: A multifaceted viral vasculopathy syndrome. Ann. Diagn. Pathol. 2021, 50, 151645. [Google Scholar] [CrossRef]
- REMAP-CAP Investigators; ACTIV-4a Investigators; ATTACC Investigators; Goligher, E.C.; Bradbury, C.A.; McVerry, B.J.; Lawler, P.R.; Berger, J.S.; Gong, M.N.; Carrier, M.; et al. Therapeutic Anticoagulation with Heparin in Critically Ill Patients with COVID-19. N. Engl. J. Med. 2021, 385, 777–789. [Google Scholar]
- Bradbury, C.A.; Lawler, P.R.; Stanworth, S.J.; McVerry, B.J.; McQuilten, Z.; Higgins, A.M.; Mouncey, P.R.; Al-Beidh, F.; Rowan, K.M.; Berry, L.R.; et al. Effect of Antiplatelet Therapy on Survival and Organ Support-Free Days in Critically Ill Patients With COVID-19: A Randomized Clinical Trial. JAMA 2022, 327, 1247–1259. [Google Scholar]
- Turecek, P.L.; Peck, R.C.; Rangarajan, S.; Reilly-Stitt, C.; Laffan, M.A.; Kazmi, R.; James, I.; Dushianthan, A.; Schrenk, G.; Gritsch, H.; et al. Recombinant ADAMTS13 reduces abnormally up-regulated von Willebrand factor in plasma from patients with severe COVID-19. Thromb. Res. 2021, 201, 100–112. [Google Scholar] [CrossRef]
- Abdel-Bakky, M.S.; Amin, E.; Ewees, M.G.; Mahmoud, N.I.; Mohammed, H.A.; Altowayan, W.M.; Abdellatif, A.A.H. Coagulation System Activation for Targeting of COVID-19: Insights into Anticoagulants, Vaccine-Loaded Nanoparticles, and Hypercoagulability in COVID-19 Vaccines. Viruses 2022, 14, 228. [Google Scholar] [CrossRef]
- Waring, W.S.; Thomson, A.J.; Adwani, S.H.; Rosseel, A.J.; Potter, J.F.; Webb, D.J.; Maxwell, S.R. Cardiovascular effects of acute oxygen administration in healthy adults. J. Cardiovasc. Pharmacol. 2003, 42, 245–250. [Google Scholar] [CrossRef]
- Griffith, D.E.; Garcia, J.G.; James, H.L.; Callahan, K.S.; Iriana, S.; Holiday, D. Hyperoxic exposure in humans. Effects of 50 percent oxygen on alveolar macrophage leukotriene B4 synthesis. Chest 1992, 101, 392–397. [Google Scholar] [CrossRef] [PubMed]
- Eastwood, G.; Bellomo, R.; Bailey, M.; Taori, G.; Pilcher, D.; Young, P.; Beasley, R. Arterial oxygen tension and mortality in mechanically ventilated patients. Intensive Care Med. 2012, 38, 91–98. [Google Scholar] [CrossRef] [PubMed]
- de Jonge, E.; Peelen, L.; Keijzers, P.J.; Joore, H.; de Lange, D.; van der Voort, P.H.; Bosman, R.J.; de Waal, R.A.; Wesselink, R.; de Keizer, N.F. Association between administered oxygen, arterial partial oxygen pressure and mortality in mechanically ventilated intensive care unit patients. Crit. Care 2008, 12, R156. [Google Scholar] [CrossRef] [PubMed]
- Velmahos, G.C.; Demetriades, D.; Shoemaker, W.C.; Chan, L.S.; Tatevossian, R.; Wo, C.C.; Vassiliu, P.; Cornwell, E.E., 3rd; Murray, J.A.; Roth, B.; et al. Endpoints of resuscitation of critically injured patients: Normal or supranormal? A prospective randomized trial. Ann. Surg. 2000, 232, 409–418. [Google Scholar] [CrossRef] [PubMed]
- Ni, Y.N.; Wang, Y.M.; Liang, B.M.; Liang, Z.A. The effect of hyperoxia on mortality in critically ill patients: A systematic review and meta analysis. BMC Pulm. Med. 2019, 19, 53. [Google Scholar] [CrossRef]
- Martin, D.S.; Grocott, M.P. Oxygen therapy in critical illness: Precise control of arterial oxygenation and permissive hypoxemia. Crit. Care Med. 2013, 41, 423–432. [Google Scholar] [CrossRef]
- Abdelsalam, M.; Cheifetz, I.M. Goal-directed therapy for severely hypoxic patients with acute respiratory distress syndrome: Permissive hypoxemia. Respir. Care 2010, 55, 1483–1490. [Google Scholar]
- Grocott, M.P.; Martin, D.S.; Levett, D.Z.; McMorrow, R.; Windsor, J.; Montgomery, H.E.; Caudwell Xtreme Everest Research Group. Arterial blood gases and oxygen content in climbers on Mount Everest. N. Engl. J. Med. 2009, 360, 140–149. [Google Scholar] [CrossRef]
- Semler, M.W.; Casey, J.D.; Lloyd, B.D.; Hastings, P.G.; Hays, M.A.; Stollings, J.L.; Buell, K.G.; Brems, J.H.; Qian, E.T.; Seitz, K.P.; et al. PILOT Investigators and the Pragmatic Critical Care Research Group Oxygen-Saturation Targets for Critically Ill Adults Receiving Mechanical Ventilation. N. Engl. J. Med. 2022, 387, 1759–1769. [Google Scholar] [CrossRef]
- He, H.W.; Liu, D.W. Permissive hypoxemia/conservative oxygenation strategy: Dr. Jekyll or Mr. Hyde? J. Thorac. Dis. 2016, 8, 748–750. [Google Scholar] [CrossRef]
- Nowroozpoor, A.; Malekmohammad, M.; Seyyedi, S.R.; Hashemian, S.M. Pulmonary Hypertension in Intensive Care Units: An Updated Review. Tanaffos 2019, 18, 180–207. [Google Scholar] [PubMed]
- Gilbert-Kawai, E.T.; Mitchell, K.; Martin, D.; Carlisle, J.; Grocott, M.P. Permissive hypoxaemia versus normoxaemia for mechanically ventilated critically ill patients. Cochrane Database Syst. Rev. 2014, 2014, CD009931. [Google Scholar] [CrossRef] [PubMed]
- Nasa, P.; Azoulay, E.; Khanna, A.K.; Jain, R.; Gupta, S.; Javeri, Y.; Juneja, D.; Rangappa, P.; Sundararajan, K.; Alhazzani, W.; et al. Expert consensus statements for the management of COVID-19-related acute respiratory failure using a Delphi method. Crit. Care 2021, 25, 106. [Google Scholar] [CrossRef] [PubMed]
- Zhong, X.; Wu, Q.; Yang, H.; Dong, W.; Wang, B.; Zhang, Z.; Liang, G. Airway pressure release ventilation versus low tidal volume ventilation for patients with acute respiratory distress syndrome/acute lung injury: A meta-analysis of randomized clinical trials. Ann. Transl. Med. 2020, 8, 1641. [Google Scholar] [CrossRef]
- Mehta, C.; Mehta, Y. Management of refractory hypoxemia. Ann. Card. Anaesth. 2016, 19, 89–96. [Google Scholar] [CrossRef]
- Moreno Franco, P.; Enders, F.; Wilson, G.; Gajic, O.; Pannu, S.R. A Comparative Effectiveness Study of Rescue Strategies in 1,000 Subjects with Severe Hypoxemic Respiratory Failure. Respir. Care 2016, 61, 127–133. [Google Scholar] [CrossRef]
- Fuller, B.M.; Mohr, N.M.; Skrupky, L.; Fowler, S.; Kollef, M.H.; Carpenter, C.R. The use of inhaled prostaglandins in patients with ARDS: A systematic review and meta-analysis. Chest 2015, 147, 1510–1522. [Google Scholar] [CrossRef]
- Garfield, B.; McFadyen, C.; Briar, C.; Bleakley, C.; Vlachou, A.; Baldwin, M.; Lees, N.; Price, S.; Ledot, S.; McCabe, C.; et al. Potential for personalised application of inhaled nitric oxide in COVID-19 pneumonia. Br. J. Anaesth. 2021, 126, e72–e75. [Google Scholar] [CrossRef]
- Sonti, R.; Pike, C.W.; Cobb, N. Responsiveness of Inhaled Epoprostenol in Respiratory Failure due to COVID-19. J. Intensive Care Med. 2021, 36, 327–333. [Google Scholar] [CrossRef]
- Matthews, L.; Baker, L.; Ferrari, M.; Sanchez, W.; Pappachan, J.; Grocott, M.P.; Dushianthan, A.; REACT COVID-19 investigators. Compassionate use of Pulmonary Vasodilators in Acute Severe Hypoxic Respiratory Failure due to COVID-19. J. Intensive Care Med. 2022, 37, 1101–1111. [Google Scholar] [CrossRef]
- Akerstrom, S.; Mousavi-Jazi, M.; Klingstrom, J.; Leijon, M.; Lundkvist, A.; Mirazimi, A. Nitric oxide inhibits the replication cycle of severe acute respiratory syndrome coronavirus. J. Virol. 2005, 79, 1966–1969. [Google Scholar] [CrossRef] [PubMed]
- Riva, C.M.; Morganroth, M.L.; Ljungman, A.G.; Schoeneich, S.O.; Marks, R.M.; Todd, R.F., 3rd; Ward, P.A.; Boxer, L.A. Iloprost inhibits neutrophil-induced lung injury and neutrophil adherence to endothelial monolayers. Am. J. Respir. Cell Mol. Biol. 1990, 3, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Burghuber, O.C.; Silberbauer, K.; Haber, P.; Sinzinger, H.; Elliott, M.; Leithner, C. Pulmonary and antiaggregatory effects of prostacyclin after inhalation and intravenous infusion. Respiration 1984, 45, 450–454. [Google Scholar] [CrossRef]
- Park, S.Y.; Kim, H.J.; Yoo, K.H.; Park, Y.B.; Kim, S.W.; Lee, S.J.; Kim, E.K.; Kim, J.H.; Kim, Y.H.; Moon, J.Y.; et al. The efficacy and safety of prone positioning in adults patients with acute respiratory distress syndrome: A meta-analysis of randomized controlled trials. J. Thorac. Dis. 2015, 7, 356–367. [Google Scholar]
- Chua, E.X.; Zahir, S.; Ng, K.T.; Teoh, W.Y.; Hasan, M.S.; Ruslan, S.R.B.; Abosamak, M.F. Effect of prone versus supine position in COVID-19 patients: A systematic review and meta-analysis. J. Clin. Anesth. 2021, 74, 110406. [Google Scholar] [CrossRef]
- Guerin, C.; Reignier, J.; Richard, J.C.; Beuret, P.; Gacouin, A.; Boulain, T.; Mercier, E.; Badet, M.; Mercat, A.; Baudin, O.; et al. Prone positioning in severe acute respiratory distress syndrome. N. Engl. J. Med. 2013, 368, 2159–2168. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Luo, J.; Pavlov, I.; Perez, Y.; Tan, W.; Roca, O.; Tavernier, E.; Kharat, A.; McNicholas, B.; Ibarra-Estrada, M.; et al. Awake prone positioning for non-intubated patients with COVID-19-related acute hypoxaemic respiratory failure: A systematic review and meta-analysis. Lancet Respir. Med. 2022, 10, 573–583. [Google Scholar] [CrossRef]
- Peek, G.J.; Mugford, M.; Tiruvoipati, R.; Wilson, A.; Allen, E.; Thalanany, M.M.; Hibbert, C.L.; Truesdale, A.; Clemens, F.; Cooper, N.; et al. Efficacy and economic assessment of conventional ventilatory support versus extracorporeal membrane oxygenation for severe adult respiratory failure (CESAR): A multicentre randomised controlled trial. Lancet 2009, 374, 1351–1363. [Google Scholar] [CrossRef]
- Noah, M.A.; Peek, G.J.; Finney, S.J.; Griffiths, M.J.; Harrison, D.A.; Grieve, R.; Sadique, M.Z.; Sekhon, J.S.; McAuley, D.F.; Firmin, R.K.; et al. Referral to an extracorporeal membrane oxygenation center and mortality among patients with severe 2009 influenza A(H1N1). JAMA 2011, 306, 1659–1668. [Google Scholar] [CrossRef]
- Camporota, L.; Meadows, C.; Ledot, S.; Scott, I.; Harvey, C.; Garcia, M.; Vuylsteke, A. Consensus on the referral and admission of patients with severe respiratory failure to the NHS ECMO service. Lancet Respir. Med. 2021, 9, e16–e17. [Google Scholar] [CrossRef]
- Investigators, I.; Sadeghipour, P.; Talasaz, A.H.; Rashidi, F.; Sharif-Kashani, B.; Beigmohammadi, M.T.; Farrokhpour, M.; Sezavar, S.H.; Payandemehr, P.; Dabbagh, A.; et al. Effect of Intermediate-Dose vs Standard-Dose Prophylactic Anticoagulation on Thrombotic Events, Extracorporeal Membrane Oxygenation Treatment, or Mortality Among Patients With COVID-19 Admitted to the Intensive Care Unit: The INSPIRATION Randomized Clinical Trial. JAMA 2021, 325, 1620–1630. [Google Scholar]
- Lopes, R.D.; de Barros, E.S.P.G.M.; Furtado, R.H.M.; Macedo, A.V.S.; Bronhara, B.; Damiani, L.P.; Barbosa, L.M.; de Aveiro Morata, J.; Ramacciotti, E.; de Aquino Martins, P.; et al. Therapeutic versus prophylactic anticoagulation for patients admitted to hospital with COVID-19 and elevated D-dimer concentration (ACTION): An open-label, multicentre, randomised, controlled trial. Lancet 2021, 397, 2253–2263. [Google Scholar] [CrossRef] [PubMed]
- NICE. COVID-19 Rapid Guideline: Managing COVID-19; National Institute for Health and Care Excellence: London, UK, 2022; Available online: https://www.nice.org.uk/guidance/ng191/resources/covid19-rapid-guideline-managing-covid19-pdf-51035553326 (accessed on 24 September 2023).
- Scully, M.; Cataland, S.R.; Peyvandi, F.; Coppo, P.; Knobl, P.; Kremer Hovinga, J.A.; Metjian, A.; de la Rubia, J.; Pavenski, K.; Callewaert, F.; et al. Caplacizumab Treatment for Acquired Thrombotic Thrombocytopenic Purpura. N. Engl. J. Med. 2019, 380, 335–346. [Google Scholar] [CrossRef] [PubMed]
- Yamaoka, S.; Kim, H.S.; Ogihara, T.; Oue, S.; Takitani, K.; Yoshida, Y.; Tamai, H. Severe Vitamin E deficiency exacerbates acute hyperoxic lung injury associated with increased oxidative stress and inflammation. Free Radic. Res. 2008, 42, 602–612. [Google Scholar] [CrossRef] [PubMed]
- Jyonouchi, H.; Sun, S.; Abiru, T.; Chareancholvanich, S.; Ingbar, D.H. The effects of hyperoxic injury and antioxidant vitamins on death and proliferation of human small airway epithelial cells. Am. J. Respir. Cell Mol. Biol. 1998, 19, 426–436. [Google Scholar] [CrossRef]
- Al-Shmgani, H.S.; Moate, R.M.; Sneyd, J.R.; Macnaughton, P.D.; Moody, A.J. Hyperoxia-induced ciliary loss and oxidative damage in an in vitro bovine model: The protective role of antioxidant vitamins E and C. Biochem. Biophys. Res. Commun. 2012, 429, 191–196. [Google Scholar] [CrossRef]
- Qiao, J.; Chen, L.; Huang, X.; Guo, F. Effects of nebulized N-acetylcystein on the expression of HMGB1 and RAGE in rats with hyperoxia-induced lung injury. J. Cell Physiol. 2019, 234, 10547–10553. [Google Scholar] [CrossRef]
- Langley, S.C.; Kelly, F.J. N-acetylcysteine ameliorates hyperoxic lung injury in the preterm guinea pig. Biochem. Pharmacol. 1993, 45, 841–846. [Google Scholar] [CrossRef]
- Kinniry, P.; Pick, J.; Stephens, S.; Jain, D.; Solomides, C.C.; Niven, R.; Segal, R.; Christofidou-Solomidou, M. KL4-surfactant prevents hyperoxic and LPS-induced lung injury in mice. Pediatr. Pulmonol. 2006, 41, 916–928. [Google Scholar] [CrossRef]
- Jain, D.; Atochina-Vasserman, E.N.; Tomer, Y.; Kadire, H.; Beers, M.F. Surfactant protein D protects against acute hyperoxic lung injury. Am. J. Respir. Crit. Care Med. 2008, 178, 805–813. [Google Scholar] [CrossRef]
- Kolliputi, N.; Shaik, R.S.; Waxman, A.B. The inflammasome mediates hyperoxia-induced alveolar cell permeability. J. Immunol. 2010, 184, 5819–5826. [Google Scholar] [CrossRef] [PubMed]
- Deng, C.H.; Li, T.Q.; Zhang, W.; Zhao, Q.; Wang, Y. Targeting Inflammasome Activation in Viral Infection: A Therapeutic Solution? Viruses 2023, 15, 1451. [Google Scholar] [CrossRef]
- Wagner, C.; Griesel, M.; Mikolajewska, A.; Mueller, A.; Nothacker, M.; Kley, K.; Metzendorf, M.I.; Fischer, A.L.; Kopp, M.; Stegemann, M.; et al. Systemic corticosteroids for the treatment of COVID-19. Cochrane Database Syst. Rev. 2021, 8, CD014963. [Google Scholar]
- Group, R.C.; Horby, P.; Lim, W.S.; Emberson, J.R.; Mafham, M.; Bell, J.L.; Linsell, L.; Staplin, N.; Brightling, C.; Ustianowski, A.; et al. Dexamethasone in Hospitalized Patients with Covid-19. N. Engl. J. Med. 2021, 384, 693–704. [Google Scholar]
- Angus, D.C.; Derde, L.; Al-Beidh, F.; Annane, D.; Arabi, Y.; Beane, A.; van Bentum-Puijk, W.; Berry, L.; Bhimani, Z.; Bonten, M.; et al. Effect of Hydrocortisone on Mortality and Organ Support in Patients With Severe COVID-19: The REMAP-CAP COVID-19 Corticosteroid Domain Randomized Clinical Trial. JAMA 2020, 324, 1317–1329. [Google Scholar] [PubMed]
- Montazersaheb, S.; Hosseiniyan Khatibi, S.M.; Hejazi, M.S.; Tarhriz, V.; Farjami, A.; Ghasemian Sorbeni, F.; Farahzadi, R.; Ghasemnejad, T. COVID-19 infection: An overview on cytokine storm and related interventions. Virol. J. 2022, 19, 92. [Google Scholar] [CrossRef] [PubMed]
- Leisman, D.E.; Ronner, L.; Pinotti, R.; Taylor, M.D.; Sinha, P.; Calfee, C.S.; Hirayama, A.V.; Mastroiani, F.; Turtle, C.J.; Harhay, M.O.; et al. Cytokine elevation in severe and critical COVID-19: A rapid systematic review, meta-analysis, and comparison with other inflammatory syndromes. Lancet Respir. Med. 2020, 8, 1233–1244. [Google Scholar] [CrossRef]
- Finney, L.J.; Glanville, N.; Farne, H.; Aniscenko, J.; Fenwick, P.; Kemp, S.V.; Trujillo-Torralbo, M.B.; Loo, S.L.; Calderazzo, M.A.; Wedzicha, J.A.; et al. Inhaled corticosteroids downregulate the SARS-CoV-2 receptor ACE2 in COPD through suppression of type I interferon. J. Allergy Clin. Immunol. 2021, 147, 510–519.e5. [Google Scholar] [CrossRef]
- van Zaane, B.; Nur, E.; Squizzato, A.; Gerdes, V.E.; Buller, H.R.; Dekkers, O.M.; Brandjes, D.P. Systematic review on the effect of glucocorticoid use on procoagulant, anti-coagulant and fibrinolytic factors. J. Thromb. Haemost. 2010, 8, 2483–2493. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Angiotensin II | Angiotensin 1-7 |
|---|---|
| Vasoconstrictor | Vasodilator |
| Potent stimulator of NADPH oxidase | Suppress the NADPH oxidase activity |
| Increase reactive oxygen and nitrogen species | Reduce reactive oxygen and nitrogen species |
| Increase oxidative stress | Reduce oxidative stress |
| Proinflammation | Anti-inflammation |
| Profibrotic | Anti-fibrotic |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dushianthan, A.; Bracegirdle, L.; Cusack, R.; Cumpstey, A.F.; Postle, A.D.; Grocott, M.P.W. Alveolar Hyperoxia and Exacerbation of Lung Injury in Critically Ill SARS-CoV-2 Pneumonia. Med. Sci. 2023, 11, 70. https://doi.org/10.3390/medsci11040070
Dushianthan A, Bracegirdle L, Cusack R, Cumpstey AF, Postle AD, Grocott MPW. Alveolar Hyperoxia and Exacerbation of Lung Injury in Critically Ill SARS-CoV-2 Pneumonia. Medical Sciences. 2023; 11(4):70. https://doi.org/10.3390/medsci11040070
Chicago/Turabian StyleDushianthan, Ahilanandan, Luke Bracegirdle, Rebecca Cusack, Andrew F. Cumpstey, Anthony D. Postle, and Michael P. W. Grocott. 2023. "Alveolar Hyperoxia and Exacerbation of Lung Injury in Critically Ill SARS-CoV-2 Pneumonia" Medical Sciences 11, no. 4: 70. https://doi.org/10.3390/medsci11040070