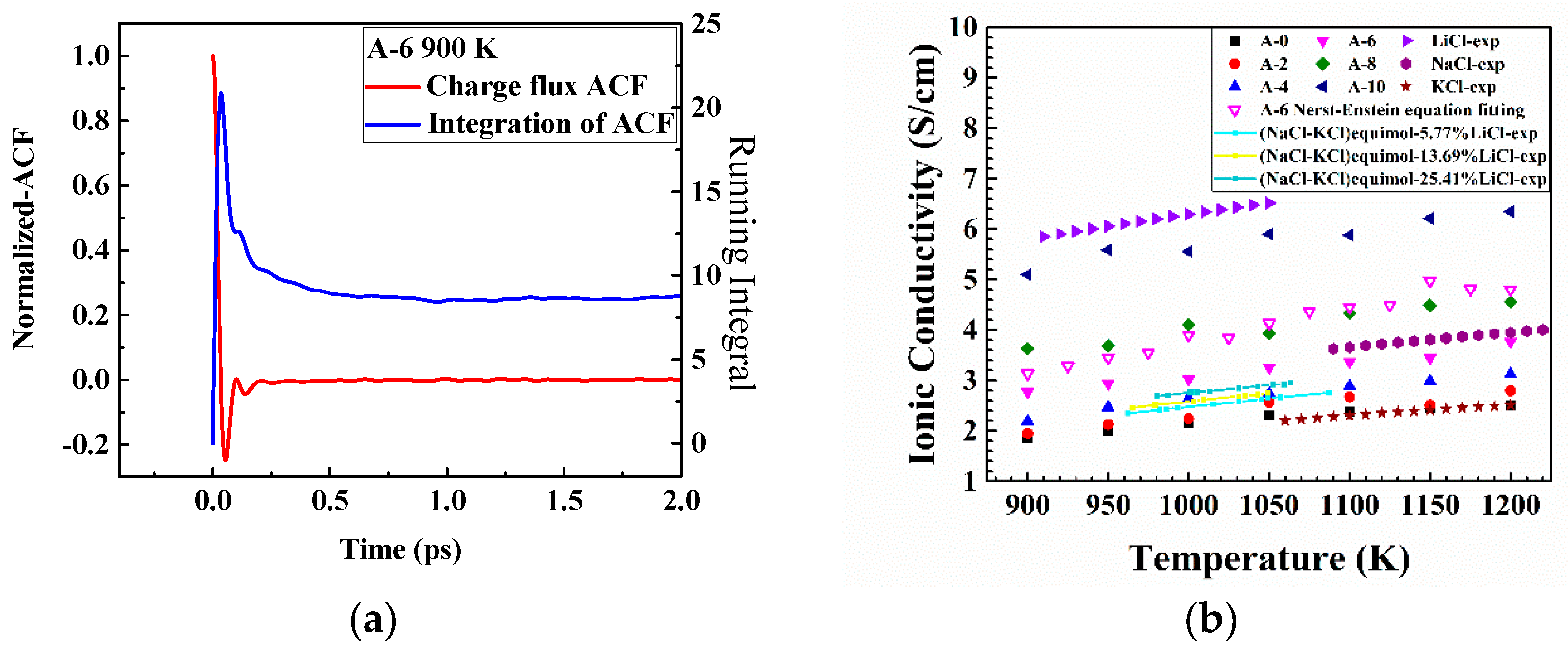
3.1.1. Partial Radial Distribution Function
(a) The LiCl concentration dependence
Partial radial distribution function (PDF) is calculated in histogram form by binning pairwise distances into 200 bins from 0.0 to the maximum force cutoff (defined as r
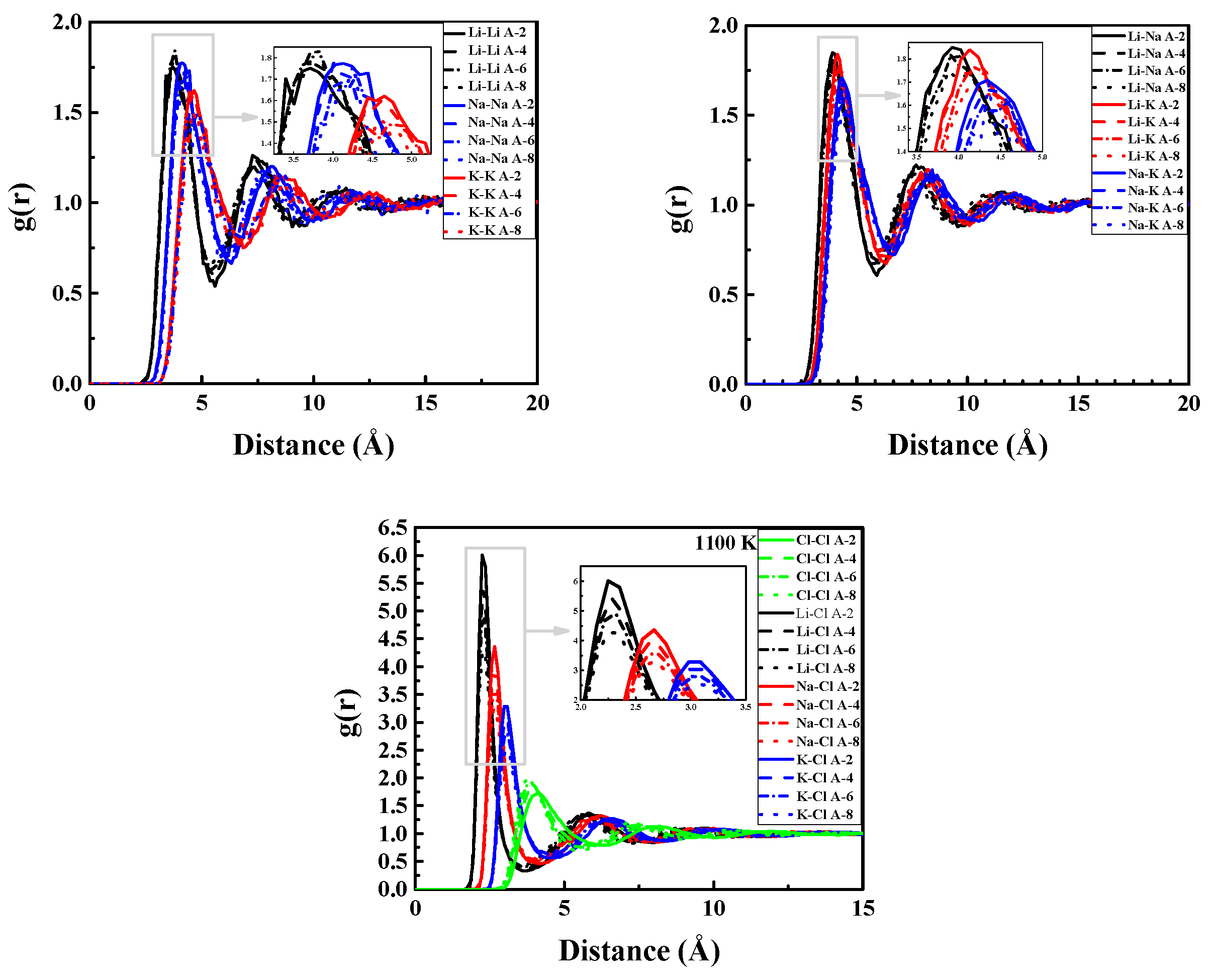
c = L/2). The bins are of uniform size in a radial distance. Thus, a single bin encompasses a thin shell of distances in 3d and a thin ring of distances in 2d. PDFs of A-2, A-4, A-6, and A-8 at 1100 K are shown in
Figure 2 to illustrate the influence of the LiCl concentration on the structure of ternary systems. The quantitatively structural information containing the first peak position, r
max, the first peak height, h
max, the first valley position, r
min, and running integral coordination number, N(r
min) are summarized in
Table 3, which were determined by fitting PDFs (R
2 = 1.0). The running integral coordination number was obtained by a spherical integration of g(r) from 0 to r
min. The structural parameters of the ternary systems were in good agreement with results of a similar composition [
22].
For cation-anion pairs, though the variations of the first peak positions of potassium are a little greater than lithium sodium ions across the concentration, the changes (maximum change was 0.05 Å) were subtle. However, the first peak heights all decreased distinctively, while the heights of the first minimum increased with an increasing LiCl. This indicated the degree of unlike charged ions’ association decreased with LiCl addition. The coordination numbers were roughly 4, 5, and 6 for Li+, Na+, and K+, respectively. With an increasing LiCl molar fraction, the coordination numbers of the cation-anion pairs all increased, which suggested that cations tended to accommodate more anions.
For cation-cation pairs, as shown in
Figure 2, the PDF curves of the cation-cation pairs are not as smooth as the ones of cation-anion pairs for the relatively weak association between like charge ions. The first peak positions shifted right and the corresponding height decreased with LiCl addition (except for Li-Li). For Li-Li, r
max showed an overall increasing tendency, but h
max increased up to 60% of the LiCl concentration then decreased. The LiCl concentration had more influence on the structure of cation-cation pairs than the cation-anion pairs. The changing rates of r
max and h
max for Li-Na and Li-K pairs were smaller than those for Na-Na, Na-K, and K-K pairs, which indicated a larger cation radius difference was a more stable structure even in cation-cation pairs.
For anion-anion pairs, the first peak positions shifted from 4.22 Å to 3.75 Å across the concentration, while the first peak heights rose from 1.73 to 2.04. The equilibrium distance of Cl-Cl was shortened and the association between Cl-Cl was strengthened. The coordination number of Cl-Cl decreased as LiCl increased, which indicated the closed pack between anions was inhibited. This could be attributed to the significant anion-anion overlap in LiCl salts relative to the other alkali chlorides [
23].
(b) Temperature dependence
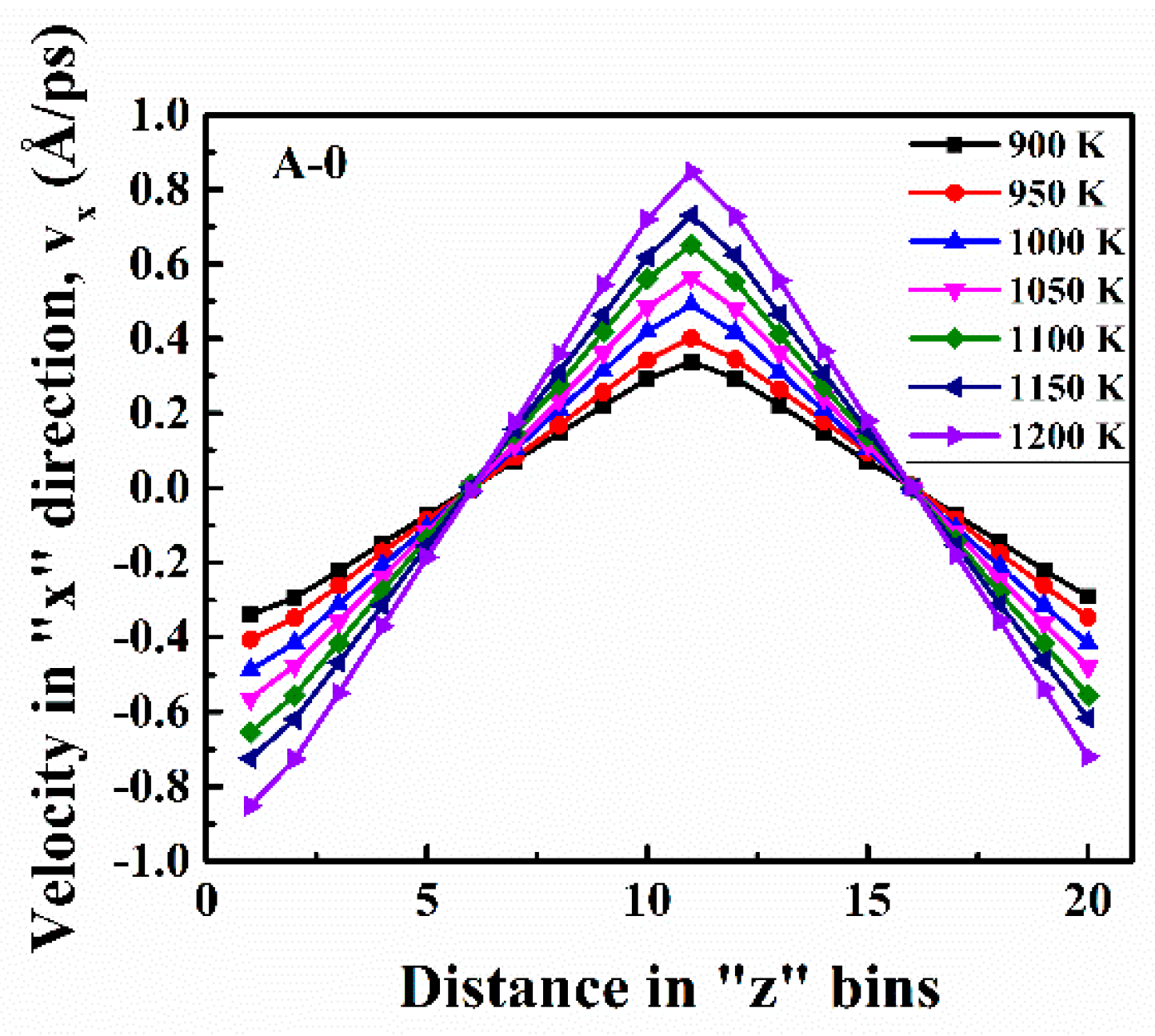
The influence of temperature for each pair at different concentrations are shown in
Appendix A (
Figure A1,
Figure A2,
Figure A3 and
Figure A4). The equilibrium distances between all pairs were essentially constant in the temperature range of 900 K to 1200 K. The increasing temperature led to a lower h
max and an increase of the minima offset, which indicated the looser coordination structure.
However, for 1500 K, the rmax of cation-cation pairs obviously shifted to the right, while the second peak became weaker, which revealed the structure of ions became more liquid-like and disordered. However, the rmax of cation-anion pairs unexpectedly shifted left. The equilibrium distances of cation-cation pairs increased, however, the cation-anion was the opposite, which means ion clusters were formed. The increasing temperature caused free space in melts to become larger, thus cations drifted apart. Meanwhile, the distances of cations and anions became shorter, thus the ion clusters were likely to be cation-anion clusters. Another possibility for this phenomenon could be that the free space became larger in melts with an ascending temperature, so less correlated, similarly charged ion pairs drifted apart, and only the more tightly coordinated anions around the cations were left.
The temperature influence on Cl-Cl pairs was similar with the cation-cation pairs. At 1500 K, the rmax of Cl-Cl pairs shifted right slightly and the corresponding hmax became weaker. However, it is interesting to note that with the LiCl concentration increasing, the Cl-Cl pairs were less affected by the temperature. We assumed that this was caused by the stronger interactions between Li-Cl pairs.
In conclusion, though the ternary system is chemically simple, there are complicated changes in the association of each ion with its first coordination shell with the composition changing. The addition of LiCl to molten ternary systems decreases the association between ion pairs, except for Li-Li and Cl-Cl pairs. It is worthy to note that the addition of LiCl strengthened the Cl-Cl association, and the closed pack between anions was inhibited. Ion clusters were formed in liquid and the distance of ion clusters became larger with an increasing temperature, which has great influence on the macro-properties. To further study the local structure, PDFs must be combined with the coordination numbers distribution and angular distribution function.
3.1.2. Coordination Numbers Distribution
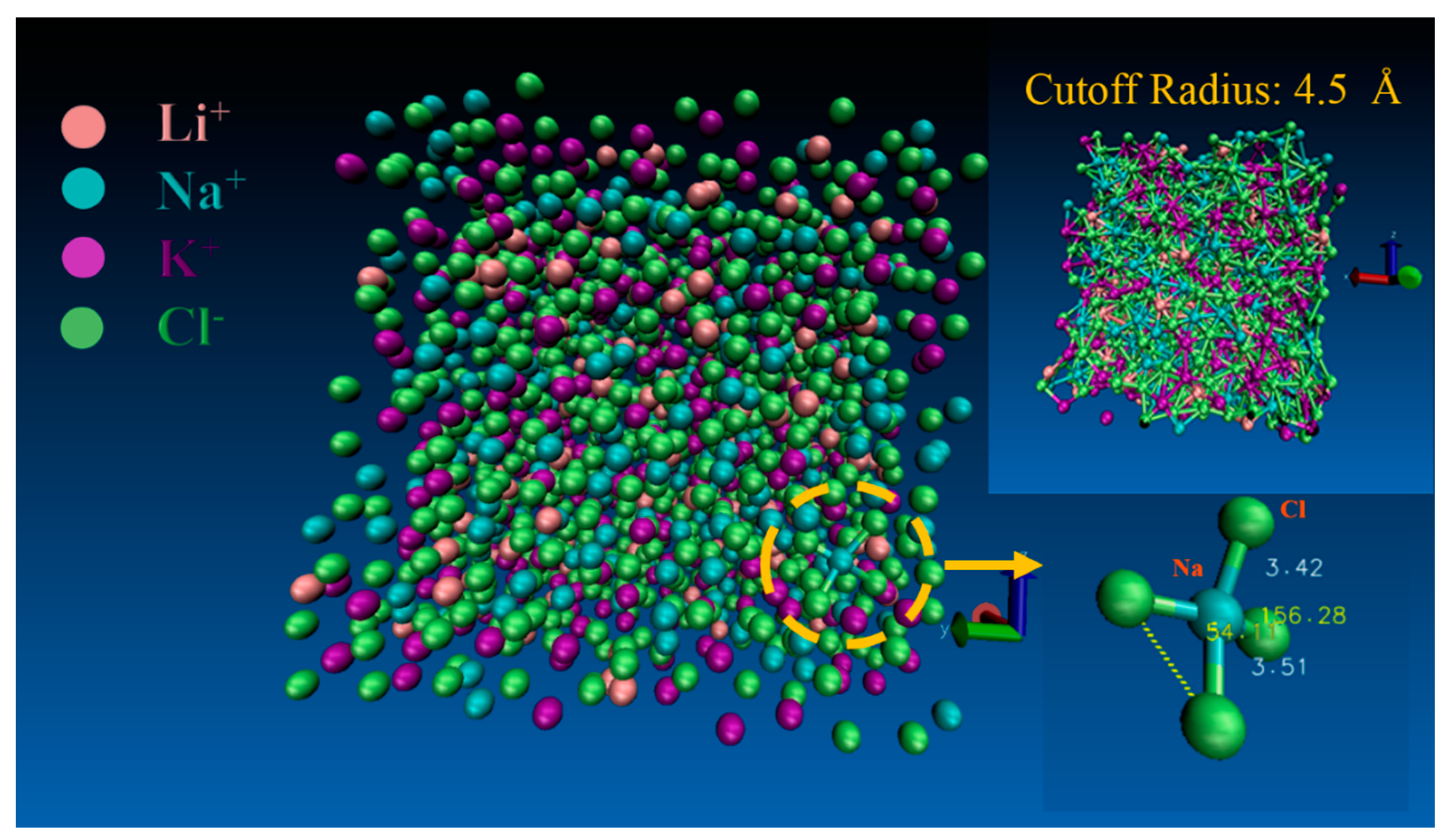
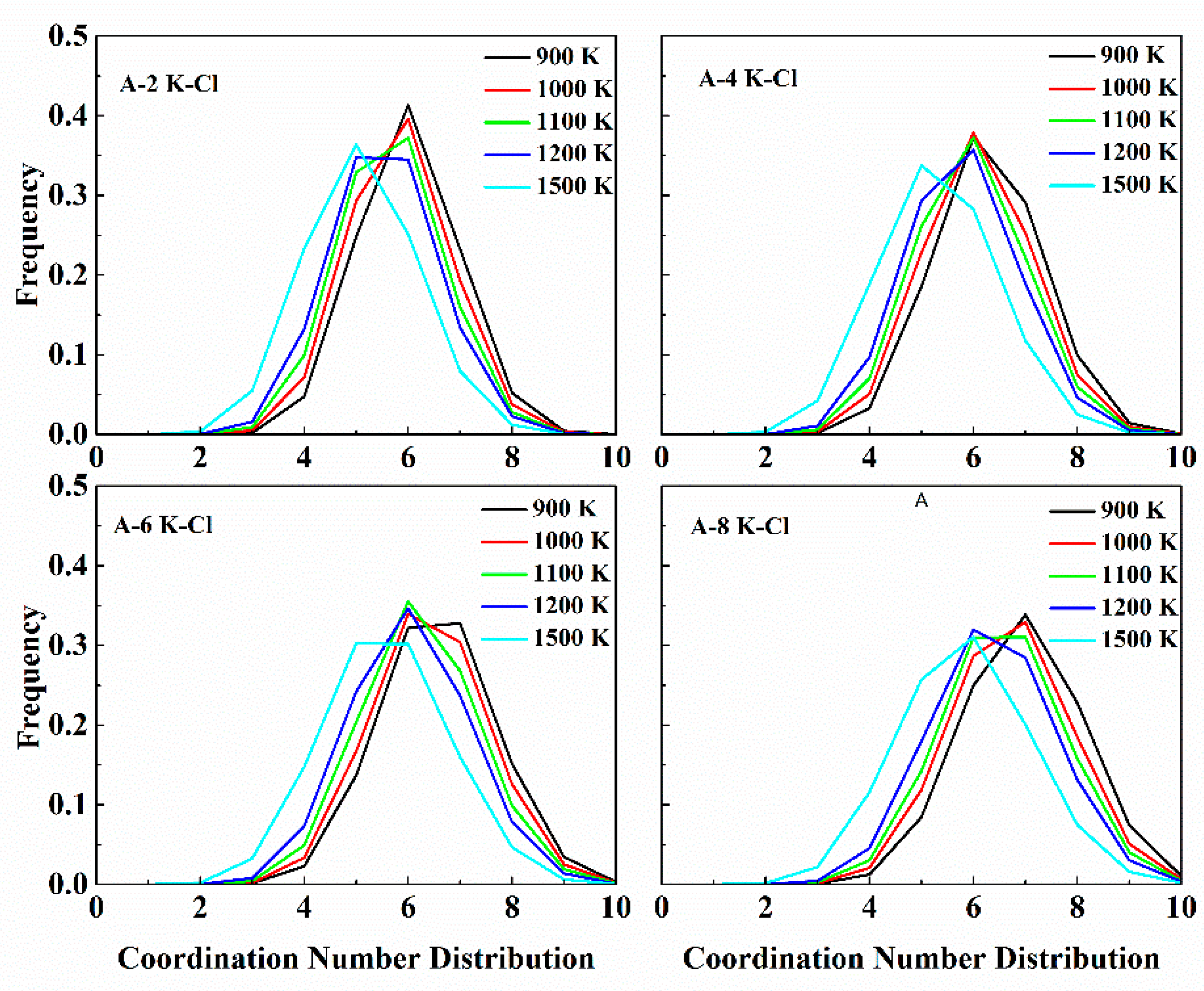
The simulation enabled deep insight into the numbers of anions in the coordination shell. We determined the number of anions within a spherical shell by collection of every step ions’ position and the shell radius was set to the position of the minimum (rmin) in the PDFs. This constitutes an instantaneous coordination number for that cation.
Figure 3 shows the coordination numbers distribution around each cation at 1100 K across the concentration. In the ternary melts, there coexisted 3- to 8-fold coordination, with different proportions. The coordination numbers distribution of Li
+ are mainly located in 4 and coexisted with 3-fold and 5-fold coordination. With an increasing LiCl composition, the mainly located 4-fold coordination of Li
+ slightly decreased. Meanwhile, the proportion of 3-fold distribution decreased, and the proportion of 5-fold coordination number increased. For Na-Cl, the coordination numbers, mainly located in 5, coexisted with 4-fold and 6-fold coordination. The 5-fold coordination almost remained constant, with no regard to the composition. However, with an increasing LiCl concentration, the 4-fold coordination largely decreased and 6-fold coordination increased. For K-Cl, the coordination numbers of K
+ were mainly 4, 5, 6, 7, and 8. When
, the coordination numbers were mainly located in 6. However, when
, the 6-fold and 7-fold dominated. With an increasing LiCl, the 4-fold and 5-fold coordination of K
+ all decreased, while the higher coordination 7-fold and 8-fold increased.
The results showed that the proportion of higher coordination numbers for each cation increased as the LiCl molar fraction increased. Combined with our previous study [
13,
15], the local structure of Li-Cl in melts is an anomalous, distorted tetrahedron, while Na-Cl and K-Cl are a distorted octahedron. It can be clearly seen that the tendency to form an octahedron became stronger for Na-Cl with an increasing LiCl addition. The anions coordination numbers distribution of K
+ were the most complex. Though sexadentate dominated, the tendency of accommodating more anions for K
+ became stronger. As for Li-Cl, though the portion of 5-fold coordination increased, the tetrahedron structure still dominated. The sharp peak in the PDF indicates a tight coordination of Li-Cl and its height decreases with LiCl addition. It can be found from
Table 3 that when the equilibrium distance between the cation and anion gets longer, the coordination numbers of the cation become greater. Combined with PDFs analysis, the LiCl addition increased the mobility of Cl
-, so the tighter coordination structure (the relatively lower coordinated structure) reduced. With the temperature increasing, the coordination became amorphous (the temperature influence on the coordination numbers distribution is provided in
Appendix A Figure A5,
Figure A6 and
Figure A7). In
Figure 4, there is a snapshot of the A-2 system at 1100 K, and the coordinates around different cations were in a dynamic equilibration.
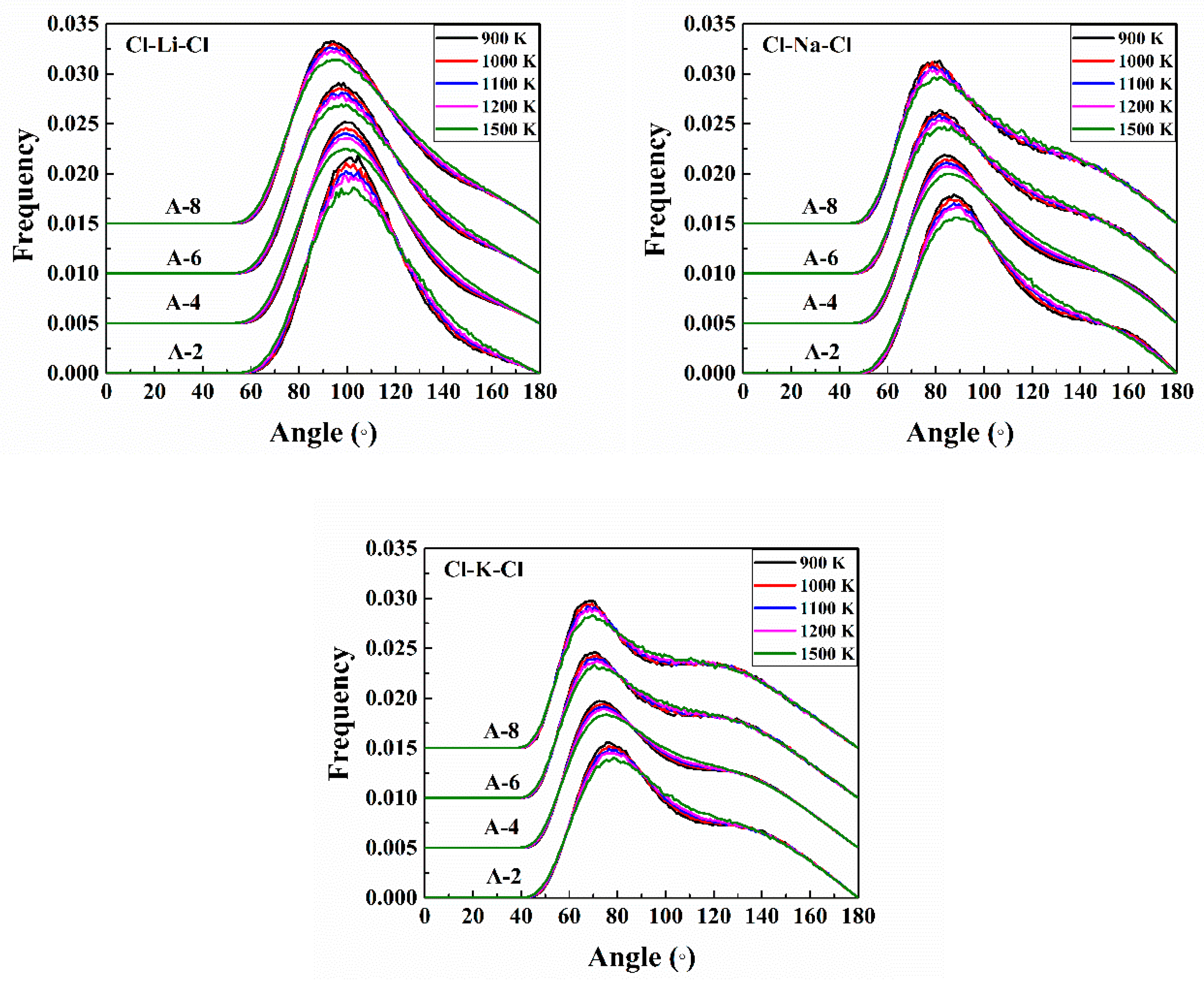
3.1.3. Angular Distribution Function
The angular distribution function (ADF) of Cl-A-Cl (A = Li, Na, K) from A-2 to A-8 at 1100 K is shown in
Figure 5. Here, “bond” does not mean a real chemical bond, but just an atom pair within some distance. In the ternary melts, the Cl-A-Cl bond angles were distributed between 60° and 150°, all showing a broad peak and a smearing phenomenon, which indicates that the coordination bonds are orientated randomly.
With an increasing LiCl addition, the peak values of the bond angle for Cl-Li-Cl shifted from 100º to 93°, for Cl-Na-Cl, shifted from 88° to 79°, and for Cl-K-Cl, shifted from 78° to 68°. The angles become smaller with an increasing LiCl concentration. The corresponding bond angles for pure melts of LiCl, NaCl, and KCl were 98.18°, 87.27°, and 83.64°, respectively [
15]. The bond angles of the corresponding pure alkali chloride crystal were mainly distributed in 90° and 180° [
24] because the three salts in the crystal type were a face-centered cubic structure and sexadentate. Though the long-range order disappears in melts, at the micro-level, there was still some tendency to keep the crystal structure. Considering the coordination numbers of each cation become larger and the bond angles become smaller, we assumed that the deforming in the micro-level structure was larger than in that of the pure [
15] and binary melt systems [
25]. The ADFs were broader and no peak shifting was observed as the temperature increased (the temperature influence on the ADFs is provided in
Appendix A Figure A8).























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}