
Impact of Glucocorticoid on a Cellular Model of Parkinson’s Disease: Oxidative Stress and Mitochondrial Function

 , , , , and
, , , , and
Abstract
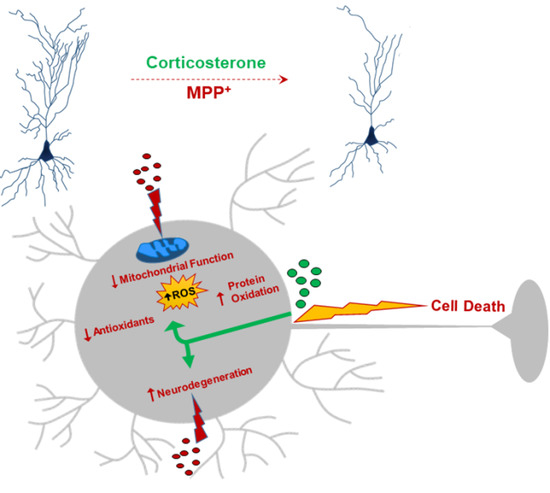
:
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Cell Viability
2.3. Determination of Mitochondrial Levels of ROS
2.4. Oxidative Stress Cellular Markers
2.5. Measurement of Mitochondrial Markers
2.5.1. Mitochondrial Membrane Potential
2.5.2. Mitochondrial Oxygen Consumption Rate
2.6. Immunocytochemistry Procedure
2.7. Neurodegeneration
2.8. Statistical Analysis
3. Results
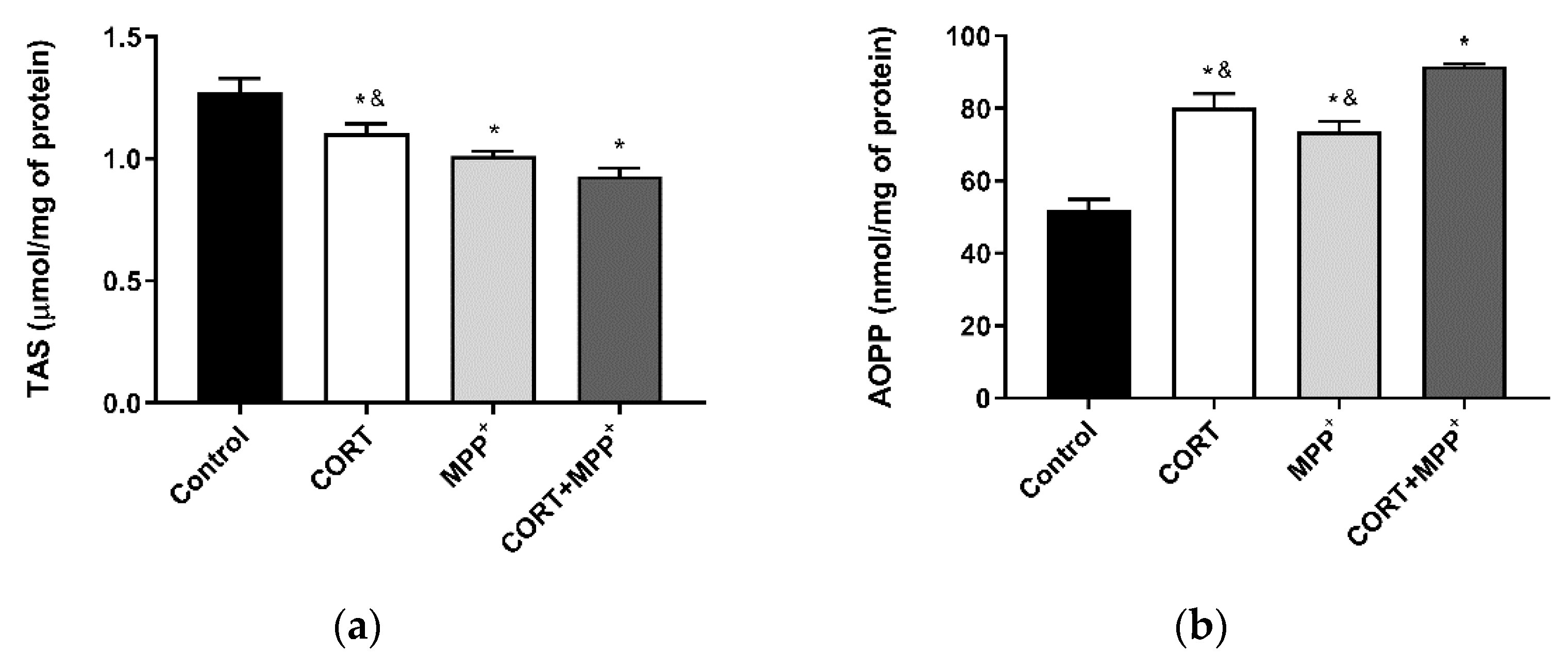
3.1. Oxidative Stress Cellular Markers
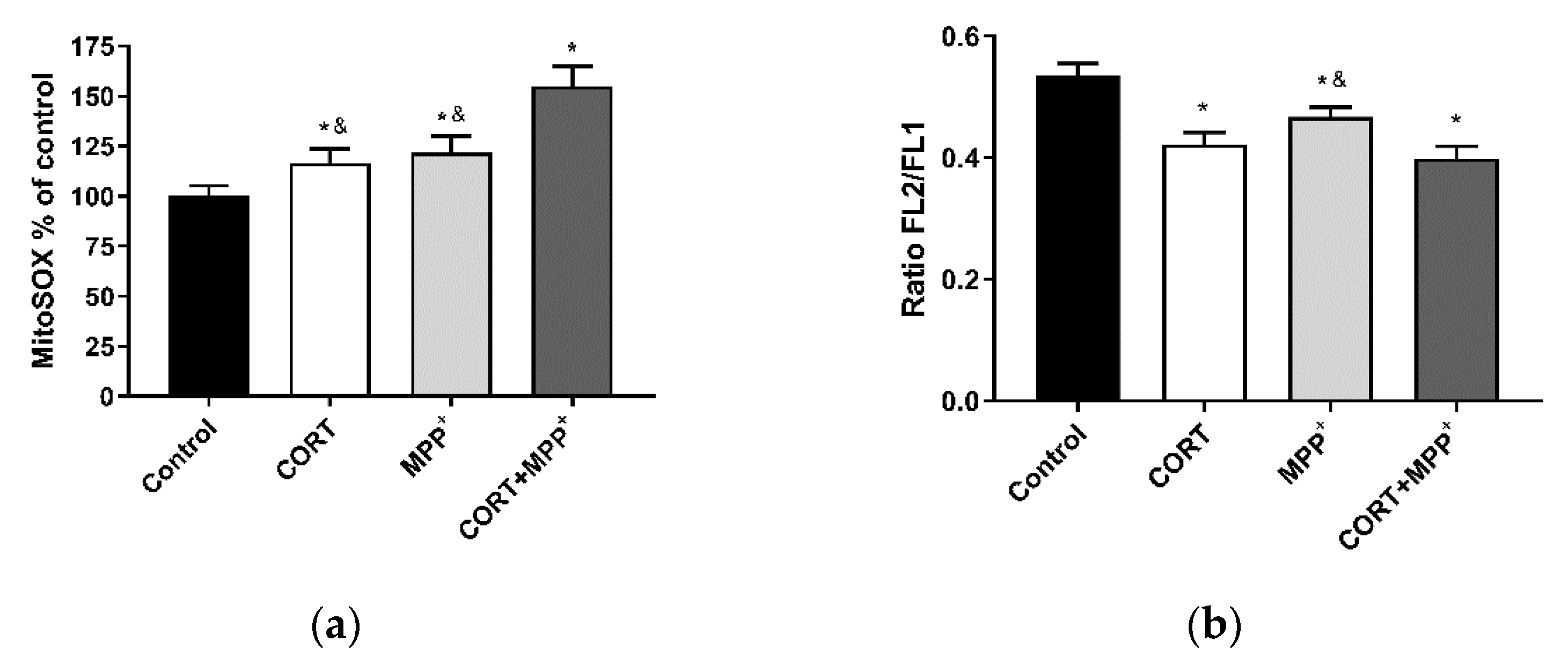
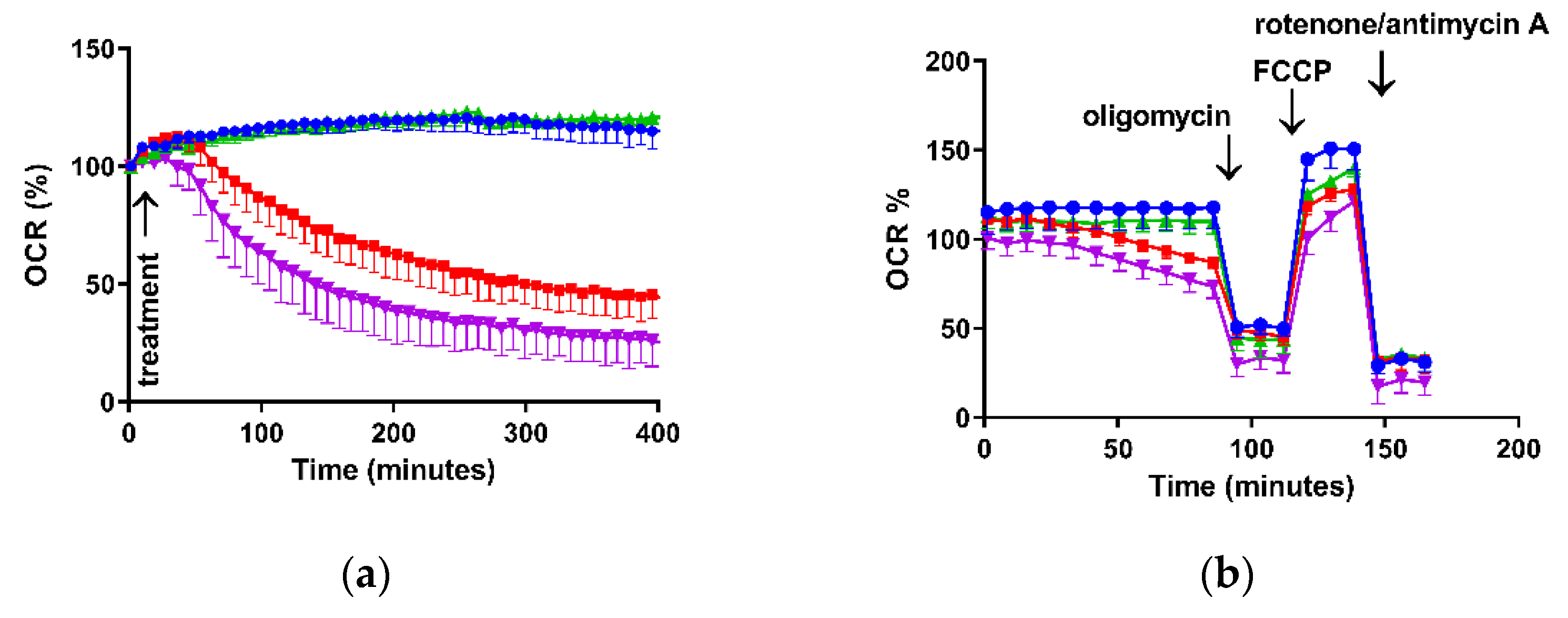
3.2. ROS Production and Measurement of Mitochondrial Function
3.3. Neurodegeneration
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tysnes, O.B.; Storstein, A. Epidemiology of Parkinson’s disease. J. Neural. Transm. 2017, 124, 901–905. [Google Scholar] [CrossRef]
- Emamzadeh, F.N.; Surguchov, A. Parkinson’s Disease: Biomarkers, treatment, and risk factors. Front. Neurosci. 2018, 12, 612. [Google Scholar] [CrossRef] [PubMed]
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.E.; Lang, A.E. Parkinson disease. Nat. Rev. Dis. Primers 2017, 3, 17013. [Google Scholar] [CrossRef] [PubMed]
- Blesa, J.; Trigo-Damas, I.; Quiroga-Varela, A.; Jackson-Lewis, V.R. Oxidative stress and Parkinson’s disease. Front. Neuroanat. 2015, 9, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schon, E.A.; Przedborski, S. Mitochondria: The next (neurode) generation. Neuron 2011, 70, 1033–1053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Djamshidian, A.; Lees, A.J. Can stress trigger Parkinson’s disease? J. Neurol. Neurosurg. Psychiatry 2014, 85, 878–881. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, D.S.; Kopin, I.J. Linking stress, catecholamine autotoxicity, and allostatic load with neurodegenerative diseases: A focused review in memory of Richard Kvetnansky. Cell. Mol. Neurobiol. 2018, 38, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Soares, N.M.; Pereira, G.M.; Altmann, V.; de Almeida, R.M.M.; Rieder, C.R.M. Cortisol levels, motor, cognitive and behavioral symptoms in Parkinson’s disease: A systematic review. J. Neural. Transm. 2019, 126, 219–232. [Google Scholar] [CrossRef]
- Johnson, W.M.; Wilson-Delfosse, A.L.; Mieyal, J.J. Dysregulation of glutathione homeostasis in neurodegenerative diseases. Nutrients 2012, 4, 1399–1440. [Google Scholar] [CrossRef] [Green Version]
- Puspita, L.; Chung, S.Y.; Shim, J.W. Oxidative stress and cellular pathologies in Parkinson’s disease. Mol. Brain 2017, 10, 53. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.D.; Zhao, X.; Li, Y.; Li, G.R.; Liu, X.L. Damage to dopaminergic neurons by oxidative stress in Parkinson’s disease (review). Int. J. Mol. Med. 2018, 41, 1817–1825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dumont, M.; Beal, M.F. Neuroprotective strategies involving ROS in Alzheimer disease. Free Radic. Biol. Med. 2011, 51, 1014–1026. [Google Scholar] [CrossRef] [Green Version]
- Yan, M.H.; Wang, X.; Zhu, X. Mitochondrial defects and oxidative stress in Alzheimer disease and Parkinson disease. Free Radic. Biol. Med. 2013, 62, 90–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richards, R.I.; Robertson, S.A.; Kastner, D.L. Neurodegenerative diseases have genetic hallmarks of autoinflammatory disease. Hum. Mol. Genet. 2018, 27, R108–R118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, G.P.; Schonhoff, A.M.; Jurkuvenaite, A.; Thome, A.D.; Standaert, D.G.; Harms, A.S. Targeting of the class II transactivator attenuates inflammation and neurodegeneration in an alpha-synuclein model of Parkinson’s disease. J. Neuroinflammation 2018, 15, 244. [Google Scholar] [CrossRef] [PubMed]
- De Pasquale, V.; Costanzo, M.; Siciliano, R.A.; Mazzeo, M.F.; Pistorio, V.; Bianchi, L.; Marchese, E.; Ruoppolo, M.; Pavone, L.M.; Caterino, M. Proteomic analysis of Mucopolysaccharidosis IIIB mouse brain. Biomolecules 2020, 10, 355. [Google Scholar] [CrossRef] [Green Version]
- Herman, J.P.; Adams, D.; Prewitt, C. Regulatory changes in neuroendocrine stress-integrative circuitry produced by a variable stress paradigm. Neuroendocrinology 1995, 61, 180–190. [Google Scholar] [CrossRef]
- Berger, I.; Werdermann, M.; Bornstein, S.R.; Steenblock, C. The adrenal gland in stress—Adaptation on a cellular level. J. Steroid Biochem. Mol. Biol. 2019, 190, 198–206. [Google Scholar] [CrossRef]
- Herman, J.P.; Cullinan, W.E. Neurocircuitry of stress: Central control of the hypothalamo-pituitary-adrenocortical axis. Trends Neurosci. 1997, 20, 78–84. [Google Scholar] [CrossRef]
- Van Craenenbroeck, K.; De Bosscher, K.; Vanden Berghe, W.; Vanhoenacker, P.; Haegeman, G. Role of glucocorticoids in dopamine-related neuropsychiatric disorders. Mol. Cell. Endocrinol. 2005, 245, 10–22. [Google Scholar] [CrossRef]
- Hemmerle, A.M.; Herman, J.P.; Seroogy, K.B. Stress, depression and Parkinson’s disease. Exp. Neurol. 2012, 233, 79–86. [Google Scholar] [CrossRef] [Green Version]
- Ros-Bernal, F.; Hunot, S.; Herrero, M.T.; Parnadeau, S.; Corvol, J.C.; Lu, L.; Alvarez-Fischer, D.; Carrillo-de Sauvage, M.A.; Saurini, F.; Coussieu, C.; et al. Microglial glucocorticoid receptors play a pivotal role in regulating dopaminergic neurodegeneration in parkinsonism. Proc. Natl. Acad. Sci. USA 2011, 108, 6632–6637. [Google Scholar] [CrossRef] [Green Version]
- Martin-Montañez, E.; Pavia, J.; Santin, L.J.; Boraldi, F.; Estivill-Torrus, G.; Aguirre, J.A.; Garcia-Fernandez, M. Involvement of IGF-II receptors in the antioxidant and neuroprotective effects of IGF-II on adult cortical neuronal cultures. Biochim. Biophys. Acta 2014, 1842, 1041–1051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin-Montañez, E.; Millon, C.; Boraldi, F.; Garcia-Guirado, F.; Pedraza, C.; Lara, E.; Santin, L.J.; Pavia, J.; Garcia-Fernandez, M. IGF-II promotes neuroprotection and neuroplasticity recovery in a long-lasting model of oxidative damage induced by glucocorticoids. Redox Biol. 2017, 13, 69–81. [Google Scholar] [CrossRef] [Green Version]
- Hemmerle, A.M.; Dickerson, J.W.; Herman, J.P.; Seroogy, K.B. Stress exacerbates experimental Parkinson’s disease. Mol. Psychiatry 2014, 19, 638–640. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Yang, X.; Zhang, Y.; Zhang, L.; Feng, L. Chronic mild stress accelerates the progression of Parkinson’s disease in A53T alpha-synuclein transgenic mice. Exp. Neurol. 2016, 285, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.K.; Jadavji, N.M.; Colwell, K.L.; Katrina Perehudoff, S.; Metz, G.A. Stress accelerates neural degeneration and exaggerates motor symptoms in a rat model of Parkinson’s disease. Eur. J. Neurosci. 2008, 27, 2133–2146. [Google Scholar] [CrossRef] [Green Version]
- Hastings, T.G.; Lewis, D.A.; Zigmond, M.J. Role of oxidation in the neurotoxic effects of intrastriatal dopamine injections. Proc. Natl. Acad. Sci. USA 1996, 93, 1956–1961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lucca, G.; Comim, C.M.; Valvassori, S.S.; Reus, G.Z.; Vuolo, F.; Petronilho, F.; Dal-Pizzol, F.; Gavioli, E.C.; Quevedo, J. Effects of chronic mild stress on the oxidative parameters in the rat brain. Neurochem. Int. 2009, 54, 358–362. [Google Scholar] [CrossRef]
- Burtscher, J.; Copin, J.C.; Rodrigues, J.; Kumar, S.T.; Chiki, A.; Guillot de Suduiraut, I.; Sandi, C.; Lashuel, H.A. Chronic corticosterone aggravates behavioral and neuronal symptomatology in a mouse model of alpha-synuclein pathology. Neurobiol. Aging 2019, 83, 11–20. [Google Scholar] [CrossRef]
- Son, J.H.; Chun, H.S.; Joh, T.H.; Cho, S.; Conti, B.; Lee, J.W. Neuroprotection and neuronal differentiation studies using substantia nigra dopaminergic cells derived from transgenic mouse embryos. J. Neurosci. Off. J. Soc. Neurosci. 1999, 19, 10–20. [Google Scholar] [CrossRef] [Green Version]
- Koh, J.Y.; Choi, D.W. Quantitative determination of glutamate mediated cortical neuronal injury in cell culture by lactate dehydrogenase efflux assay. J. Neurosci. Methods 1987, 20, 83–90. [Google Scholar] [CrossRef]
- Kauffman, M.E.; Kauffman, M.K.; Traore, K.; Zhu, H.; Trush, M.A.; Jia, Z.; Li, Y.R. MitoSOX-Based Flow Cytometry for Detecting Mitochondrial ROS. React. Oxyg. Species 2016, 2, 361–370. [Google Scholar] [CrossRef] [Green Version]
- Martin-Montañez, E.; Pavia, J.; Valverde, N.; Boraldi, F.; Lara, E.; Oliver, B.; Hurtado-Guerrero, I.; Fernandez, O.; Garcia-Fernandez, M. The S1P mimetic fingolimod phosphate regulates mitochondrial oxidative stress in neuronal cells. Free Radic. Biol. Med. 2019, 137, 116–130. [Google Scholar] [CrossRef] [Green Version]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Raha, S.; Robinson, B.H. Mitochondria, oxygen free radicals, disease and ageing. Trends Biochem. Sci. 2000, 25, 502–508. [Google Scholar] [CrossRef]
- Wu, M.; Neilson, A.; Swift, A.L.; Moran, R.; Tamagnine, J.; Parslow, D.; Armistead, S.; Lemire, K.; Orrell, J.; Teich, J.; et al. Multiparameter metabolic analysis reveals a close link between attenuated mitochondrial bioenergetic function and enhanced glycolysis dependency in human tumor cells. Am. J. Physiol. Cell Physiol. 2007, 292, C125–C136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gil, A.; Martin-Montañez, E.; Valverde, N.; Lara, E.; Boraldi, F.; Claros, S.; Romero-Zerbo, S.Y.; Fernandez, O.; Pavia, J.; Garcia-Fernandez, M. Neuronal metabolism and neuroprotection: Neuroprotective effect of fingolimod on menadione-induced mitochondrial damage. Cells 2020, 10, 34. [Google Scholar] [CrossRef] [PubMed]
- Schmuck, G.; Kahl, R. The use of Fluoro-Jade in primary neuronal cell cultures. Arch. Toxicol. 2009, 83, 397–403. [Google Scholar] [CrossRef]
- Mudo, G.; Makela, J.; Di Liberto, V.; Tselykh, T.V.; Olivieri, M.; Piepponen, P.; Eriksson, O.; Malkia, A.; Bonomo, A.; Kairisalo, M.; et al. Transgenic expression and activation of PGC-1alpha protect dopaminergic neurons in the MPTP mouse model of Parkinson’s disease. Cell. Mol. Life Sci. CMLS 2012, 69, 1153–1165. [Google Scholar] [CrossRef]
- Wang, J.; Gu, J.; Wu, H.; Zhu, G.; Feng, D.; Li, Y.; Guo, W.; Tian, K.; Gao, G.; Gao, L. Pentazocine protects SN4741 cells against MPP(+)-induced cell damage via up-regulation of the canonical wnt/beta-catenin signaling pathway. Front. Aging Neurosci. 2017, 9, 196. [Google Scholar] [CrossRef]
- Du, J.; Wang, Y.; Hunter, R.; Wei, Y.; Blumenthal, R.; Falke, C.; Khairova, R.; Zhou, R.; Yuan, P.; Machado-Vieira, R.; et al. Dynamic regulation of mitochondrial function by glucocorticoids. Proc. Natl. Acad. Sci. USA 2009, 106, 3543–3548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, K.; Guo, W.; Tang, G.; Zheng, B.; Zheng, Z. A Case of early onset Parkinson’s disease after major stress. Neuropsychiatr. Dis. Treat. 2013, 9, 1067–1069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haglin, L.; Backman, L. Covariation between plasma phosphate and daytime cortisol in early Parkinson’s disease. Brain Behav. 2016, 6, e00556. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C.; Fan, W.; Procaccio, V. Mitochondrial energetics and therapeutics. Annu. Rev. Pathol. 2010, 5, 297–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Küçükdoğru, R.; Turkez, H.; Arslan, M.E.; Tozlu, O.O.; Sonmez, E.; Mardinoglu, A.; Cacciatore, I.; Di Stefano, A. Neuroprotective effects of boron nitride nanoparticles in the experimental Parkinson’s disease model against MPP+ induced apoptosis. Metab. Brain Dis. 2020, 35, 947–957. [Google Scholar] [CrossRef]
- Giordano, S.; Lee, J.; Darley-Usmar, V.M.; Zhang, J. Distinct effects of rotenone, 1-methyl-4-phenylpyridinium and 6-hydroxydopamine on cellular bioenergetics and cell death. PLoS ONE 2012, 7, e44610. [Google Scholar] [CrossRef]
- Dranka, B.P.; Benavides, G.A.; Diers, A.R.; Giordano, S.; Zelickson, B.R.; Reily, C.; Zou, L.; Chatham, J.C.; Hill, B.G.; Zhang, J.; et al. Assessing bioenergetic function in response to oxidative stress by metabolic profiling. Free Radic. Biol. Med. 2011, 51, 1621–1635. [Google Scholar] [CrossRef] [Green Version]
- Boeck, C.; Gumpp, A.M.; Calzia, E.; Radermacher, P.; Waller, C.; Karabatsiakis, A.; Kolassa, I.T. The association between cortisol, oxytocin, and immune cell mitochondrial oxygen consumption in postpartum women with childhood maltreatment. Psychoneuroendocrinology 2018, 96, 69–77. [Google Scholar] [CrossRef]
- Brand, M.D.; Nicholls, D.G. Assessing mitochondrial dysfunction in cells. Biochem. J. 2011, 435, 297–312. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Michaelis, E.K. Selective neuronal vulnerability to oxidative stress in the brain. Front. Aging Neurosci. 2010, 2, 12. [Google Scholar] [CrossRef]
- Du, X.; Pang, T.Y. Is Dysregulation of the HPA-Axis a core pathophysiology mediating co-morbid depression in neurodegenerative diseases? Front. Psychiatry 2015, 6, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa, C.M.; Oliveira, G.L.; Fonseca, A.C.S.; Lana, R.C.; Polese, J.C.; Pernambuco, A.P. Levels of cortisol and neurotrophic factor brain-derived in Parkinson’s disease. Neurosci. Lett. 2019, 708, 134359. [Google Scholar] [CrossRef] [PubMed]
- van den Heuvel, L.L.; du Plessis, S.; Stalder, T.; Acker, D.; Kirschbaum, C.; Carr, J.; Seedat, S. Hair glucocorticoid levels in Parkinson’s disease. Psychoneuroendocrinology 2020, 117, 104704. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.D.; Castro, S.L.; Zigmond, M.J. Stress-induced Parkinson’s disease: A working hypothesis. Physiol. Behav. 2002, 77, 527–531. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Conditions | Control | CORT | MPP+ | MPP+ + CORT |
|---|---|---|---|---|
| Basal respiration | 20.9 ± 1.8 | 21.8 ± 1.1 | 12.8 ± 1.3 * | 9.1 ± 2.1 * |
| ATP Production | 16.0 ± 1.7 | 17.9 ± 0.9 | 10.1 ± 1.5 * | 8.9 ± 2.1 * |
| Proton leak | 5.4 ± 0.5 | 4.6 ± 1.1 | 3.6 ± 0.9 | 7.4 ± 1.6 * |
| Spare respiratory capacity | 20.2 ± 1.2 | 8.5 ± 1.4 * | 10.4 ± 0.9 * | 9.9 ± 3.1* |
| Maximal respiration | 41.1 ± 1.2 | 30.2 ± 1.8 * | 23.1 ± 0.6 *,& | 20.5 ± 1.9 *,& |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Claros, S.; Gil, A.; Martinelli, M.; Valverde, N.; Lara, E.; Boraldi, F.; Pavia, J.; Martín-Montañez, E.; Garcia-Fernandez, M. Impact of Glucocorticoid on a Cellular Model of Parkinson’s Disease: Oxidative Stress and Mitochondrial Function. Brain Sci. 2021, 11, 1106. https://doi.org/10.3390/brainsci11081106
Claros S, Gil A, Martinelli M, Valverde N, Lara E, Boraldi F, Pavia J, Martín-Montañez E, Garcia-Fernandez M. Impact of Glucocorticoid on a Cellular Model of Parkinson’s Disease: Oxidative Stress and Mitochondrial Function. Brain Sciences. 2021; 11(8):1106. https://doi.org/10.3390/brainsci11081106
Chicago/Turabian StyleClaros, Silvia, Antonio Gil, Mauro Martinelli, Nadia Valverde, Estrella Lara, Federica Boraldi, Jose Pavia, Elisa Martín-Montañez, and María Garcia-Fernandez. 2021. "Impact of Glucocorticoid on a Cellular Model of Parkinson’s Disease: Oxidative Stress and Mitochondrial Function" Brain Sciences 11, no. 8: 1106. https://doi.org/10.3390/brainsci11081106