
Neurological Insights into Sleep Disorders in Parkinson’s Disease
 , , ,
, , ,  and
and
Abstract
:
1. Introduction
1.1. Parkinson’s Disease
1.2. Sleep Disorders
2. Sleep Disturbances in Parkinson’s Disease
2.1. Excessive Daytime Sleepiness (EDS)
2.2. Insomnia
2.3. Rapid-Eye-Movement (REM) Sleep Behavior Disorder (RBD)
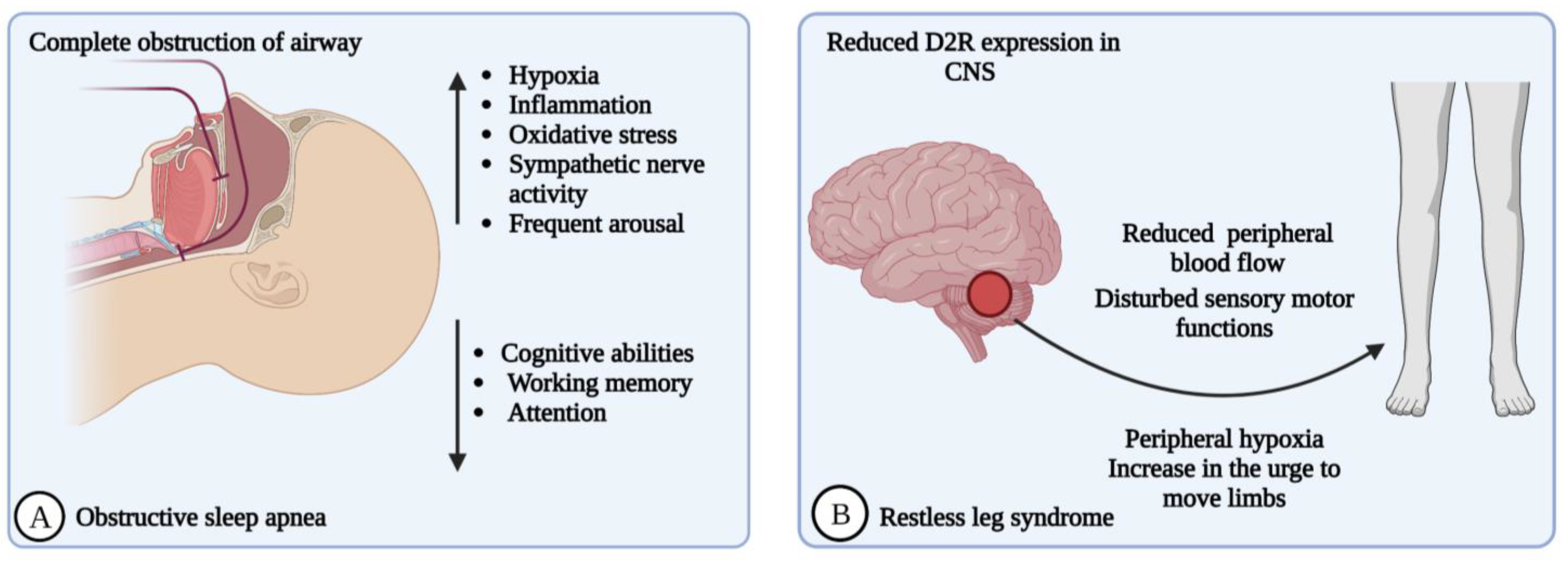
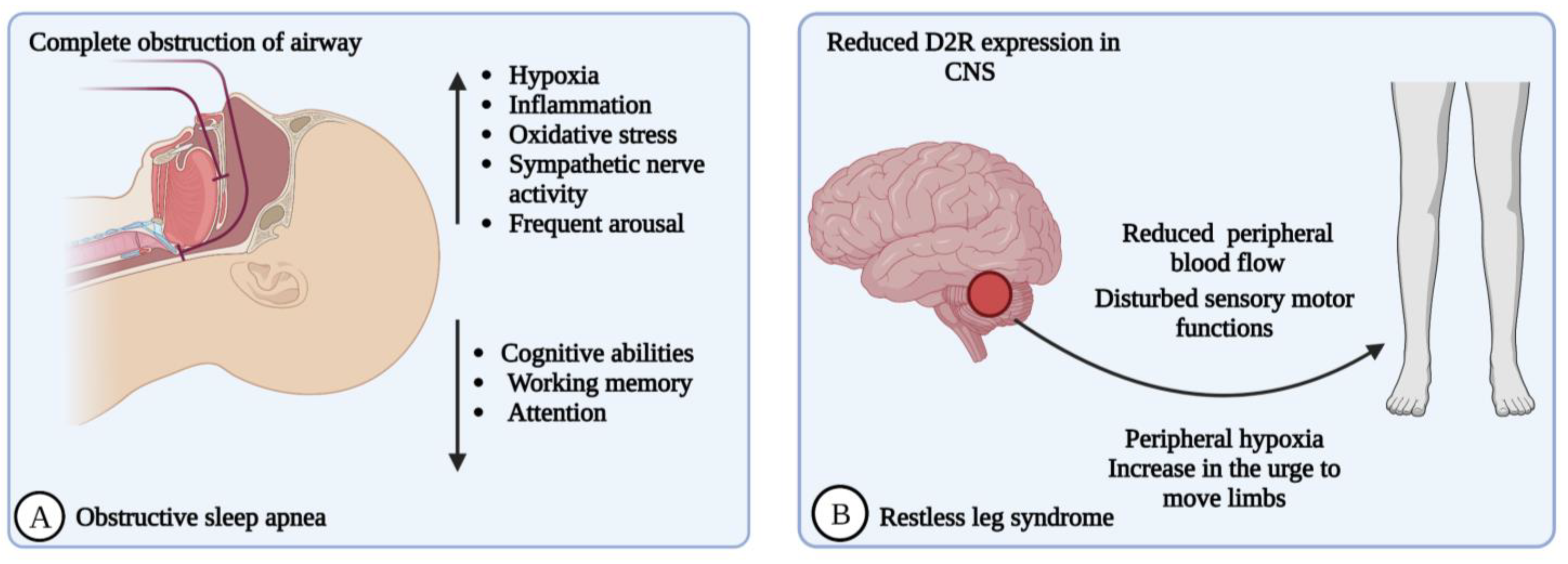
2.4. Obstructive Sleep Apnea (OSA)
2.5. Restless Legs Syndrome (RLS)
3. Neurological Changes in Sleep Disturbances
4. Circadian-Rhythm Dysfunction in PD
5. Genetic Heterogeneity of Sleep Disorders in Patients with PD
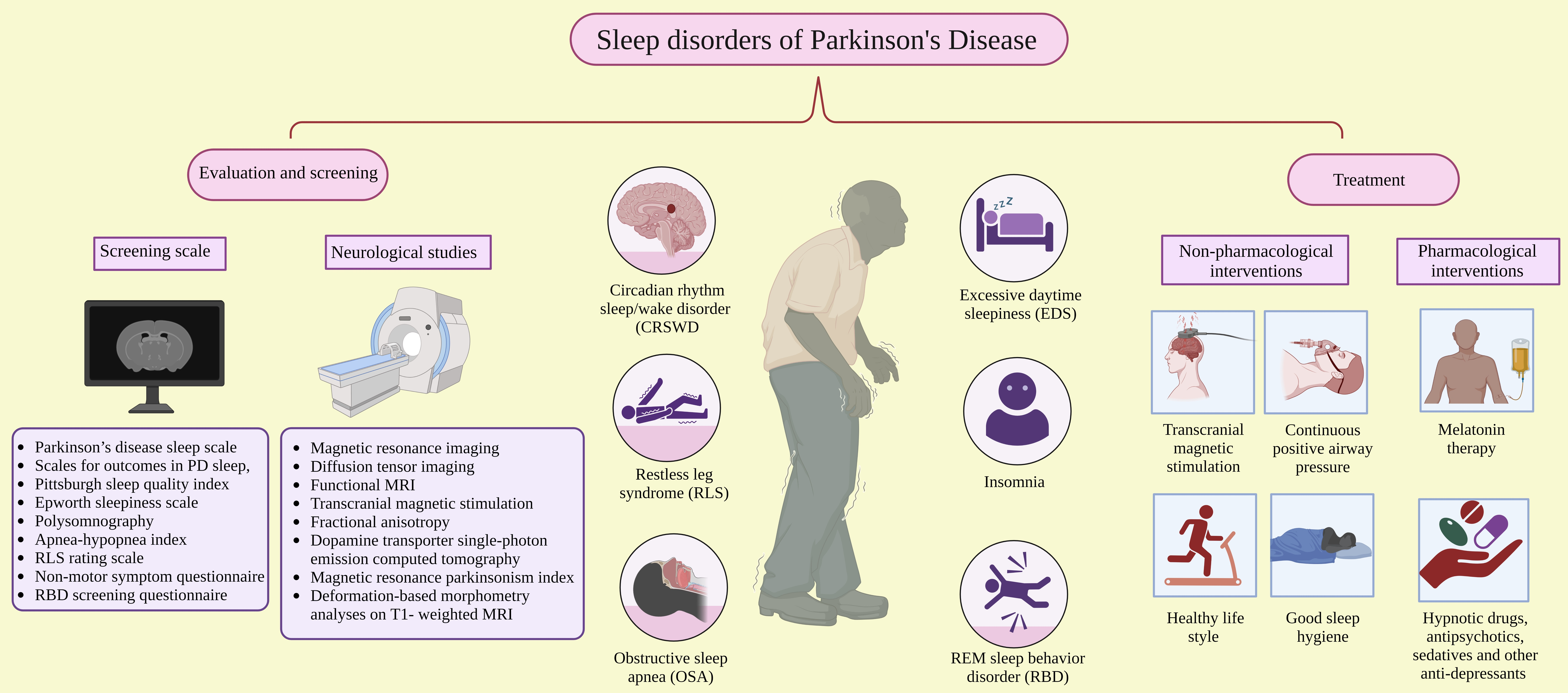
6. Evaluation of Sleep Disorders
6.1. Evaluation Scales and Screening Methods
6.2. Neuroimaging Studies
6.3. Challenges in Diagnosis and Evaluation
7. Treatment for PD-Associated Sleep Disorders
7.1. Non-Pharmacological Interventions
7.2. Pharmacological Interventions
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| PD | Parkinson’s disease |
| iPD | idiopathic Parkinson’s disease |
| AD | Alzheimer’s disease |
| CNS | Central nervous system |
| REM | Rapid eye movement |
| NREM | Non-rapid eye movement |
| RBD | Rapid eye movement sleep behavior disorder |
| iRBD | idiopathic Rapid eye movement sleep behavior disorder |
| EDS | Excessive daytime sleeping |
| RLS | Restless leg syndrome |
| OSA | Obstructive sleep apnea |
| SRBD | Sleep-related breathing disorder |
| CRSWD | Circadian rhythm sleep–wake disorder |
| DSWPD | Delayed sleep–wake phase disorder |
| α-SYN | Alpha-synuclein |
| SCNA | Alpha-synuclein |
| PARK2 | Parkin 2 |
| LRRK2 | Leucine-rich repeat kinase 2 |
| PINK | PTEN-induced putative kinase |
| PRKN | Parkin RBR E3 ubiquitin-protein ligase |
| GBA | Glucocerebrosidase genes |
| ATP13A2 | ATPase type 13A2 |
| ICSD | International Classification of sleep disorders |
| CSF | Cerebrospinal fluid |
| LBD | Lewy body dementia |
| SNPc | Substantia nigra pars compacta |
| SN | Substantia nigra |
| VTA | Ventral tegmental area |
| PVN | Paraventricular nucleus |
| MPN | Medial preoptic nucleus |
| MPTP | 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine |
| DA | Dopamine |
| GABA | Gamma-aminobutyric acid |
| IL-1β | Interleukin-1β |
| TNF-α | Tumour necrosis factor- α |
| ESS | Epworth sleepiness scale |
| DaTSCAN | Dopamine transporter scan |
| CLOCK | Circadian locomotor output cycles kaput |
| HCRT2-SAP | Hypocretin-2-sap |
| CPAP | Continuous positive airway pressure |
| EMG | Electromyographic |
| MoCA | Montreal cognitive assessments |
| MMSE | Mini-mental State Examination |
| D2R | Dopamine 2 receptor |
| PLMS | Periodic leg movements in sleeping |
| ALS | Amyotrophic lateral sclerosis |
| ARAS | Ascending reticular activating system |
| SCN | Suprachiasmatic nucleus |
| PER3 | Period circadian regulator 3 |
| CRY1 | Cryptochrome |
| Nr1d1 | Nuclear receptor subfamily 1 group D member 1 |
| Nr1d2 | Nuclear receptor subfamily 1 group D member 2 |
| PDSS | Parkinson’s disease sleep scale |
| SCOPA | Scales for outcomes in PD sleep |
| PSQI | Pittsburgh sleep quality index |
| RBDSQ | RBD screening questionnaire |
| RBD-I | Innsbruck RBD inventory |
| RBDQ-HK | Hong Kong RBD questionnaire |
| RBD-IQ | RBD-single question screen |
| MSGBs | Minor salivary gland biopsies |
| CSHQ | Cleveland sleep habits questionnaire |
| MRPI | Magnetic resonance parkinsonism index |
| MRI | Magnetic resonance imaging |
| DTI | Diffusion tensor imaging |
| fMRI | Functional magnetic resonance imaging |
| FA | Fractional anisotropy |
| TMS | Transcranial magnetic stimulation |
| DAT-SPECT | Dopamine transporter single-photon emission computed tomography |
| BLT | Bright light therapy |
| CBT | Cognitive behavioral therapy |
| PBM | Photobiomodulation |
| tDCS | Transcranial direct current stimulation |
| LCIG | Levodopa/carbidopa intestinal gel |
| LCE | Levodopa/carbidopa/entacapone |
References
- Radhakrishnan, D.M.; Goyal, V. Parkinson’s disease. A review. Neurol. India 2018, 66, 26–35. [Google Scholar]
- Shulman, L.M. Understanding disability in Parkinson’s disease. Mov. Disord. 2010, 25, 131–135. [Google Scholar] [CrossRef]
- Braak, H.; Bohl, J.R.; Muller, C.M.; Rub, U.; de Vos, R.A.; Del Tredici, K. Stanley Fahn Lecture 2005: The staging procedure for the inclusion body pathology associated with sporadic Parkinson’s disease reconsidered. Mov. Disord. 2006, 21, 2042–2051. [Google Scholar] [CrossRef]
- Leng, Y.; Blackwell, T.; Cawthon, P.M.; Ancoli-Israel, S.; Stone, K.L.; Yaffe, K. Association of Circadian Abnormalities in Older Adults with an Increased Risk of Developing Parkinson’s Disease. JAMA Neurol. 2020, 77, 1270–1278. [Google Scholar] [CrossRef] [PubMed]
- Baldereschi, M.; Di Carlo, A.; Rocca, W.A.; Vanni, P.; Maggi, S.; Perissinotto, E. Parkinson’s disease and Parkinsonism in a longitudinal study: Two-fold higher incidence in men. ILSA Working Group. Italian Longitudinal Study on Aging. Neurology 2000, 55, 1358–1363. [Google Scholar] [CrossRef]
- Van Den Eeden, S.K.; Tanner, C.M.; Bernstein, A.L.; Fross, R.D.; Leimpeter, A. Incidence of Parkinson’s disease: Variation by age, gender, and race/ethnicity. Am. J. Epidemiol. 2003, 157, 1015–1022. [Google Scholar] [CrossRef] [Green Version]
- Thangaleela, S.; Sivamaruthi, B.S.; Kesika, P.; Bharathi, M.; Chaiyasut, C. Role of the Gut-Brain Axis, Gut Microbial Composition, Diet, and Probiotic Intervention in Parkinson’s Disease. Microorganisms 2022, 10, 1544. [Google Scholar] [CrossRef] [PubMed]
- Bloem, B.; Okun, M.S.; Klein, C. Parkinson’s disease. Lancet 2021, 397, 2284–2303. [Google Scholar] [CrossRef]
- Warner, T.T.; Schapira, A.H. Genetic and environmental factors in the cause of Parkinson’s disease. Ann. Neurol. 2003, 53, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Cheng, Y.; Li, C.; Shang, H. Genetic heterogeneity on sleep disorders in Parkinson’s disease: A systematic review and meta-analysis. Transl. Neurodegener. 2022, 8, 11–21. [Google Scholar]
- Cherian, A.; Divya, K.P. Genetics of Parkinson’s disease. Acta Neurol. Belg. 2020, 120, 1297–1305. [Google Scholar] [CrossRef] [PubMed]
- Polymeropoulos, M.H.; Higgins, J.J.; Golbe, L.I.; Johnson, W.G.; Ide, S.E.; Di Iorio, G.; Sanges, G.; Stenroos, E.S.; Pho, L.T.; Schaffer, A.A.; et al. Mapping of a gene for Parkinson’s disease to chromosome 4q21-q23. Science 1996, 274, 1197–1199. [Google Scholar] [CrossRef] [Green Version]
- Mata, I.F.; Leverenz, J.B.; Weintraub, D.; Trojanowski, J.Q.; Chen-Plotkin, A.; Van Deerlin, V.M.; Ritz, B.; Rausch, R.; Factor, S.A.; Wood-Siverio, C.; et al. GBA variants are associated with a distinct pattern of cognitive deficits in Parkinson’s disease. Mov. Disord. 2016, 31, 95–102. [Google Scholar] [CrossRef] [Green Version]
- Cilia, R.; Tunesi, S.; Marotta, G.; Cereda, E.; Siri, C.; Tesei, S.; Zecchinelli, A.L.; Canesi, M.; Mariani, C.B.; Meucci, N.; et al. Survival and dementia in GBA-associated Parkinson’s disease: The mutation matters. Ann. Neurol. 2016, 80, 662–673. [Google Scholar] [CrossRef] [PubMed]
- Jankovic, J. Parkinson’s disease: Clinical features and diagnosis. J. Neurol. Neurosurg. Psychiatry 2008, 79, 368–376. [Google Scholar] [CrossRef] [Green Version]
- Tolosa, E.; Garrido, A.; Scholz, S.W.; Poewe, W. Challenges in the diagnosis of Parkinson’s disease. Lancet Neurol. 2021, 20, 385–397. [Google Scholar] [CrossRef]
- Mahlknecht, P.; Stockner, H.; Marini, K.; Gasperi, A.; Djamshidian, A.; Willeit, P.; Kiechl, S.; Willeit, J.; Rungger, G.; Poewe, W.; et al. Midbrain hyperechogenicity, hyposmia, mild parkinsonian signs and risk for incident Parkinson’s disease over 10 years: A prospective population-based study. Park. Relat. Disord. 2020, 70, 51–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armstrong, M.J.; Okun, M.S. Diagnosis and Treatment of Parkinson Disease: A Review. JAMA 2020, 323, 548–560. [Google Scholar] [CrossRef]
- Postuma, R.B.; Berg, D.; Stern, M.; Poewe, W.; Olanow, C.W.; Oertel, W.; Obeso, J.; Marek, K.; Litvan, I.; Lang, A.E.; et al. MDS clinical diagnostic criteria for Parkinson’s disease. Mov. Disord. 2015, 30, 1591–1601. [Google Scholar] [CrossRef]
- Ohtsuka, C.; Sasaki, M.; Konno, K.; Koide, M.; Kato, K.; Takahashi, J.; Takahashi, S.; Kudo, K.; Yamashita, F.; Terayama, Y. Changes in substantia nigra and locus coeruleus in patients with early-stage Parkinson’s disease using neuromelanin-sensitive MR imaging. Neurosci. Lett. 2013, 541, 93–98. [Google Scholar] [CrossRef]
- Langkammer, C.; Schweser, F.; Krebs, N.; Deistung, A.; Goessler, W.; Scheurer, E.; Sommer, K.; Reishofer, G.; Yen, K.; Fazekas, F.; et al. Quantitative susceptibility mapping (QSM) as a means to measure brain iron? A post mortem validation study. Neuroimage 2012, 62, 1593–1599. [Google Scholar] [PubMed] [Green Version]
- Goetz, C.G.; Tilley, B.C.; Shaftman, S.R.; Stebbins, G.T.; Fahn, S.; Martinez-Martin, P.; Poewe, W.; Sampaio, C.; Stern, M.B.; Dodel, R. Movement Disorder Society-sponsored revision of the Unified Parkinson’s Disease Rating Scale (MDS-UPDRS): Scale presentation and clinimetric testing results. Mov. Disord. 2008, 23, 2129–2170. [Google Scholar] [CrossRef]
- Hoehn, M.M.; Yahr, M.D. Parkinsonism: Onset, progression and mortality. Neurology 1967, 17, 427–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwab, J.; England, A. Projection technique for evaluating surgery in Parkinson’s disease. In Third Symposium on Parkinson’s Disease; Gillingham, F., Donaldson, M., Eds.; E & S Livingstone: Edinburgh, Scotland, 1969; pp. 152–157. [Google Scholar]
- Stephenson, R.; Siderowf, A.; Stern, M.B. Premotor Parkinson’s disease: Clinical features and detection strategies. Mov. Disord. 2009, 24, S665–S670. [Google Scholar] [CrossRef]
- Weiner, W.J. There is no Parkinson disease. Arch. Neurol. 2008, 65, 705–708. [Google Scholar] [CrossRef] [Green Version]
- Tarazi, F.I.; Sahli, Z.T.; Wolny, M.; Mousa, S.A. Emerging therapies for Parkinson’s disease: From bench to bedside. Pharmacol. Ther. 2014, 144, 123–133. [Google Scholar] [CrossRef]
- Jagadeesan, A.J.; Murugesan, R.; Vimala Devi, S.; Meera, M.; Madhumala, G.; Vishwanathan Padmaja, M.; Ramesh, A.; Banerjee, A.; Sushmitha, S.; Khokhlov, A.N.; et al. Current trends in etiology, prognosis and therapeutic aspects of Parkinson’s disease: A review. Acta Biomed. 2017, 88, 249–262. [Google Scholar]
- Brooks, D.J.; Leinonen, M.; Kuoppamaki, M.; Nissinen, H. Five-year efficacy and safety of levodopa/DDCI and entacapone in patients with Parkinson’s disease. J. Neural. Transm. 2008, 115, 843–849. [Google Scholar] [CrossRef]
- Leegwater-Kim, J.; Waters, C. Tolcapone in the management of Parkinson’s disease. Expert Opin. Pharmacother. 2006, 7, 2263–2270. [Google Scholar] [CrossRef]
- Leinenga, G.; Langton, C.; Nisbet, R.; Götz, J. Ultrasound treatment of neurological diseases-current and emerging applications. Nat. Rev. Neurol. 2016, 12, 161–174. [Google Scholar] [CrossRef]
- Abbruzzese, G.; Marchese, R.; Avanzino, L.; Pelosin, E. Rehabilitation for Parkinson’s disease: Current outlook and future challenges. Parkinsonism Relat Disord. 2016, 1, S60–S64. [Google Scholar] [CrossRef]
- Grandner, M.A. Sleep, Health, and Society. Sleep Med. Clin. 2017, 12, 1–22. [Google Scholar] [PubMed]
- O’Brien, L.M. The neurocognitive effects of sleep disruption in children and adolescents. Child Adolesc. Psychiatr. Clin. N. Am. 2009, 18, 813–823. [Google Scholar] [CrossRef]
- Benca, R.M.; Teodorescu, M. Sleep physiology and disorders in aging and dementia. Handb. Clin. Neurol. 2019, 167, 477–493. [Google Scholar]
- Thorpy, M. Classification of Sleep Disorders. In Sleep Disorders Medicine; Chokroverty, S., Ed.; Butterworth Heinemann: Woburn, MA, USA, 1999; pp. 287–300. [Google Scholar]
- Reite, M.; Ruddy, J.; Nagel, K. Concise Guide to Evaluation and Management of Sleep Disorders; American Psychiatric Publishing: Washington, DC, USA, 2002; pp. 1–273. [Google Scholar]
- Borbely, A. Sleep: Circadian rhythm vs recovery process. In Functional States of the Brain: Their Determinants; Koukou-Lehman, M., Ed.; Elsevier/North Holland: Amsterdam, The Netherlands, 1980; pp. 151–161. [Google Scholar]
- Zee, P.; Harsanyi, K. Highlights of sleep neuroscience. In Review of Sleep Medicine; Bowman, T., Ed.; Butterworth Heinemann: Burlington, MA, USA, 2003; pp. 19–39. [Google Scholar]
- Silber, M.H. Neurologic treatment sleep disorders. Neurol. Clin. 2001, 19, 173–186. [Google Scholar] [CrossRef]
- Elsenbruch, S.; Thompson, J.J.; Hamish, M.J.; Exton, M.S.; Orr, W.C. Behavioral and physiological sleep characteristics in women with irritable bowel syndrome. Am. J. Gastroenterol. 2002, 97, 2306–2314. [Google Scholar] [CrossRef] [PubMed]
- Moldofsky, H. Management of sleep disorders in fibromyalgia. Rheum Dis. Clin. N. Am. 2002, 28, 173–186. [Google Scholar] [CrossRef]
- Chokroverty, S. Diagnosis and treatment of sleep disorders caused by comorbid disease. Neurology 2000, 54, S8–S15. [Google Scholar] [PubMed]
- Neubauer, D. Sleep problems in the elderly. Am. Fam. Physician 1999, 59, 2551–2560. [Google Scholar]
- Sateia, M.J. International classification of sleep disorders-third edition: Highlights and modifications. Chest 2014, 146, 1387–1394. [Google Scholar] [CrossRef]
- Krueger, J.M.; Rector, D.M.; Roy, S.; Van Dongen, H.P.; Belenky, G.; Panksepp, J. Sleep as a fundamental property of neuronal assemblies. Nat. Rev. Neurosci. 2008, 9, 910–919. [Google Scholar] [CrossRef] [PubMed]
- Saper, C.B.; Scammell, T.E.; Lu, J. Hypothalamic regulation of sleep and circadian rhythms. Nature 2005, 43, 1257–1263. [Google Scholar] [CrossRef] [PubMed]
- Datta, S.; Maclean, R.R. Neurobiological mechanisms for the regulation of mammalian sleep-wake behavior: Reinterpretation of historical evidence and inclusion of contemporary cellular and molecular evidence. Neurosci. Biobehav. Rev. 2007, 31, 775–824. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Jhou, T.C.; Saper, C.B. Identification of wake-active dopaminergic neurons in the ventral periaqueductal gray matter. J. Neurosci. 2006, 26, 193–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fifel, K.; Videnovic, A. Circadian and Sleep Dysfunctions in Neurodegenerative Disorders-An Update. Front. Neurosci. 2021, 14, 627330. [Google Scholar] [CrossRef]
- Chesson, A.L., Jr.; Ferber, R.A.; Fry, J.M.; Grigg-Damberger, M.; Hartse, K.M.; Hurwitz, T.D.; Johnson, S.; Kader, G.A.; Littner, M.; Rosen, G.; et al. Practice parameters for the indications for polysomnography and related procedures. Sleep 1997, 20, 406–422. [Google Scholar] [CrossRef] [Green Version]
- Chesson, A.L., Jr.; Ferber, R.A.; Fry, J.M.; Grigg-Damberger, M.; Hartse, K.M.; Hurwitz, T.D.; Johnson, S.; Kader, G.A.; Littner, M.; Rosen, G.; et al. The indications for polysomnography and related procedures. Sleep 1997, 20, 423–487. [Google Scholar] [CrossRef] [Green Version]
- Abad, V.C.; Guilleminault, C. Diagnosis and treatment of sleep disorders: A brief review for clinicians. Dialogues Clin. Neurosci. 2003, 5, 371–388. [Google Scholar] [CrossRef]
- Bohnen, N.I.; Hu, M.T.M. Sleep Disturbance as Potential Risk and Progression Factor for Parkinson’s Disease. J. Park. Dis. 2019, 9, 603–614. [Google Scholar] [CrossRef]
- Montanaro, E.; Romagnolo, A.; Fabbri, M.; Artusi, C.A.; Imbalzano, G.; Rizzone, M.G.; Lopiano, L.; Zibetti, M. Association between sleep disorders and cognitive dysfunctions in non-demented patients with advanced Parkinson’s disease. J. Neurol. 2022, 269, 1538–1545. [Google Scholar] [CrossRef]
- Stefani, A.; Högl, B. Sleep in Parkinson’s disease. Neuropsychopharmacology 2020, 45, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, A.; Kamble, N.; Yadav, R.; Stezin, A.; Pal, P.K. Abnormal Intracortical Functions in Parkinson’s Disease with Rapid Eye Movement Sleep Behaviour Disorder. Can. J. Neurol. Sci. 2022, 49, 672–677. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.E.; Jeon, B.S. Clinicalimplication of REM sleep behavior disorder in Parkinson’s disease. J. Park. Dis. 2014, 4, 237–244. [Google Scholar]
- Miglis, M.G.; Adler, C.H.; Antelmi, E.; Arnaldi, D.; Baldelli, L.; Boeve, B.F.; Cesari, M.; Dall’Antonia, I.; Diederich, N.J.; Doppler, K. Biomarkers of conversion to α-synucleinopathy in isolated rapid-eye-movement sleep behaviour disorder. Lancet Neurol. 2021, 20, 671–684. [Google Scholar] [CrossRef] [PubMed]
- Hawken, J.; Robertson, N. Sleep disorders in Parkinson’s disease. J. Neurol. 2022, 269, 6685–6687. [Google Scholar] [CrossRef]
- Wang, X.T.; Yu, H.; Liu, F.T.; Zhang, C.; Ma, Y.H.; Wang, J.; Dong, Q.; Tan, L.; Wang, H.; Yu, J.T. Associations of sleep disorders with cerebrospinal fluid α-synuclein in prodromal and early Parkinson’s disease. J. Neurol. 2022, 269, 2469–2478. [Google Scholar] [CrossRef]
- Lerche, S.; Wurster, I.; Roeben, B.; Zimmermann, M.; Riebenbauer, B.; Deuschle, C.; Hauser, A.K.; Schulte, C.; Berg, D.; Maetzler, W.; et al. Parkinson’s disease: Glucocerebrosidase 1 mutation severity is associated with CSF alpha-synuclein profiles. Mov. Disord. 2020, 35, 495–499. [Google Scholar]
- Mallampalli, M.P.; Carter, C.L. Exploring sex and gender differences in sleep health: A Society for Women’s Health Research Report. J. Womens Health 2014, 23, 553–562. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.A.; Kryger, M.H. Women and sleep. J Womens Health 2008, 17, 1189–1190. [Google Scholar]
- Zhu, K.; van Hilten, J.J.; Marinus, J. Course and risk factors for excessive daytime sleepiness in Parkinson’s disease. Parkinsonism Relat. Disord. 2016, 24, 34–40. [Google Scholar] [CrossRef]
- Abbott, R.D.; Ross, G.W.; White, L.R.; Tanner, C.M.; Masaki, K.H.; Nelson, J.S.; Curb, J.D.; Petrovitch, H. Excessive daytime sleepiness and subsequent development of Parkinson’s disease. Neurology 2005, 65, 1442–1446. [Google Scholar]
- Fifel, K.; Piggins, H.; Deboer, T. Modeling sleep alterations in Parkinson’s disease: How close are we to valid translational animal models? Sleep Med. Rev. 2016, 25, 95–111. [Google Scholar] [CrossRef]
- Yousaf, T.; Pagano, G.; Niccolini, F.; Politis, M. Excessive daytime sleepiness may be associated with caudate denervation in Parkinson disease. J. Neurol. Sci. 2018, 387, 220–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medeiros, D.C.; Lopes Aguiar, C.; Moraes, M.F.D.; Fisone, G. Sleep disorders in rodent models of Parkinson’s disease. Front. Pharmacol. 2019, 10, 1414. [Google Scholar] [CrossRef] [Green Version]
- Amara, A.W.; Chahine, L.M.; Caspell-Garcia, C.; Long, J.D.; Coffey, C.; Högl, B.; Videnovic, A.; Iranzo, A.; Mayer, G.; Foldvary-Schaefer, N.; et al. Longitudinal assessment of excessive daytime sleepiness in early Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry. 2017, 88, 653–662. [Google Scholar] [PubMed]
- Naismith, S.L.; Terpening, Z.; Shine, J.M.; Lewis, S.J.G. Neuropsychological functioning in Parkinson’s disease: Differential relationships with self-reported sleep-wake disturbances. Mov. Disord. 2011, 26, 1537–1541. [Google Scholar] [CrossRef]
- De Cock, V.C.; Vidailhet, M.; Arnulf, I. Sleep disturbances in patients with parkinsonism. Nat. Clin. Pract. Neurol. 2008, 4, 254–266. [Google Scholar] [CrossRef] [PubMed]
- Morin, C.M.; Drake, C.L.; Harvey, A.G.; Krystal, A.D.; Manber, R.; Riemann, D.; Spiegelhalder, K. Insomnia disorder. Nat. Rev. Dis. Primers 2015, 1, 15–26. [Google Scholar] [CrossRef]
- Schütz, L.; Sixel-Döring, F.; Hermann, W. Management of Sleep Disturbances in Parkinson’s Disease. J. Parkinsons Dis. 2022, 12, 2029–2058. [Google Scholar] [CrossRef]
- Tholfsen, L.K.; Larsen, J.P.; Schulz, J.; Tysnes, O.B.; Gjerstad, M.D. Changes in insomnia subtypes in early Parkinson disease. Neurology 2017, 88, 352–358. [Google Scholar] [CrossRef]
- Gerashchenko, D.; Blanco-Centurion, C.A.; Miller, J.D.; Shiromani, P.J. Insomnia following hypocretin2-saporin lesions of the substantia nigra. Neuroscience 2006, 137, 29–36. [Google Scholar] [CrossRef]
- Boucetta, S.; Salimi, A.; Dadar, M.; Jones, B.E.; Collins, D.L.; Vu, T.T.D. Structural Brain Alterations Associated with Rapid Eye Movement Sleep Behavior Disorder in Parkinson’s Disease. Sci. Rep. 2016, 6, 26782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnulf, I. REM sleep behavior disorder: Motor manifestations and pathophysiology. Mov. Disord. 2012, 27, 677–689. [Google Scholar] [CrossRef]
- Oltra, J.; Campabadal, A.; Segura, B.; Uribe, C.; Marti, M.J.; Compta, Y.; Valldeoriola, F.; Bargallo, N.; Iranzo, A.; Junque, C. Disrupted functional connectivity in PD with probable RBD and its cognitive correlates. Sci. Rep. 2021, 11, 24351. [Google Scholar] [CrossRef] [PubMed]
- Jia, X.; Fan, W.; Wang, Z.; Liu, Y.; Li, Y.; Li, H.; Li, H.; Ma, T.; Wang, J.; Yang, Q. Progressive Prefrontal Cortex Dysfunction in Parkinson’s Disease with Probable REM Sleep Behavior Disorder: A 3-Year Longitudinal Study. Front. Aging Neurosci. 2022, 1, e750767. [Google Scholar] [CrossRef]
- Chen, Y.; Xu, Q.; Wu, L.; Zhou, M.; Lin, Y.; Jiang, Y.; He, Q.; Zhao, L.; Dong, Y.; Liu, J.; et al. REM sleep behavior disorder correlates with constipation in de novo Chinese Parkinson’s disease patients. Neurol. Sci. 2023, 44, 191–197. [Google Scholar] [CrossRef]
- Burn, D.J.; Anderson, K. To sleep, perchance to dement: RBD and cognitive decline in Parkinson’s disease. Mov. Disord. 2012, 27, 671–673. [Google Scholar] [CrossRef]
- Mahmood, Z.; Van Patten, R.; Nakhla, M.Z.; Twamley, E.W.; Filoteo, J.V.; Schiehser, D.M. REM Sleep Behavior Disorder in Parkinson’s Disease: Effects on Cognitive, Psychiatric, and Functional outcomes. J. Int. Neuropsychol. Soc. 2020, 26, 894–905. [Google Scholar] [CrossRef]
- Meng, L.; Benedetti, A.; Lafontaine, A.L.; Mery, V.; Robinson, A.R.; Kimoff, J.; Gros, P.; Kaminska, M. Obstructive sleep apnea, CPAP therapy and Parkinson’s disease motor function: A longitudinal study. Park. Relat Disord. 2020, 70, 45–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ju, Y.S.; Videnovic, A.; Vaughn, B.V. Comorbid Sleep Disturbances in Neurologic Disorders. Continuum 2017, 23, 1117–1131. [Google Scholar] [CrossRef]
- Spicuzza, L.; Caruso, D.; Di Maria, G. Obstructive sleep apnoea syndrome and its management. Ther. Adv. Chronic. Dis. 2015, 6, 273–285. [Google Scholar] [CrossRef] [Green Version]
- Hwang, O. Role of oxidative stress in Parkinson’s disease. Exp. Neurobiol. 2013, 22, 11–17. [Google Scholar] [CrossRef] [Green Version]
- Crosta, F.; Desideri, G.; Marini, C. Obstructive sleep apnea syndrome in Parkinson’s disease and other parkinsonisms. Funct. Neurol. 2017, 32, 137–141. [Google Scholar] [CrossRef] [PubMed]
- Elfil, M.; Bahbah, E.I.; Attia, M.M.; Eldokmak, M.; Koo, B.B. Impact of Obstructive Sleep Apnea on Cognitive and Motor Functions in Parkinson’s Disease. Mov. Disord. 2021, 36, 570–580. [Google Scholar] [CrossRef] [PubMed]
- Sun, A.P.; Liu, N.; Zhang, Y.S.; Zhao, H.Y.; Liu, X.L. The relationship between obstructive sleep apnea and Parkinson’s disease: A systematic review and meta-analysis. Neurol. Sci. 2020, 41, 1153–1162. [Google Scholar] [CrossRef]
- Bahia, C.M.C.S.; Pereira, J.S.; Lopes, A.J. Laryngopharyngeal motor dysfunction and obstructive sleep apnea in Parkinson’s disease. Sleep Breath. 2019, 23, 543–550. [Google Scholar] [CrossRef]
- Shen, Y.; Shen, Y.; Dong, Z.F.; Pan, P.L.; Shi, H.C.; Liu, C.F. Obstructive sleep apnea in Parkinson’s disease: A study in 239 Chinese patients. Sleep Med. 2020, 67, 237–243. [Google Scholar] [CrossRef]
- Endo, T.; Matsumura, R.; Tokuda, I.T.; Yoshikawa, T.; Shigeyoshi, Y.; Node, K.; Sakoda, S.; Akashi, M. Bright light improves sleep in patients with Parkinson’s disease: Possible role of circadian restoration. Sci. Rep. 2020, 10, 79–82. [Google Scholar] [CrossRef]
- Setó-Salvia, N.; Pagonabarraga, J.; Houlden, H.; Pascual-Sedano, B.; Dols-Icardo, O.; Tucci, A.; Paisán-Ruiz, C.; Campolongo, A.; Antón-Aguirre, S.; Martín, I.; et al. Glucocerebrosidase mutations confer a greater risk of dementia during Parkinson’s disease course. Mov. Disord. 2012, 27, 393–399. [Google Scholar] [CrossRef]
- Xiao, C.M.; Zhuang, Y.C. Efect of health Baduanjin Qigong for mild to moderate Parkinson’s disease. Geriatr. Gerontol. Int. 2016, 16, 909–911. [Google Scholar] [CrossRef] [PubMed]
- Amara, A.W.; Wood, K.H.; Joop, A.; Memon, R.A.; Pilkington, J.; Tuggle, S.C.; Reams, J.; Barrett, M.J.; Edwards, D.A.; Weltman, A.L.; et al. Randomized, controlled trial of exercise on objective and subjective sleep in Parkinson’s disease. Mov. Disord. 2020, 35, 947–958. [Google Scholar] [CrossRef]
- Zibetti, M.; Rizzone, M.; Merola, A.; Angrisano, S.; Rizzi, L.; Montanaro, E.; Cicolin, A.; Lopiano, L. Sleep improvement with levodopa/carbidopa intestinal gel infusion in Parkinson disease. Acta Neurol. Scand. 2013, 127, 28–32. [Google Scholar] [CrossRef] [PubMed]
- Serin, Y.; Tek, N.K. Effect of Circadian Rhythm on Metabolic Processes and the Regulation of Energy Balance. Ann. Nutr. Metab. 2019, 74, 322–330. [Google Scholar] [CrossRef] [PubMed]
- Mery, V.P.; Gros, P.; Anne-Louise, L.; Robinson, A.; Andrea Benedetti, R.; Kimoff, J.; Kaminska, M. Reduced cognitive function in patients with Parkinson disease and obstructive sleep apnea. Neurology 2017, 88, 1120–1128. [Google Scholar] [CrossRef]
- Alcalay, R.N.; Mejia-Santana, H.; Tang, M.X.; Rakitin, B.; Rosado, L.; Ross, B.; Verbitsky, M.; Kisselev, S.; Louis, E.D.; Comella, C.L.; et al. Self-report of cognitive impairment and mini-mental state examination performance in PRKN, LRRK2, and GBA carriers with early onset Parkinson’s disease. J. Clin. Exp. Neuropsychol. 2010, 32, 775–779. [Google Scholar] [CrossRef] [PubMed]
- You, S.; Jeon, S.M.; Do, S.Y.; Cho, Y.W. Restless Legs Syndrome in Parkinson’s Disease Patients: Clinical Features Including Motor and Nonmotor Symptoms. J. Clin. Neurol. 2019, 15, 321–327. [Google Scholar] [CrossRef]
- Fereshtehnejad, S.M.; Shafieesabet, M.; Shahidi, G.A.; Delbari, A.; Lökk, J. Restless legs syndrome in patients with Parkinson’s disease: A comparative study on prevalence, clinical characteristics, quality of life and nutritional status. Acta Neurol. Scand. 2015, 131, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Oh, Y.S.; Kim, J.S.; Park, I.S.; Song, I.U.; Son, Y.M.; Park, J.W.; Yang, D.W.; Kim, H.T.; Lee, K.S. Association between nocturnal/supine hypertension and restless legs syndrome in patients with Parkinson’s disease. J. Neurol. Sci. 2014, 344, 186–189. [Google Scholar] [CrossRef]
- Gjerstad, M.D.; Tysnes, O.B.; Larsen, J.P. Increased risk of leg motor restlessness but not RLS in early Parkinson disease. Neurology 2011, 77, 1941–1946. [Google Scholar] [CrossRef]
- Allen, R.P.; Picchietti, D.L.; Garcia-Borreguero, D.; Ondo, W.G.; Walters, A.S.; Winkelman, J.W.; Zucconi, M.; Ferri, R.; Trenkwalder, C.; Lee, H.B. International Restless Legs Syndrome Study Group Restless legs syndrome/Willis-Ekbom disease diagnostic criteria: Updated International Restless Legs Syndrome Study Group (IRLSSG) consensus criteria–history, rationale, description, and significance. Sleep Med. 2014, 15, 860–873. [Google Scholar] [CrossRef]
- Trenkwalder, C.; Paulus, W.; Walters, A.S. The restless leg syndrome. Lancet Neurol. 2005, 4, 465–475. [Google Scholar] [CrossRef]
- Mitchell, U.H.; Obray, J.D.; Hunsaker, E.; Garcia, B.T.; Clarke, T.J.; Hope, S.; Steffensen, S.C. Peripheral Dopamine in Restless Legs Syndrome. Front. Neurol. 2018, 15, 149–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kushida, C.; Martin, M.; Nikam, P.; Blaisdell, B.; Wallenstein, G.; Ferini-Strambi, L.; Ware, J.E., Jr. Burden of restless leg syndrome on health-related quality of life. Qual. Life Res. 2007, 16, 617–624. [Google Scholar] [CrossRef]
- Michaud, M.; Dumont, M.; Selmaoui, B.; Paquet, J.; Fantini, M.L.; Montplaisir, J. Circadian rhythm of restless leg syndrome: Relationship with biological markers. Ann. Neurol. 2004, 55, 372–380. [Google Scholar] [CrossRef]
- Barriere, G.; Cazalets, J.R.; Bioulac, B.; Tison, F.; Ghorayeb, I. The restless leg syndrome. Prog. Neurobiol. 2005, 77, 139–165. [Google Scholar] [CrossRef]
- Maestri, M.; Romigi, A.; Schirru, A.; Fabbrini, M.; Gori, S.; Bonuccelli, U.; Bonanni, E. Excessive daytime sleepiness and fatigue in neurological disorders. Sleep Breath 2020, 24, 413–424. [Google Scholar] [CrossRef] [PubMed]
- Verbaan, D.; van Rooden, S.M.; Visser, M.; Marinus, J.; van Hilten, J.J. Nighttime sleep problems and daytime sleepiness in Parkinson’s disease. Mov. Disord. 2008, 23, 35–41. [Google Scholar] [CrossRef]
- Wen, M.C.; Ng, S.Y.E.; Heng, H.S.E.; Chao, Y.X.; Chan, L.L.; Tan, E.K.; Tan, L.C. Neural substrates of excessive daytime sleepiness in early drug naïve Parkinson’s: A resting state functional MRI study. Park. Relat. Disord. 2016, 24, 63–68. [Google Scholar] [CrossRef]
- Sulzer, D.; Surmeier, D.J. Neuronal vulnerability, pathogenesis, and Parkinson’s disease. Mov. Disord. 2013, 28, 715–724. [Google Scholar] [CrossRef] [PubMed]
- Swick, T.J. Parkinson’s disease and sleep/wake disturbances. Park. Dis. 2012, 15, e205471. [Google Scholar] [CrossRef]
- Diederich, N.J.; Comella, C.L. Sleep disturbances in Parkinson’s disease. In Sleep and Movement Disorders; Chokroverty, S., Hening, W.A., Walters, A.S., Eds.; Oxford University Press: Hong Kong, China, 2003; pp. 478–488. [Google Scholar]
- Burgess, C.; Lai, D.; Siegel, J.; Peever, J. An endogenous glutamatergic drive onto somatic motoneurons contributes to the stereotypical pattern of muscle tone across the sleep-wake cycle. J. Neurosci. 2008, 28, 4649–4660. [Google Scholar] [CrossRef] [Green Version]
- Boeve, B.F. REM sleep behavior disorder: Updated review of the core features, the REM sleep behavior disorder-neurodegenerative disease association, evolving concepts, controversies, and future directions. Ann. N. Y. Acad. Sci. 2010, 1184, 15–54. [Google Scholar] [CrossRef] [Green Version]
- Espana, R.A.; Scammell, T.E. Sleep neurobiology from a clinical perspective. Sleep 2011, 34, 845–858. [Google Scholar]
- Diederich, N.J.; McIntyre, D.J. Sleep disorders in Parkinson’s disease: Many causes, few therapeutic options. J. Neurol. Sci. 2012, 314, 9–12. [Google Scholar] [CrossRef]
- Torterolo, P.; Lagos, P.; Monti, J. Melanin-concentrating hormone: A new sleep factor. Front. Neurol. 2011, 2, 14. [Google Scholar] [CrossRef] [Green Version]
- Meloni, M.; Figorilli, M.; Carta, M.; Tamburrino, L.; Cannas, A.; Sanna, F.; Defazio, G.; Puligheddu, M. Preliminary finding of a randomized, double-blind, placebo-controlled, crossover study to evaluate the safety and efficacy of 5-hydroxytryptophan on REM sleep behavior disorder in Parkinson’s disease. Sleep Breath. 2022, 26, 1023–1031. [Google Scholar] [CrossRef]
- Hipólide, D.C.; Moreira, K.M.; Barlow, K.B.; Wilson, A.A.; Nobrega, J.N.; Tufik, S. Distinct effects of sleep deprivation on binding to norepinephrine and serotonin transporters in rat brain. Prog. Neuropsychopharmacol. Biol. Psychiatry. 2005, 29, 297–303. [Google Scholar] [CrossRef]
- Lima, M.M. Sleep disturbances in Parkinson’s disease: The contribution of dopamine in REM sleep regulation. Sleep Med. Rev. 2013, 17, 367–375. [Google Scholar] [CrossRef]
- Détári, L.; Rasmusson, D.D.; Semba, K. The role of basal forebrain neurons in tonic and phasic activation of the cerebral cortex. Prog. Neurobiol. 1999, 58, 249–277. [Google Scholar] [CrossRef]
- Weber, F.; Dan, Y. Circuit-based interrogation of sleep control. Nature 2016, 538, 51–59. [Google Scholar] [CrossRef] [Green Version]
- Sherin, J.E.; Shiromani, P.J.; McCarley, R.W.; Saper, C.B. Activation of ventrolateral preoptic neurons during sleep. Science 1996, 271, 216–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, H.; McGinty, D.; Guzman-Marin, R.; Chew, K.T.; Stewart, D.; Szymusiak, R. Activation of c-fos in GABAergic neurones in the preoptic area during sleep and in response to sleep deprivation. J. Physiol. 2004, 556, 935–946. [Google Scholar] [CrossRef] [PubMed]
- Feenstra, M.G.; Botterblom, M.H.; Mastenbroek, S. Dopamine and noradrenaline efflux in the prefrontal cortex in the light and dark period: Effects of novelty and handling and comparison to the nucleus accumbens. Neuroscience 2000, 100, 741–748. [Google Scholar] [CrossRef]
- Léna, I.; Parrot, S.; Deschaux, O.; Muffat-Joly, S.; Sauvinet, V.; Renaud, B.; Suaud-Chagny, M.F.; Gottesmann, C. Variations in extracellular levels of dopamine, noradrenaline, glutamate, and aspartate across the sleep-wake cycle in the medial prefrontal cortex and nucleus accumbens of freely moving rats. J. Neurosci. Res. 2005, 81, 891–899. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Ren, R.; Sanford, L.D.; Yang, L.; Zhou, J.; Tan, L.; Li, T.; Zhang, J.; Wing, Y.K.; Shi, J.; et al. Sleep in Parkinson’s disease: A systematic review and meta-analysis of polysomnographic findings. Sleep Med. Rev. 2020, 51, e101281. [Google Scholar] [CrossRef]
- Steele, T.A.; St Louis, E.K.; Videnovic, A.; Auger, R.R. Circadian Rhythm Sleep-Wake Disorders: A Contemporary Review of Neurobiology, Treatment, and Dysregulation in Neurodegenerative Disease. Neurotherapeutics 2021, 18, 53–74. [Google Scholar] [CrossRef]
- Welsh, D.K.; Takahashi, J.S.; Kay, S.A. Suprachiasmatic nucleus: Cell autonomy and network properties. Annu. Rev. Physiol. 2010, 72, 551–577. [Google Scholar] [CrossRef] [Green Version]
- Sun, S.Y.; Chen, G.H. Treatment of Circadian Rhythm Sleep-Wake Disorders. Curr. Neuropharmacol. 2022, 20, 1022–1034. [Google Scholar] [CrossRef]
- Wyatt, J.K. Delayed sleep phase syndrome: Pathophysiology and treatment options. Sleep 2004, 27, 1195–1203. [Google Scholar] [CrossRef]
- Boscolo, R.A.; Esteves, A.M.; de Santana, M.G.; Viana, V.A.R.; Grassmann, V.; Tufik, S.; de Mello, M.T. Is there an association between body composition, basal metabolic rate, and sleep in elderly patients with and without obstructive sleep apnea? Sleep Sci. 2013, 6, 129–134. [Google Scholar]
- Harvey, J.; Plante, A.E.; Meredith, A.L. Ion channels controlling circadian rhythms in suprachiasmatic nucleus excitability. Physiol. Rev. 2020, 100, 1415–1454. [Google Scholar] [CrossRef]
- Borbély, A.A.; Daan, S.; Wirz-Justice, A.; Deboer, T. The two-process model of sleep regulation: A reappraisal. J. Sleep Res. 2016, 25, 131–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crowley, S.J.; Acebo, C.; Carskadon, M.A. Sleep, circadian rhythms, and delayed phase in adolescence. Sleep Med. 2007, 8, 602–612. [Google Scholar] [CrossRef] [PubMed]
- Videnovic, A.; Lazar, A.S.; Barker, R.A.; Overeem, S. ‘The clocks that time us’—Circadian rhythms in neurodegenerative disorders. Nat. Rev. Neurol. 2014, 10, 683–693. [Google Scholar] [CrossRef] [Green Version]
- Hunt, J.; Coulson, E.J.; Rajnarayanan, R.; Oster, H.; Videnovic, A.; Rawashdeh, O. Sleep and circadian rhythms in Parkinson’s disease and preclinical models. Mol. Neurodegener. 2022, 17, 2. [Google Scholar] [CrossRef]
- Sohail, S.; Yu, L.; Schneider, J.A.; Bennett, D.A.; Buchman, A.S.; Lim, A.S.P. Sleep fragmentation and Parkinson’s disease pathology in older adults without Parkinson’s disease. Mov. Disord. 2017, 32, 1729–1737. [Google Scholar] [CrossRef]
- Lauretti, E.; Di Meco, A.; Merali, S.; Pratico, D. Circadian rhythm dysfunction: A novel environmental risk factor for Parkinson’s disease. Mol. Psychiatry 2017, 22, 280–286. [Google Scholar] [CrossRef]
- Bordet, R.; Devos, D.; Brique, S.; Touitou, Y.; Guieu, J.D.; Libersa, C.; Destée, A. Study of circadian melatonin secretion pattern at different stages of Parkinson’s disease. Clin. Neuropharmacol. 2003, 26, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Breen, D.P.; Vuono, R.; Nawarathna, U.; Fisher, K.; Shneerson, J.N.; Reddy, A.B.; Barker, R.A. Sleep and circadian rhythm regulation in early Parkinson disease. JAMA Neurol. 2014, 71, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Grippo, R.M.; Purohit, A.M.; Zhang, Q.; Zweifel, L.S.; Guler, A.D. Direct Midbrain Dopamine Input to the Suprachiasmatic Nucleus Accelerates Circadian Entrainment. Curr. Biol. 2017, 27, 2465–2475. [Google Scholar] [CrossRef]
- Winder-Rhodes, S.E.; Evans, J.R.; Ban, M.; Mason, S.L.; Williams-Gray, C.H.; Foltynie, T.; Duran, R.; Mencacci, N.E.; Sawcer, S.J.; Barker, R.A. Glucocerebrosidase mutations influence the natural history of Parkinson’s disease in a community-based incident cohort. Brain 2013, 136, 392–399. [Google Scholar] [CrossRef] [PubMed]
- Malek, N.; Weil, R.S.; Bresner, C.; Lawton, M.A.; Grosset, K.A.; Tan, M. Features of GBA-associated Parkinson’s disease at presentation in the UK Tracking Parkinson’s study. J. Neurol. Neurosurg. Psychiatry 2018, 89, 702–709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krohn, L.; Ruskey, J.A.; Rudakou, U.; Leveille, E.; Asayesh, F.; Hu, M.T.M.; Arnulf, I.; Dauvilliers, Y.; Högl, B.; Stefani, A.; et al. GBA variants in REM sleep behavior disorder: A multicenter study. Neurology 2020, 95, 1008–1016. [Google Scholar] [CrossRef]
- Tayebi, N.; Walker, J.; Stubblefield, B.; Orvisky, E.; LaMarca, M.E.; Wong, K.; Rosenbaum, H.; Schiffmann, R.; Bembi, B.; Sidranksy, E. Gaucher disease with parkinsonian manifestations: Does glucocerebrosidase deficiency contribute to a vulnerability to Parkinsonism? Mol. Genet. Metab. 2003, 79, 104–109. [Google Scholar] [CrossRef]
- Boeve, B.F.; Silber, M.H.; Ferman, T.J.; Lin, S.C.; Benarroch, E.E.; Schmeichel, A.M.; Ahlskog, J.E.; Caselli, R.J.; Jacobson, S.; Sabbagh, M.; et al. Clinicopathologic correlations in 172 cases of rapid eye movement sleep behavior disorder with or without a coexisting neurologic disorder. Sleep Med. 2013, 14, 754–762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gan-Or, Z.; Girard, S.L.; Noreau, A.; Leblond, C.S.; Gagnon, J.F.; Arnulf, I.; Mirachi, C.; Dauvilliers, Y.; Desautels, A.; Mitterling, T.; et al. Parkinson’s disease genetic loci in rapid eye movement sleep behavior disorder. J. Mol. Neurosci. 2015, 56, 617–622. [Google Scholar] [CrossRef] [PubMed]
- Simuni, T.; Brumm, M.C.; Uribe, L.; Caspell-Garcia, C.; Cofey, C.S.; Siderowf, A.; Alcalay, R.N.; Trojanowski, J.Q.; Shaw, L.M.; Seibyl, J.; et al. Clinical and dopamine transporter imaging characteristics of leucine rich repeat kinase 2 (LRRK2) and glucosylceramidase beta (GBA) Parkinson’s disease participants in the Parkinson’s progression markers initiative: A cross-sectional study. Mov. Disord. 2020, 35, 833–844. [Google Scholar] [CrossRef] [Green Version]
- Yahalom, G.; Greenbaum, L.; Israeli-Korn, S.; Fay-Karmon, T.; Livneh, V.; Ruskey, J.A.; Roncière, L.; Alam, A.; Gan-Or, Z.; Hassin-Baer, S. Carriers of both GBA and LRRK2 mutations, compared to carriers of either, in Parkinson’s disease: Risk estimates and genotype-phenotype correlations. Park. Relat. Disord. 2019, 62, 179–184. [Google Scholar] [CrossRef]
- Kumru, H.; Santamaria, J.; Tolosa, E.; Valldeoriola, F.; Muñoz, E.; Marti, M.J.; Iranzo, A. Rapid eye movement sleep behavior disorder in parkinsonism with parkin mutations. Ann. Neurol. 2004, 56, 599–603. [Google Scholar] [CrossRef]
- Gelegen, C.; Cash, D.; Ilic, K.; Sander, M.; Kim, E.; Simmons, C.; Bernanos, M.; Lama, J.; Randall, K.; Brown, J.T.; et al. Relevance of sleep and associated structural changes in GBA1 mouse to human rapid eye movement behavior disorder. Sci. Rep. 2022, 12, 73–79. [Google Scholar] [CrossRef]
- Kalia, L.V.; Lang, A.E.; Hazrati, L.N.; Fujioka, S.; Wszolek, Z.K.; Dickson, D.W.; Ross, O.A.; Van Deerlin, V.M.; Trojanowski, J.Q.; Hurtig, H.I.; et al. Clinical correlations with Lewy body pathology in LRRK2-related Parkinson disease. JAMA Neurol. 2015, 72, 100–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ehrminger, M.; Leu-Semenescu, S.; Cormier, F.; Corvol, J.C.; Vidailhet, M.; Debellemaniere, E.; Brice, A.; Arnulf, I. Sleep aspects on video-polysomnography in LRRK2 mutation carriers. Mov. Disord. 2015, 30, 1839–1843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pont-Sunyer, C.; Iranzo, A.; Gaig, C.; Fernández-Arcos, A.; Vilas, D.; Valldeoriola, F.; Compta, Y.; Fernández-Santiago, R.; Fernández, M.; Bayés, A.; et al. Sleep Disorders in Parkinsonian and Nonparkinsonian LRRK2 Mutation Carriers. PLoS ONE 2015, 15, e0132368. [Google Scholar] [CrossRef] [PubMed]
- Ouled Amar Bencheikh, B.; Ruskey, J.A.; Arnulf, I.; Dauvilliers, Y.; Monaca, C.C.; De Cock, V.C.; Gagnon, J.F.; Spiegelman, D.; Hu, M.T.M.; Högl, B.; et al. LRRK2 protective haplotype and full sequencing study in REM sleep behavior disorder. Park. Relat. Disord. 2018, 52, 98–101. [Google Scholar] [CrossRef] [PubMed]
- Limousin, N.; Konofal, E.; Karroum, E.; Lohmann, E.; Theodorou, I.; Dürr, A.; Arnulf, I. Restless legs syndrome, rapid eye movement sleep behavior disorder, and hypersomnia in patients with two parkin mutations. Mov. Disord. 2009, 24, 1970–1976. [Google Scholar] [CrossRef]
- Valadas, J.S.; Esposito, G.; Vandekerkhove, D.; Miskiewicz, K.; Deaulmerie, L.; Raitano, S.; Seibler, P.; Klein, C.; Verstreken, P. ER lipid defects in neuro peptidergic neurons impair sleep patterns in Parkinson’s disease. Neuron 2018, 98, 1155–1169. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Ruskey, J.A.; Arnulf, I.; Dauvilliers, Y.; Hu, M.T.M.; Högl, B.; Leblond, C.S.; Zhou, S.; Ambalavanan, A.; Ross, J.P.; et al. Full sequencing and haplotype analysis of MAPT in Parkinson’s disease and rapid eye movement sleep behavior disorder. Mov. Disord. 2018, 33, 1016–1020. [Google Scholar] [CrossRef]
- Ling, Y.; Zhu, J.; Yan, F.; Tse, L.A.; Kinra, S.; Jiang, M. Sleep behaviors and Parkinson’s disease: A bidirectional Mendelian randomization analysis. Behav. Brain Res. 2023, 441, 114281. [Google Scholar] [CrossRef]
- Cullell, N.; Cárcel-Márquez, J.; Gallego-Fábrega, C.; Muiño, E.; Llucià-Carol, L.; Lledós, M.; Amaut, K.E.U.; Krupinski, J.; Fernández-Cadenas, I. Sleep/wake cycle alterations as a cause of neurodegenerative diseases: A Mendelian randomization study. Neurobiol. Aging. 2021, 106, 320.e1–320.e12. [Google Scholar] [CrossRef]
- Li, J.; Zhao, L.; Ding, X.; Cui, X.; Qi, L.; Chen, Y. Obstructive sleep apnea and the risk of Alzheimer’s disease and Parkinson disease: A Mendelian randomization study OSA, Alzheimer’s disease and Parkinson disease. Sleep Med. 2022, 97, 55–63. [Google Scholar] [CrossRef]
- Kim, H.J.; Kim, H.J.; Lee, J.Y.; Yun, J.Y.; Kim, S.Y.; Park, S.S.; Jeon, B.S. Phenotype analysis in patients with early onset Parkinson’s disease with and without parkin mutations. J. Neurol. 2011, 258, 2260–2267. [Google Scholar] [CrossRef] [PubMed]
- Belarbi, S.; Hecham, N.; Lesage, S.; Kediha, M.I.; Smail, N.; Benhassine, T.; Ysmail-Dahlouk, F.; Lohman, E.; Benhabyles, B.; Hamadouche, T.; et al. LRRK2 G2019S mutation in Parkinson’s disease: A neuropsychological and neuropsychiatric study in a large Algerian cohort. Park. Relat Disord. 2010, 16, 676–679. [Google Scholar] [CrossRef] [PubMed]
- Gan-Or, Z.; Mirelman, A.; Postuma, R.B.; Arnulf, I.; Bar-Shira, A.; Dauvilliers, Y.; Desautels, A.; Gagnon, J.F.; Leblond, C.S.; Frauscher, B.; et al. GBA mutations are associated with rapid eye movement sleep behavior disorder. Ann. Clin. Transl. Neurol. 2015, 2, 941–945. [Google Scholar] [CrossRef] [PubMed]
- Alcalay, R.N.; Caccappolo, E.; Mejia-Santana, H.; Tang, M.; Rosado, L.; Orbe Reilly, M.; Ruiz, D.; Ross, B.; Verbitsky, M.; Kisselev, S.; et al. Cognitive performance of GBA mutation carriers with early-onset PD: The CORE-PD study. Neurology 2012, 78, 1434–1440. [Google Scholar] [CrossRef] [Green Version]
- Trenkwalder, C.; Kohnen, R.; Högl, B.; Metta, V.; Sixel-Döring, F.; Frauscher, B.; Hülsmann, J.; Martinez-Martin, P.; Chaudhuri, K.L. Parkinson’s Disease Sleep scale—Validation of the revised version PDSS-2. Mov. Disord. 2011, 26, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Martin, P.; Visser, M.; Rodriguez-Blazquez, C.; Marinus, J.; Chaudhuri, K.R.; van Hilten, J.J. SCOPA-sleep and PDSS: Two scales for assessment of sleep disorder in Parkinson’s disease. Mov. Disord. 2008, 23, 1681–1688. [Google Scholar] [CrossRef]
- Shrivastava, D.; Jung, S.; Saadat, M.; Sirohi, R.; Crewson, K. How to interpret the results of a sleep study. J. Community Hosp. Intern. Med. Perspect. 2014, 4, 24983. [Google Scholar] [CrossRef] [Green Version]
- Prabhudesai, P.; Patankar, M.; Vardhan, A. Sleep Study Interpretation in Obstructive Sleep Apnea. Int. J. Head Neck Surg. 2019, 10, 42–46. [Google Scholar] [CrossRef]
- Carskadon, M.A.; Dement, W.C. Normal Human Sleep: An Overview. In Principles and Practice of Sleep Medicine; Kryger, M.H., Roth, T., Dement, W.C., Eds.; Elsevier Saunders: St. Louis, MI, USA, 2011; pp. 16–26. [Google Scholar]
- Schmidt, C.; Peigneux, P.; Cajochen, C. Age-related changes in sleep and circadian rhythms: Impact on cognitive performance and underlying neuroanatomical networks. Front. Neurol. 2012, 3, 118. [Google Scholar] [CrossRef] [Green Version]
- Kapur, V.K.; Auckley, D.H.; Choudhuri, S.; Kuhlmann, D.C.; Mehra, R.; Ramar, K.; Harrod, C.G. Clinical practice guideline for diagnostic testing for adult obstructive sleep apnea: An American Academy of Sleep Medicine clinical practice guideline. J. Clin. Sleep Med. 2017, 13, 479–504. [Google Scholar] [CrossRef]
- Collop, N.A.; Anderson, W.M.; Boehlecke, B.; Claman, D.; Goldberg, R.; Gottlieb, D.J.; Hudgel, D.; Sateia, M.; Schwab, R. Clinical guidelines for the Portable Monitoring Task Force of the American Academy of Sleep Medicine. Clinical guidelines for the use of unattended portable monitors in the diagnosis of obstructive sleep apnea in adult patients. J. Clin. Sleep Med. 2007, 3, 737–747. [Google Scholar] [PubMed]
- Chou, K.L.; Amick, M.M.; Brandt, J.; Camicioli, R.; Frei, K.; Gitelman, D.; Goldman, J.; Growdon, J.; Hurtig, H.I.; Levin, B.; et al. A recommended scale for cognitive screening in clinical trials of Parkinson’s disease. Mov. Disord. 2010, 25, 2501–2507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stefani, A.; Heidbreder, A.; Brandauer, E.; Guaita, M.; Neier, L.M.; Mitterling, T.; Santamaria, J.; Iranzo, A.; Videnovic, A.; Trenkwalder, C.; et al. Screening for idiopathic REM sleep behavior disorder: Usefulness of actigraphy. Sleep 2018, 41, zsy053. [Google Scholar] [CrossRef]
- Stiasny-Kolster, K.; Mayer, G.; Schäfer, S.; Möller, J.C.; Heinzel-Gutenbrunner, M.; Oertel, W.H. The REM sleep behavior disorder screening questionnaire–a new diagnostic instrument. Mov. Disord. 2007, 22, 2386–2393. [Google Scholar] [CrossRef]
- Frauscher, B.; Ehrmann, L.; Zamarian, L.; Auer, F.; Mitterling, T.; Gabelia, D.; Brandauer, E.; Delazer, M.; Poewe, W.; Högl, B. Validation of the Innsbruck REM sleep behavior disorder inventory. Mov. Disord. 2012, 27, 1673–1678. [Google Scholar] [CrossRef]
- Li, S.X.; Wing, Y.K.; Lam, S.P.; Zhang, J.; Yu, M.W.M.; Ho, C.K.W.; Tsoh, J.; Mok, V. Validation of a new REM sleep behavior disorder questionnaire (RBDQ-HK). Sleep Med. 2010, 11, 43–48. [Google Scholar] [CrossRef]
- Boeve, B.F.; Molano, J.R.; Ferman, T.J.; Lin, S.C.; Bieniek, K.; Tippmann-Peikert, M.; Boot, B.; St. Louis, E.K.; Knopman, D.S.; Petersen, R.C.; et al. Validation of the Mayo Sleep Questionnaire to screen for REM sleep behavior disorder in an aging and dementia cohort. Sleep Med. 2011, 12, 445–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Postuma, R.B.; Arnulf, I.; Hogl, B.; Iranzo, A.; Miyamoto, T.; Dauvilliers, Y.; Oertel, W.; Ju, Y.E.; Puligheddu, M.; Jennum, P.; et al. A single-question screen for rapid eye movement sleep behavior disorder: A multicenter validation study. Mov. Disord. 2012, 27, 913–916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mangone, G.; Houot, M.; Gaurav, R.; Boluda, S.; Pyatigorskaya, N.; Chalancon, A.; Seilhean, D.; Prigent, A.; Lehéricy, S.; Arnulf, I.; et al. Relationship between Substantia Nigra Neuromelanin Imaging and Dual Alpha-Synuclein Labeling of Labial Minor in Salivary Glands in Isolated Rapid Eye Movement Sleep Behavior Disorder and Parkinson’s Disease. Genes 2022, 24, 1715. [Google Scholar] [CrossRef]
- Chong-Wen, W.; Sha-Sha, L.; Xu, E. Predictors of rapid eye movement sleep behavior disorder in patients with Parkinson’s disease based on random forest and decision tree. PLoS ONE 2022, 17, e0269392. [Google Scholar] [CrossRef]
- Senthilvel, E.; Auckley, D.; Dasarathy, J. Evaluation of sleep disorders in the primary care setting: History taking compared to questionnaires. J. Clin. Sleep Med. 2011, 15, 41–48. [Google Scholar] [CrossRef]
- Jiang, Y.; An, H.; Xi, Q.; Yang, W.; Xie, H.; Li, Y.; Huang, D. Diffusion Tensor Imaging Reveals Deep Brain Structure Changes in Early Parkinson’s Disease Patients with Various Sleep Disorders. Brain Sci. 2022, 30, 463. [Google Scholar] [CrossRef]
- Iranzo, A.; Molinuevo, J.L.; Santamaría, J.; Serradell, M.; Martí, M.J.; Valldeoriola, F.; Tolosa, E. Rapid-eye-movement sleep behaviour disorder as an early marker for a neurodegenerative disorder: A descriptive study. Lancet Neurol. 2006, 5, 572–577. [Google Scholar] [CrossRef] [PubMed]
- MacKinnon, C.D.; Gilley, E.A.; Weis-McNulty, A.; Simuni, T. Pathways mediating abnormal intracortical inhibition in Parkinson’s disease. Ann. Neurol. 2005, 58, 516–524. [Google Scholar] [CrossRef]
- Pierpaoli, C.; Jezzard, P.; Basser, P.J.; Barnett, A.; Chiro, G.D. Diffusion tensor MR imaging of the human brain. Radiology 1996, 201, 637–648. [Google Scholar] [CrossRef] [PubMed]
- Basser, P.J.; Pierpaoli, C. Microstructural and physiological features of tissues elucidated by quantitative-diffusion-tensor MRI. J. Magn. Reson. B 1996, 111, 209–219. [Google Scholar] [CrossRef]
- Darcourt, J.; Booij, J.; Tatsch, K.; Varrone, A.; Vander Borght, T.; Kapucu, O.L.; Någren, K.; Nobili, F.; Walker, Z.; Laere, K.V. EANM procedure guidelines for brain neurotransmission SPECT using (123) I-labelled dopamine transporter ligands, version 2. Eur. J. Nucl. Med. Mol. Imaging 2010, 37, 443–450. [Google Scholar] [CrossRef]
- Urso, D.; Nigro, S.; Tafuri, B.; Gnoni, V.; Filardi, M.; De Blasi, R.; Ray Chaudhuri, K.; Logroscino, G. Magnetic Resonance Parkinsonism Index Is Associated with REM Sleep Behavior Disorder in Parkinson’s Disease. Brain Sci. 2022, 31, 202. [Google Scholar] [CrossRef]
- Borsoi, L.; Armeni, P.; Donin, G.; Costa, F.; Ferini-Strambi, L. The invisible costs of obstructive sleep apnea (OSA): Systematic review and cost-of-illness analysis. PLoS ONE 2022, 17, e0268677. [Google Scholar] [CrossRef]
- Wenning, G.K.; Litvan, I.; Tolosa, E. Milestones in atypical and secondary Parkinsonisms. Mov. Disord. 2011, 26, 1083–1095. [Google Scholar] [CrossRef]
- Höglinger, G.U.; Respondek, G.; Stamelou, M.; Kurz, C.; Josephs, K.A.; Lang, A.E.; Mollenhauer, B.; Müller, U.; Nilsson, C.; Whitwell, J.L.; et al. Clinical diagnosis of progressive supranuclear palsy: The movement disorder society criteria. Mov. Disord. 2017, 32, 853–864. [Google Scholar] [CrossRef] [PubMed]
- Morin, C.M.; Belleville, G.; Bélange, L.; Ivers, H. Insomnia severity index: Psychometric indicators to detect insomnia cases and evaluate treatment response. Sleep 2011, 34, 601–608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.H.; Wang, Y.Q.; Ye, S.Q.; Cheng, Y.G.; Chen, Y.; Feng, X.Z. The efects of group-based versus individual-based tai chi training on nonmotor symptoms in patients with mild to moderate Parkinson’s disease: A randomized controlled pilot trial. Park. Dis. 2017, 2017, 2856–2867. [Google Scholar]
- Wassom, D.J.; Lyons, K.E.; Pahwa, R.; Liu, W. Qigong exercise may improve sleep quality and gait performance in Parkinson’s disease: A pilot study. Int. J. Neurosci. 2015, 125, 578–584. [Google Scholar] [CrossRef]
- Auger, R.R.; Burgess, H.J.; Emens, J.S.; Deriy, L.V.; Thomas, S.M.; Sharkey, K.M. Clinical practice guideline for the treatment of intrinsic circadian rhythm sleep-wake disorders: Advanced sleep-wake phase disorder (ASWPD), delayed sleep-wake phase disorder (DSWPD), non-24-hour sleep-wake rhythm disorder (N24SWD), and irregular sleep-wake rhythm disorder (ISWRD). An American academy of sleep medicine clinical practice guideline. J. Clin. Sleep Med. 2015, 11, 1199–1236. [Google Scholar]
- Rios Romenets, S.; Creti, L.; Fichten, C.; Bailes, S.; Libman, E.; Pelletier, A.; Postuma, R.B. Doxepin and cognitive behavioural therapy for insomnia in patients with Parkinson’s disease—A randomized study. Park. Relat. Disord. 2013, 19, 670–675. [Google Scholar] [CrossRef] [PubMed]
- Gradisar, M.; Dohnt, H.; Gardner, G.; Paine, S.; Starkey, K.; Menne, A.; Slater, A.; Wright, H.; Hudson, J.L.; Weaver, E.; et al. A randomized controlled trial of cognitive-behavior therapy plus bright light therapy for adolescent delayed sleep phase disorder. Sleep 2011, 34, 1671–1680. [Google Scholar] [CrossRef] [Green Version]
- Videnovic, A.; Klerman, E.B.; Wang, W.; Marconi, A.; Kuhta, T.; Zee, P.C. Timed Light Therapy for Sleep and Daytime Sleepiness Associated with Parkinson Disease: A Randomized Clinical Trial. JAMA Neurol. 2017, 74, 411–418. [Google Scholar] [CrossRef]
- Hamblin, M.R. Shining light on the head: Photobiomodulation for brain disorders. BBA Clin. 2016, 6, 113–124. [Google Scholar] [CrossRef] [Green Version]
- Tian, F.; Hase, S.N.; Gonzalez-Lima, F.; Liu, H. Transcranial laser stimulation improves human cerebral oxygenation. Lasers Surg. Med. 2016, 48, 343–349. [Google Scholar] [CrossRef] [Green Version]
- El Massri, N.; Lemgruber, A.P.; Rowe, I.J.; Moro, C.; Torres, N.; Reinhart, F.; Chabrol, C.; Benabid, A.L.; Mitrofanis, J. Photobiomodulation-induced changes in a monkey model of Parkinson’s disease: Changes in tyrosine hydroxylase cells and GDNF expression in the striatum. Exp. Brain Res. 2017, 235, 1861–1874. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Lv, Q.K.; Xie, W.Y.; Gong, S.Y.; Zhuang, S.; Liu, J.Y.; Mao, C.J.; Liu, C.F. Circadian disruption and sleep disorders in neurodegeneration. Transl. Neurodegener. 2023, 13, 8–12. [Google Scholar] [CrossRef]
- Zhang, X.; Zhuang, S.; Wu, J.; Wang, L.; Mao, C.; Chen, J.; Liu, C.F. Effects of repetitive transcranial magnetic stimulation over right dorsolateral prefrontal cortex on excessive daytime sleepiness in patients with Parkinson’s disease. Sleep Med. 2022, 100, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Howell, M.J.; Arneson, P.A.; Schenck, C.H. A novel therapy for REM sleep behavior disorder (RBD). J. Clin. Sleep Med. 2011, 7, 639–644. [Google Scholar] [CrossRef] [Green Version]
- Stocchi, F.; Torti, M. Adjuvant therapies for Parkinson’s disease: Critical evaluation of safinamide. Drug Des. Devel. Ther. 2016, 10, 609–618. [Google Scholar] [CrossRef] [Green Version]
- Paff, M.; Loh, A.; Sarica, C.; Lozano, A.M.; Fasano, A. Update on Current Technologies for Deep Brain Stimulation in Parkinson’s Disease. J. Mov. Disord. 2020, 13, 185–198. [Google Scholar] [CrossRef] [PubMed]
- Rye, D.B.; Jankovic, J. Emerging views of dopamine in modulating sleep/wake state from an unlikely source: PD. Neurology 2002, 58, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Park, K.W.; Jo, S.; Lee, S.H.; Hwang, Y.S.; Lee, D.; Ryu, H.S.; Chung, S.J. Therapeutic Effect of Levodopa/Carbidopa/Entacapone on Sleep Disturbance in Patients with Parkinson’s Disease. J. Mov. Disord. 2020, 13, 205–212. [Google Scholar] [CrossRef]
- Kunz, D.; Mahlberg, R. A two-part, double-blind, placebo-controlled trial of exogenous melatonin in REM sleep behaviour disorder. J. Sleep Res. 2010, 19, 591–596. [Google Scholar] [CrossRef]
- McCarter, S.J.; Boswell, C.L.; St Louis, E.K.; Dueffert, L.G.; Slocumb, N.; Boeve, B.F.; Silber, M.H.; Olson, E.J.; Tippmann-Peikert, M. Treatment outcomes in REM sleep behavior disorder. Sleep Med. 2013, 14, 237–242. [Google Scholar] [CrossRef] [Green Version]
- Shin, C.; Park, H.; Lee, W.W.; Kim, H.J.; Kim, H.J.; Jeon, B. Clonazepam for probable REM sleep behavior disorder in Parkinson’s disease: A randomized placebo-controlled trial. J. Neurol. Sci. 2019, 401, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Menza, M.; Dobkin, R.D.; Marin, H.; Gara, M.; Bienfait, K.; Dicke, A.; Comella, C.L.; Cantor, C.; Hyer, L. Treatment of insomnia in Parkinson’s disease: A controlled trial of eszopi-clone and placebo. Mov. Disord. 2010, 25, 1708–1714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, S.A.; Kayumov, L.; Shapiro, C.M. Antidepressant action of melatonin in the treatment of delayed sleep phase syndrome. Sleep Med. 2010, 11, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Sletten, T.L.; Magee, M.; Murray, J.M.; Gordon, C.J.; Lovato, N.; Kennaway, D.J.; Gwini, S.M.; Bartlett, D.J.; Lockley, S.W.; Lack, L.C.; et al. Efficacy of melatonin with behavioural sleep-wake scheduling for delayed sleep-wake phase disorder: A double-blind, randomised clinical trial. PLoS Med. 2018, 15, e1002587. [Google Scholar]
- Gerrard, P.; Malcolm, R. Mechanisms of modafinil: A review of current research. Neuropsychiatr. Dis. Treat. 2007, 3, 349–364. [Google Scholar]
- Wisor, J.P.; Eriksson, K.S. Dopaminergic-adrenergic interactions in the wake promoting mechanism of modafinil. Neuroscience 2005, 132, 1027–1034. [Google Scholar] [CrossRef]
- Wisor, J.P.; Nishino, S.; Sora, I.; Uhl, G.H.; Mignot, E.; Edgar, D.M. Dopaminergic role in stimulant-induced wakefulness. J. Neurosci. 2001, 21, 1787–1794. [Google Scholar] [CrossRef]
- Trotti, L.M.; Bliwise, D.L. Treatment of the sleep disorders associated with Parkinson’s disease. Neurotherapeutics 2014, 11, 68–77. [Google Scholar] [CrossRef] [Green Version]
- O’Callaghan, F.; Muurlink, O.; Reid, N. Effects of caffeine on sleep quality and daytime functioning. Risk Manag. Healthc. Policy 2018, 11, 263–271. [Google Scholar] [CrossRef] [Green Version]
- Postuma, R.B.; Lang, A.E.; Munhoz, R.P.; Charland, K.; Pelletier, A.; Moscovich, M.; Filla, L.; Zanatta, D.; Rios Romenets, S.; Altman, R.; et al. Caffeine for treatment of Parkinson disease: A randomized controlled trial. Neurology 2012, 79, 651–658. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Borreguero, D.; Silber, M.H.; Winkelman, J.W.; Högl, B.; Bainbridge, J.; Buchfuhrer, M.; Hadjigeorgiou, G.; Inoue, Y.; Manconi, M.; Oertel, W.; et al. Guidelines for the first-line treatment of restless legs syndrome/Willis–Ekbom disease, prevention, and treatment of dopaminergic augmentation: A combined task force of the IRLSSG, EURLSSG, and the RLS-foundation. Sleep Med. 2016, 21, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winkelman, J.W.; Armstrong, M.J.; Allen, R.P.; Chaudhuri, K.R.; Ondo, W.; Trenkwalder, C.; Zee, P.C.; Gronseth, G.S.; Gloss, D.; Zesiewicz, T. Practice guideline summary: Treatment of restless legs syndrome in adults: Report of the Guideline Development, Dissemination, and Implementation Subcommittee of the American Academy of Neurology. Neurology 2016, 87, 2585–2593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winkelmann, J.; Allen, R.P.; Högl, B.; Inoue, Y.; Oertel, W.; Salminen, A.V.; Winkelman, J.W.; Trenkwalder, C.; Sampaio, C. Treatment of restless legs syndrome: Evidence-based review and implications for clinical practice. Mov. Disord. 2018, 33, 1077–1091. [Google Scholar] [CrossRef] [Green Version]
- Allen, R.P.; Picchietti, D.L.; Auerbach, M.; Cho, Y.W.; Connor, J.R.; Earley, C.J.; Garcia-Borreguero, D.; Kotagal, S.; Manconi, M.; Ondo, W.; et al. Evidence-based and consensus clinical practice guidelines for the iron treatment of restless legs syndrome/ Willis–Ekbom disease in adults and children: An IRLSSG task force report. Sleep Med. 2018, 41, 27–44. [Google Scholar] [CrossRef]
- Kakar, R.S.; Kushida, C.A. Ropinirole in the treatment of restless legs syndrome. Expert Rev. Neurother. 2005, 5, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Allen, R. Dopamine and iron in restless legs syndrome (RLS) pathophysiology. Sleep Med. 2004, 5, 385–391. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | No. of Subjects | Age | Study Conditions | Study Findings | Ref. |
|---|---|---|---|---|---|
| Rapid-eye-movement sleep behavior disorder (RBD) | |||||
| 1 | PD-RBD (n = 20); PD without RBD (n = 20) | Age- and gender-matched with controls | PD patients with and without RBD were evaluated for neurophysiological abnormalities with single- and paired-pulse TMS, and RMT, CMCT, SP, SICI, and ICF were recorded. | PD-RBD patients showed reduced intracortical facilitation, reduced glutaminergic transmission, and enhanced GABAergic transmission. | [57] |
| 2 | PD subjects (n = 360); prodromal PD subjects ((n = 46); subjects displaying RBD behaviors); controls (n = 169) | Mean age: 61.24 years for controls, 61.31 years for PD patients, 68.20 years for prodromal PD subjects | The association of RBD and the level of CSF alpha-synuclein was evaluated. | PD individuals with probable RBD had significantly lower alpha-synuclein levels in CSF. No significant association between daytime sleepiness and CSF alpha-synuclein levels was found. | [61] |
| 3 | PDGBA (n = 80); PDGBA-wildtype (n = 80); controls (n = 39) | 59 ± 12 years for controls, 64 ± 10 years for PD-GBA patients, 66 ± 10 years for PD-GBA risk-variant patients | PD patients with and without GBA1 mutation were screened for total CSF alpha-synuclein. | PDGBA patients showed early-onset cognitive decline, high chance of RBD development, and reduced total CSF alpha-synuclein. | [62] |
| 4 | Idiopathic RBD patients (n = 1061); controls (n = 3086) | - | The role of GBA variants in the risk of developing idiopathic RBD and development of neurodegeneration was studied. | Individuals with GBA variants had increased risk of idiopathic RBD, and the rate of neurodegeneration also increased in GBA-variant individuals. | [93] |
| 5 | RBD patients (n = 261); controls (n = 379) | 67.2 ± 9.2 years for RBD patients, 58.9 ± 12.3 years for controls | RBD patients and controls were screened for PD-associated SNPs and their effects on RBD and progression of synucleinopathies. | Data from 56 RBD patients showed that 19 developed neurodegeneration during the follow-up period, 9 were diagnosed with PD, and 10 had DLB. The SCARB2 rs6812193 SNP and the MAPT rs12185268 SNP were associated with RBD, and the carriers of these SNPs progressed to synucleinopathies. A few patients with the USP25 rs2823357 SNP demonstrated faster progression to synucleinopathy from RBD. | [94] |
| Excessive daytime sleepiness (EDS) | |||||
| 6 | PD patients (n = 400) | - | Five-year hospital-based cohort study to analyze the risk factors of EDS in PD using SCOPA-SLEEP-DS scores. | The proportion of EDS in PD increased with longer follow-up. In total, 43% of PD patients had EDS at baseline. A total of 46% of patients without EDS at baseline developed EDS during follow-up. | [65] |
| 7 | Unmedicated PD patients (n = 423); Controls (n = 196) | - | EDS was assessed using the ESS. Clinical, biological, and imaging variables were assessed. | EDS was developed during the follow-up. EDS in PD was associated with autonomic dysfunction, depression, and anxiety. EDS was also associated with presynaptic dopaminergic dysfunction. | [70] |
| 8 | Idiopathic PD patients (n = 101); unmedicated (n = 12); Patients with levodopa monotherapy (n = 29); Patients with dopamine agonist monotherapy (n = 5); Patients with levodopa plus adjuvant agent therapy (n = 55); Patients, who taking anti-depressants (n = 26), Patients, who taking benzodiazepines (n = 15) | 67.3 ± 8.0 years for controls, 65.9 ± 9.5 years for all PD patients, 67.9 ± 9.0 years for PD-RBD patients, and 62.8 ± 9.6 years for PD-non-RBD patients | All patients’ neuropsychological functioning was assessed with standard tests using Wechsler Adult Intelligent Scale-III, Cambridge Neuropsychological Test Automated Battery, and Wechsler Memory Scale-III; daytime sleepiness was assessed with the SCOPA-day, and EDS, with the ESS. | Patients with greater levodopa dose equivalents showed greater nocturnal disturbances and daytime sleepiness but not RBD symptoms. EDS was a significant predictor of slow processing speed, working memory, and verbal frequency performance. | [71] |
| 9 | Patients with EDS receiving stable dopaminergic therapy without cognitive impairment or primary sleep disorder (n = 31) | - | Safety and efficacy of light therapy on EDS were evaluated. Participants were randomly assigned in a 1:1 ratio to receive bright light and dim light (as control) twice daily in 1-hour intervals for 14 days. | Bright-light therapy significantly improved EDS scores. Bright and dim light improved sleep quality based on the Pittsburg Sleep Quality Index. Bright-light therapy improved mean sleep metrics and sleep quality. | [95] |
| Insomnia | |||||
| 10 | Drug naïve PD patients (n = 182); Controls (n = 202). | 67.5 ± 9.2 years for patients and 66.2± 9.6 for controls | Participants were assessed for insomnia with the Stavanger Sleepiness Questionnaire and Parkinson’s Disease Sleep Scale before treatment initiation and after 1, 3, and 5 years. | Insomnia prevalence was not higher in PD patients at the 5-year follow-up. Sleep-maintenance problems increased, and solitary-sleep-initiation problems decreased after 5 years. | [75] |
| 11 | PD Patients with insomnia randomized for three-arm six-week randomized pilot study (n = 18); Placebo (n = 6); CBT with BLT (n = 6); Doxepin (10 mg/daily) (n = 6). | - | This three-arm, six-week randomized pilot study assessed non-pharmacological and pharmacological treatment outputs in PD patients with insomnia. Sleep outcomes were measured using insomnia scales, sleep diaries, actigraphy, and clinical global impression. | Doxepin improved the scores in Insomnia Severity Index, SCOPA-night, and Pittsburgh Sleep Quality Index-Sleep Disturbances Subscale. Doxepin reduced the score on the Fatigue Severity Scale and improved the scores on the Montreal Cognitive Assessment. Non-pharmacological treatment reduced the Insomnia Severity Index. | [96] |
| 12 | Patients under 65 received 3 mg eszopiclone or matching placebo at night. Patients 65 or older received 2 mg of eszopiclone or placebo at night (n = 30). | 35 to 85 years | Patients were equally randomized to eszopiclone and placebo for 6 weeks. Patients with other primary sleep disorders were excluded. Total sleep time, wake after sleep onset, and number of awakenings were measured. | Significant differences were found in the number of awakenings, sleep quality, and wake after sleep onset, favoring eszopiclone. Eszopiclone did not increase the total sleep time but improved the sleep quality compared with the placebo group. | [97] |
| Obstructive sleep apnea (OSA) | |||||
| 13 | PD patients with OSA (n = 67). | 64.7 years | Patients were treated with CPAP, and motor symptoms were assessed using the MDS-UPDRS and TUG with a follow-up time of 3, 6, and 12 months. | CPAP treatment stabilized the motor function over 12 months of follow-up treatment. | [84] |
| 14 | PD patients (n = 239); PD (n = 66) with OSA including mild (n = 34), moderate (n = 16), severe sleep apnea (n = 16); PD without OSA (n = 173). | n = 66 PD patients with OSA had a mean age of 45 years; n = 173 PD patients without OSA had a mean age of 81 years | Participants underwent assessments to examine disease severity, polysomnography characteristics, and non-motor symptoms. | Binary logistic regression analysis showed that age and male gender were risk factors for OSA. RBD and higher levodopa equivalent dose were protective factors against OSA. Thus, OSA in PD was lower in PD patients with RBD. And OSA could increase excessive day sleeping in PD patients. | [92] |
| 15 | Subjects were divided into OSA and non-OSA groups (n = 95). | 69.1 ± 3.4 years | Subjects were evaluated with protocols that included polysomnography, BMR, and body composition. BMR was evaluated in the morning after polysomnography. | Patients with OSA had higher values in weight, fat mass, arousal, and AHI. The OSA group had lower REM sleep. | [98] |
| 16 | Idiopathic PD patients (n = 67) | Mean age of 64.4 years | Idiopathic PD patients were recruited from a movement-disorder clinic. OSA was defined using the AHI. The H&Y Scale and MDS-UPDRS were used to assess PD severity. And NMSs were assessed with the MoCA, ESS, Fatigue Severity Scale, Apathy Scale, BDI, HDAS, and PDSS. | OSA in PD was associated with sleepiness and cognitive dysfunction. Treatment for OSA could improve excessive sleepiness and cognitive dysfunction in PD. | [99] |
| Restless legs syndrome (RLS) | |||||
| 17 | PD patients with parkin mutations (n = 11); Sex matched IPD patients (n = 11) | PD patients with parkin mutations were aged 35–60 years and were from seven families; IPD patients were aged 51–65 years. | Patients with parkin mutations and IPD patients were compared to evaluate the sleep–wake phenotype using the UPDRS, ESS, MMSE, and RLS Study Group Rating Scale; a sleep specialist interview; and video-polysomnography. | Parkin patients showed sleep phenotypes like insomnia and RLS, and neuronal loss. Parkin-mutation patients had all polygraphical abnormalities reported in IPD. Two Parkin siblings had central hypersomnia and normal night-time sleep. | [100] |
| 18 | PD patients (n = 74); Drug-I patients (n = 16); Patients treated with levodopa/aromatic L-amino acid decarboxylase inhibitor, monoamine oxidase B inhibitor and amantadine (n = 58) | 65.5 ± 9.1 years | Patients were assessed for RLS based on the diagnostic criteria of the International RLS Study Group revised in 2003. | The frequency of RLS in PD patients was higher than the general RLS population. PD patients with RLS had worse sleep quality, anxiety, depression, and autonomic disturbances. | [101] |
| 19 | Idiopathic PD patients (n = 108); Matched controls (n = 424) | ≥35 years | This comparative study analyzed the prevalence of RLS in PD patients and investigated the quality of life, nutritional status, and clinical characteristics using IRLSSG, PD severity scales, psychiatric features, nutritional status, and quality of life. | RLS was significantly more common in IPD patients than controls. PD patients with RLS suffered from more anxiety, and worse nutritional status and quality of life. RLS was found to be correlated with psychiatric problems and cognitive impairment. | [102] |
| 20 | PD patients (n = 225) | - | RLS was diagnosed using IRLSSG criteria. Orthostatic vital signs and blood pressure were monitored. | PD patients with RLS showed nocturnal/supine hypertension and fluctuations in blood pressure and some sleep dysfunctions. RLS could be a determinant for neurocirculatory abnormalities. | [103] |
| 21 | Drug naïve early, unmedicated PD patients (n = 200); Controls (n = 173) | Age- and gender-matched controls | Subjects were assessed for RLS with structured interviews, clinical examinations, and blood samples. RLS was diagnosed using IRLSSG criteria. | PD patients reported leg restlessness, which was 3-fold greater in patients than in controls, which could indicate a relative risk for RLS. | [104] |
| S. No. | No. of Subjects | Age, Gender, and Other Details of the Subjects | Study Conditions | Study Findings | Ref. |
|---|---|---|---|---|---|
| 1 | Patients with early-onset PD (n = 124); Patients completed the assessments (n = 84). | Age at onset of 34.1 ± 5.7 years. Male-to-female ratio was 66:58. | Native Korean patients with early-onset PD clinically examined for PD according to UKPDSBB criteria. | Among 84 patients, 23 carried Parkin mutations. Further, 1 patient was homozygote; 13 patients were heterozygotes; and 6 patients were single heterozygotes. Among 13 heterozygotes, 11 had exon rearrangements, 2 carried point mutations (p.Gly284Arg with exon 2-3-4 del, p.Leu272Ile, and p.Ala398Thr), and 1 had a frameshift mutation (p.His200ThrfsX6 with exon 4 del). | [167] |
| 2 | Patients with familial parkinsonism (n = 106) | The two groups (G2019S-mutation carriers and non-carriers) of patients had similar age at onset and age at examination. | Patients were clinically and genetically evaluated for LRRK2 G2019S mutation and underwent cognitive and neuropsychiatric testing. | G2019S mutation was identified in 34 out of 106 patients. A total of 71 patients gave consent for cognitive and neuropsychiatric testing. Among 71 patients (45 men, 26 women), 23 (11 men, 12 women) were G2019S-mutation carriers, and 48 (34 men, 14 women) were non-carriers. Cognitive functions were similarly affected in both carriers and non-carriers. Behavioral abnormalities, depression, and hallucinations were frequent in LRRK2 G2019S carriers. | [168] |
| 3 | Idiopathic RBD patients of European ancestry (n = 265); Controls of European origin, including 189 controls who did not have PD at recruitment (n = 2240); 120 subjects formed an independent PD cohort, including 120 Ashkenazi-Jewish patients from Tel-Aviv, Israel. | The cohort had 79.6% men, age at enrollment of 67.2 ± 9.8 years. | Patients were diagnosed according to the International Classification of Sleep Disorders (ICSD-2) criteria. The independent PD cohort was analyzed for founder GBA mutations, and 5 were screened for RBD using the RBD Screening Questionnaire (RBDSQ). | GBA mutations were significantly more frequent among RBD patients. In the cohort of 120 patients, 19 were GBA mutation carriers. Of these 19, 9 patients had RBD. The results demonstrate that rapid-eye-movement sleep behavior disorder is associated with GBA mutations. | [169] |
| 4 | RBD patients (n = 261); Controls (n = 379) | Of the RBD patients, 80% were men, aged 67.2 ± 9.2 years; data on gender and age were available for 250 and 142 individuals, respectively. Of the controls, 50% were men, aged 58.9 ± 12.3 years, and data on gender and age available were for 369 and 183 individuals, respectively. | All the RBD patients were recruited through the International RBD Study Group and were diagnosed with RBD according to the International Classification of Sleep Disorders (ICSD-2) criteria. | Before adjusting for sex and age variables, the SCARB2 rs6812193 SNP was associated with RBD with odds ratio of 0.67 and 95% confidence interval. After adjusting for sex and age variables, the SCARB2 rs6812193 SNP and the MAPT rs12185268 SNP were associated with RBD with odds ratio of 0.23 and 95% confidence interval. Data for progression from RBD to synucleinopathies (n = 56) showed that 7 carriers of the MAPT rs12185268 SNP progressed to synucleinopathy, 11 carriers of the SCARB2 rs6812193 SNP progressed to synucleinopathy, a few patients with the USP25 rs2823357 SNP in the recessive model demonstrated faster progression from RBD to any synucleinopathy. | [152] |
| 5 | Individuals with early-onset PD. GBA carrier (n = 33); Non-carriers (n = 114). | Age at onset of PD < 51 years | Participants were screened for mutations in SNCA, PARKIN, PINK-1, DJ-1, LRRK2, and GBA. Given the higher frequency of GBA mutations among Ashkenazi Jews, participants who self-reported Ashkenazi-Jewish ancestry were further screened for an additional 6 common GBA mutations (V394L, D409G, A456P, R496H, 84GG, and exon 2 IVS2 + 1) with direct sequencing. | Among 147 participants, 33 were GBA carriers, while 60 did not have any mutations in the other genes tested. Among 33 GBA mutation carriers, 7 were heterozygous L444P carriers; a total of 16 heterozygous were N370S carriers; only 1 was N370S homozygote; in total, 2 were 84GG carriers; and 1 was an R496H carrier. And 3 had both GBA and PARKIN mutations, while 3 had both GBA and LRRK2 G2019S mutations. | [170] |
| 6 | PD patients (n = 1893) | A total of 142 PD cases had a variant detected in the GBA gene, and their mean age was 65.6 years. Cases carrying GD-causing variants in the GBA gene were younger (mean age of 62.9 years and non-carriers mean age of 67.6 years. | The GBA gene was fully sequenced, and cognitive and motor features were assessed using the MoCA and MDS-UPDRS part 3. | In total, 48 were heterozygous carriers for Gaucher’s disease, while 117 had non-synonymous variants, previously associated with PD, and patients carried variants of the GBA gene of unknown significance. L444P was the most common pathogenic GBA mutation. Patients with GBA mutations were more likely to present with PIGD and showed advanced scores on the H&Y Scale. In the early disease stage, there were no differences in cognitive function between carriers and non-carriers of GBA mutation. | [148] |
| 7 | Patients with iRBD (n = 1061); Controls (n = 3086) | Controls had a mean age of 46.5 ± 15.0 years, and 46.6% were men; patients had a mean age of 60.5 ± 9.9 years, and 81% were men. | GBA was fully sequenced using molecular inversion probes and Sanger sequencing. | In total, 9.5% of iRBD patients and 4.1% of controls had GBA variants. The mild p.N370S variant of GBA was found in 1.9% of iRBD patients and 0.5% of controls. Severe variants, like p.L444P, p.D409H, p.W291X, p.H255Q, and p.R131L, were found in 0.6% of iRBD patients and in 0.03% of controls. The p.E326K variant was associated with iRBD (4.4%) and controls (1.5%). The carrier frequency of variant p.T369M was only slightly elevated in iRBD patients (1.9%) compared with controls (1.7%) without any statistical significance. | [149] |
| 8 | n = 158 LRRK2 patients, n = 80 GBA and n = 361 sporadic-PD participants | Mean age: LRRK2 PD participants, 63.8 ± 9.2; GBA PD, 62.7 ± 9.9; sPD, 63.8 ± 9.7. | Participants were evaluated annually with a battery of motor and non-motor scales and 123-I Ioflupane DAT imaging. Genetic testing for LRRK2 (for G2019S, R1441G/C, and N1437H mutations) and GBA (N370S, L483P, L444P, IVS2 + 1, and 84GG mutations) were performed. | Compared with sPD patients, GBA PD patients were likely women. In total, 89% of GBA patients carried the N370S mutation. Among LRRK2 patients, 89% carried the G2019S mutation. DAT imaging results showed higher specific binding ratios in contralateral putamen and putamen in both LRRK2 and GBA PD patients compared with sPD patients. LRRK2 had higher hyposmic scores. GBA PD patients had higher RBDSQ scores. MoCA scores or neurocognitive battery scores showed no difference among the groups. | [153] |
| 9 | PD patients categorized into four groups: LRRK2 PD (n = 80); GBA PD (n = 78); GBA-LRRK2 PD (n =12); and mutation-negative PD (n = 80). | - | Odds ratios were estimated using published data on frequencies of GBA and LRRK2 mutations. Clinical data were collected from medical records. | Probable RBD was significantly more common in GBA-PD than in LRRK2-PD. None of the GBA-LRRK2-PD patients reported RBD. Compared with LRRK2 and MNPD participants, GBA PD patients showed significantly high dementia. And psychosis was the most common in GBA-PD and the least common in LRRK2-GBA-PD. | [154] |
| 10 | 2764 unrelated consecutive PD patients, of which 123 were GBA carriers (67 mild-p.N370S and 56 severe p.L444P) and 2641 were non-carriers. | - | Brain perfusion and DAT imaging analysis were performed, including dementia and DLB. | GBA carriers were at greater risk of dementia and death than PD non carriers. GBA carriers had worse motor symptoms, and reduced posterior parietal and occipital cortical synaptic activity, and nigrostriatal function than PD non-carriers. | [14] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thangaleela, S.; Sivamaruthi, B.S.; Kesika, P.; Mariappan, S.; Rashmi, S.; Choeisoongnern, T.; Sittiprapaporn, P.; Chaiyasut, C. Neurological Insights into Sleep Disorders in Parkinson’s Disease. Brain Sci. 2023, 13, 1202. https://doi.org/10.3390/brainsci13081202
Thangaleela S, Sivamaruthi BS, Kesika P, Mariappan S, Rashmi S, Choeisoongnern T, Sittiprapaporn P, Chaiyasut C. Neurological Insights into Sleep Disorders in Parkinson’s Disease. Brain Sciences. 2023; 13(8):1202. https://doi.org/10.3390/brainsci13081202
Chicago/Turabian StyleThangaleela, Subramanian, Bhagavathi Sundaram Sivamaruthi, Periyanaina Kesika, Subramanian Mariappan, Subramanian Rashmi, Thiwanya Choeisoongnern, Phakkharawat Sittiprapaporn, and Chaiyavat Chaiyasut. 2023. "Neurological Insights into Sleep Disorders in Parkinson’s Disease" Brain Sciences 13, no. 8: 1202. https://doi.org/10.3390/brainsci13081202