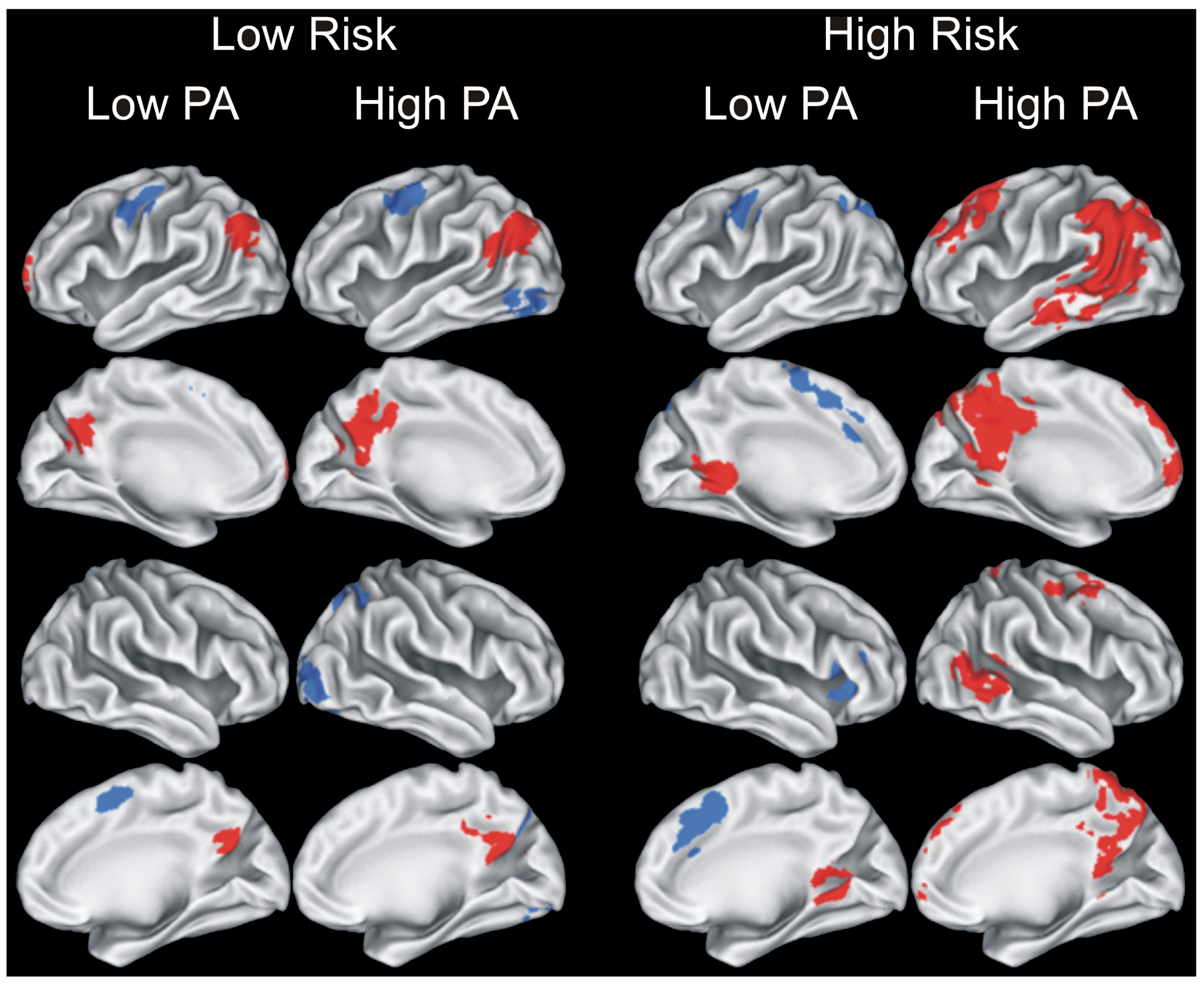
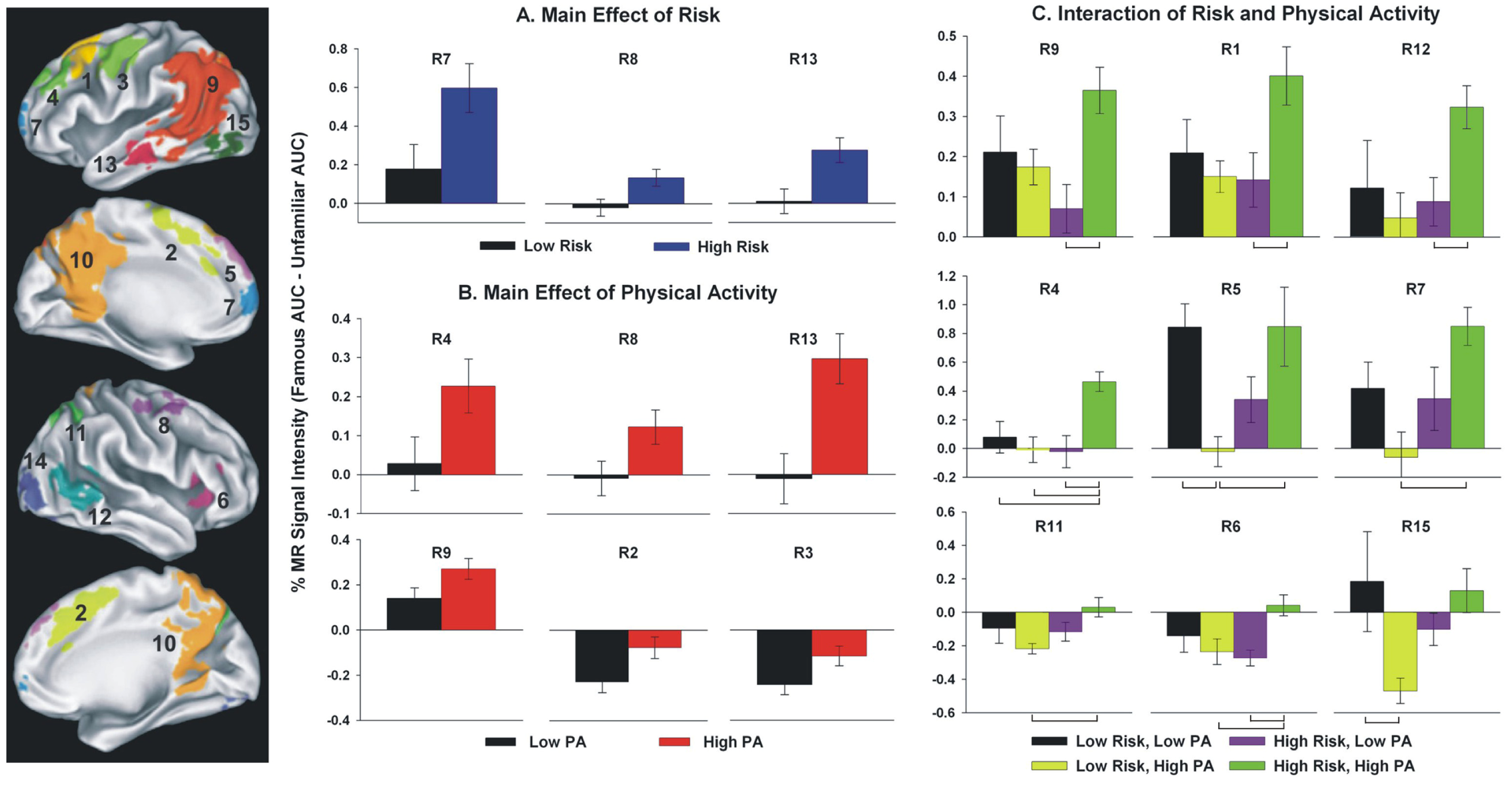
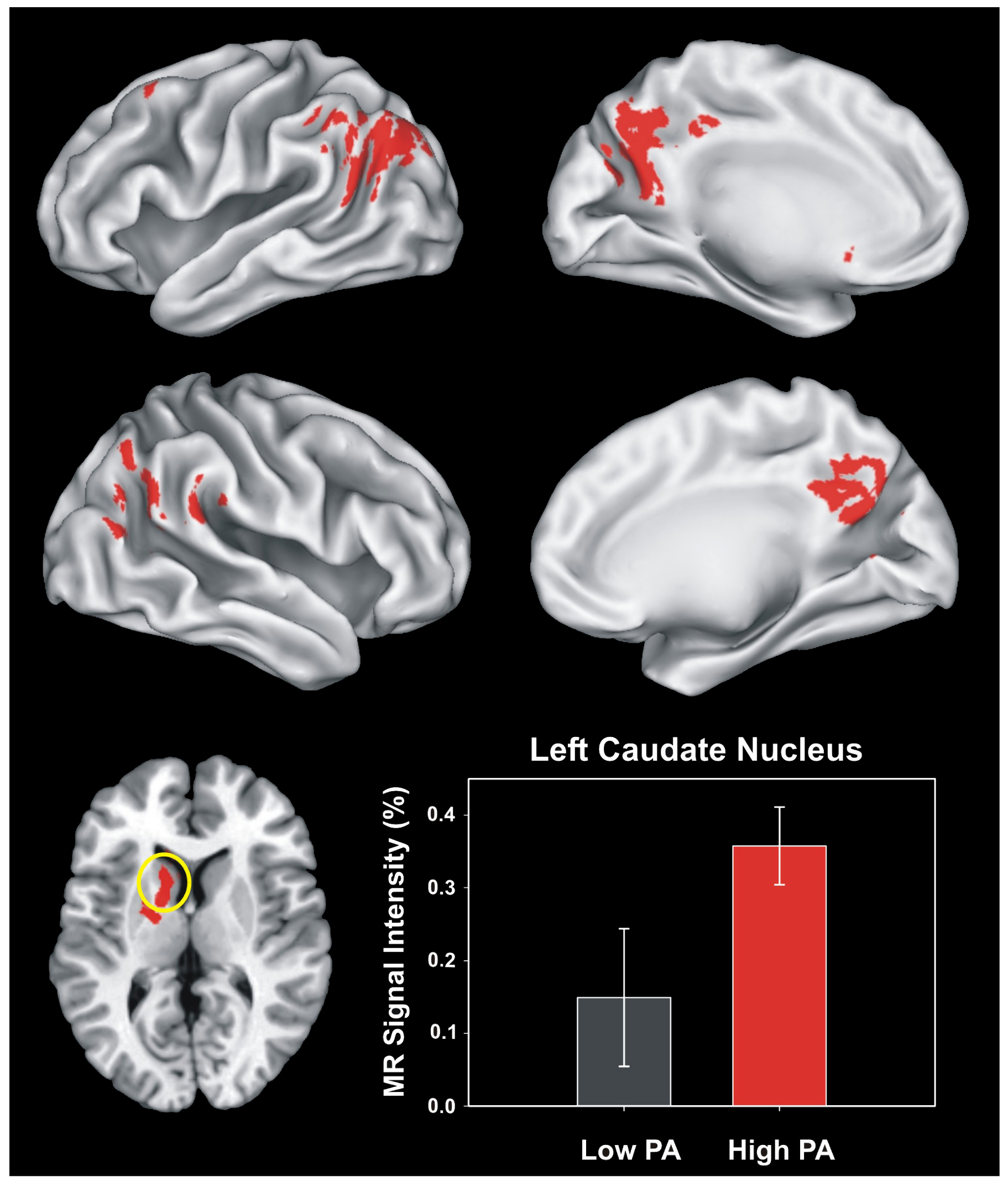
Physical Activity and Brain Function in Older Adults at Increased Risk for Alzheimer’s Disease

{kind=link}
{kind=link}
{kind=link}
Abstract
Share and Cite
Smith, J.C.; Nielson, K.A.; Woodard, J.L.; Seidenberg, M.; Rao, S.M. Physical Activity and Brain Function in Older Adults at Increased Risk for Alzheimer’s Disease. Brain Sci. 2013, 3, 54-83. https://doi.org/10.3390/brainsci3010054
Smith JC, Nielson KA, Woodard JL, Seidenberg M, Rao SM. Physical Activity and Brain Function in Older Adults at Increased Risk for Alzheimer’s Disease. Brain Sciences. 2013; 3(1):54-83. https://doi.org/10.3390/brainsci3010054
Chicago/Turabian StyleSmith, J. Carson, Kristy A. Nielson, John L. Woodard, Michael Seidenberg, and Stephen M. Rao. 2013. "Physical Activity and Brain Function in Older Adults at Increased Risk for Alzheimer’s Disease" Brain Sciences 3, no. 1: 54-83. https://doi.org/10.3390/brainsci3010054
APA StyleSmith, J. C., Nielson, K. A., Woodard, J. L., Seidenberg, M., & Rao, S. M. (2013). Physical Activity and Brain Function in Older Adults at Increased Risk for Alzheimer’s Disease. Brain Sciences, 3(1), 54-83. https://doi.org/10.3390/brainsci3010054