Long-Lasting Neural Circuit Dysfunction Following Developmental Ethanol Exposure

Abstract
:
{kind=link}
{kind=link}
1. Overview of FASD and Its Impact on Society
2. Assays of Basic Neural Circuit Function
2.1. Background of Neural Circuit Function
2.1.1. Spatial Information Coding
2.1.2. Temporal Information Coding
2.2. Early Circuit Function and Early Ethanol Exposure: A Hypothesis
3. Animal Models of Early Alcohol Exposure
4. Findings of Neuropathology in Acute Ethanol Exposure Animal Models of FASD
4.1. Neuroanatomical Effects of Acute Ethanol Exposure
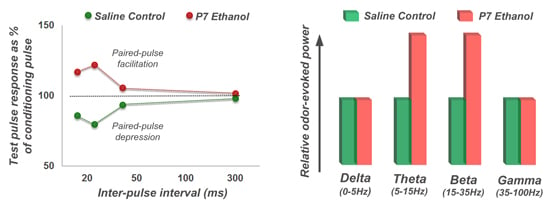
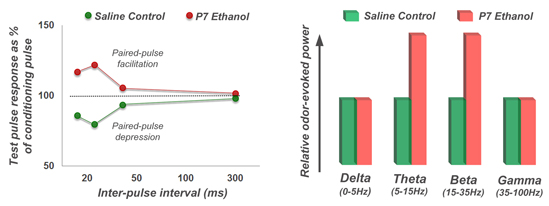
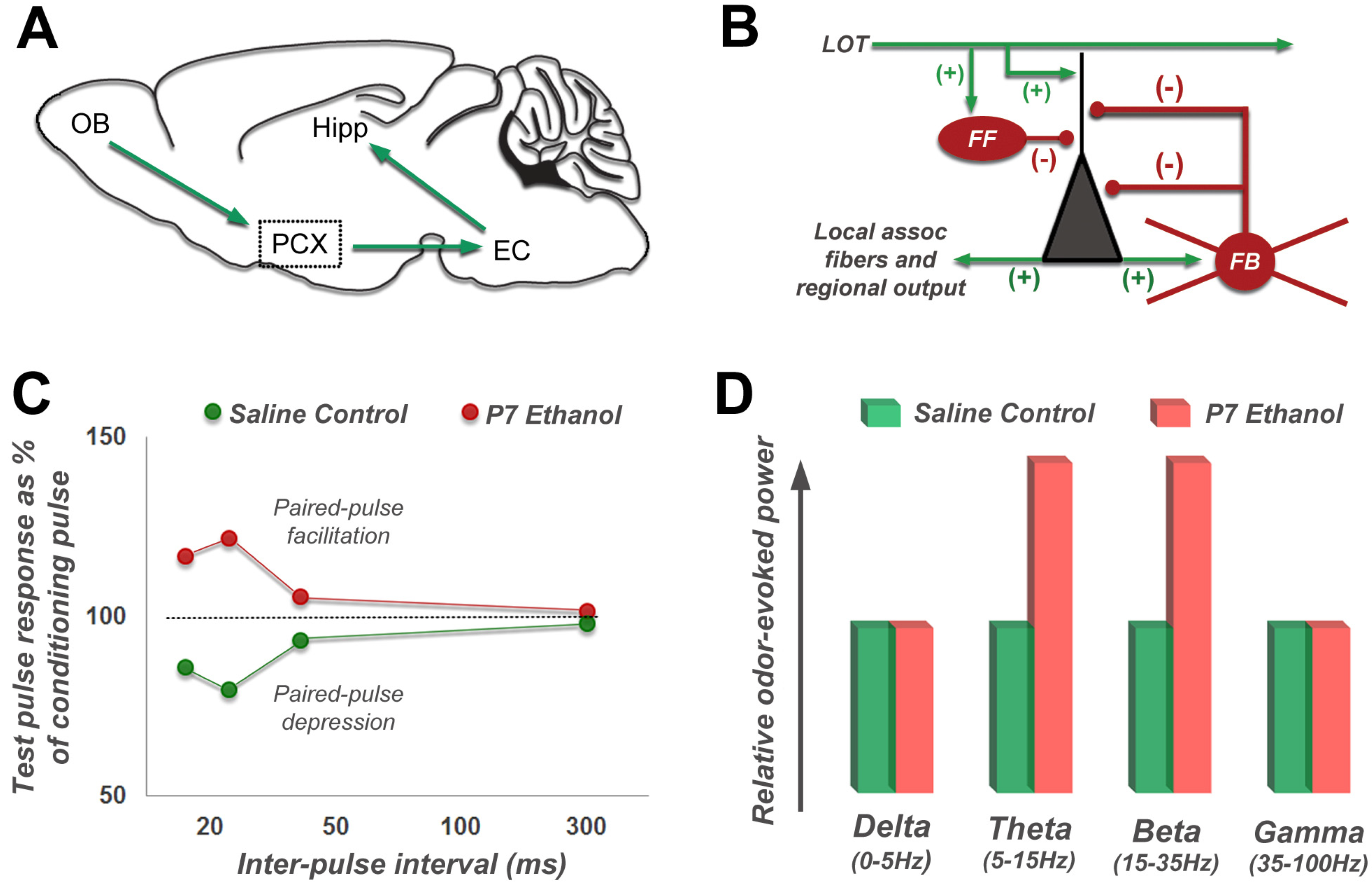
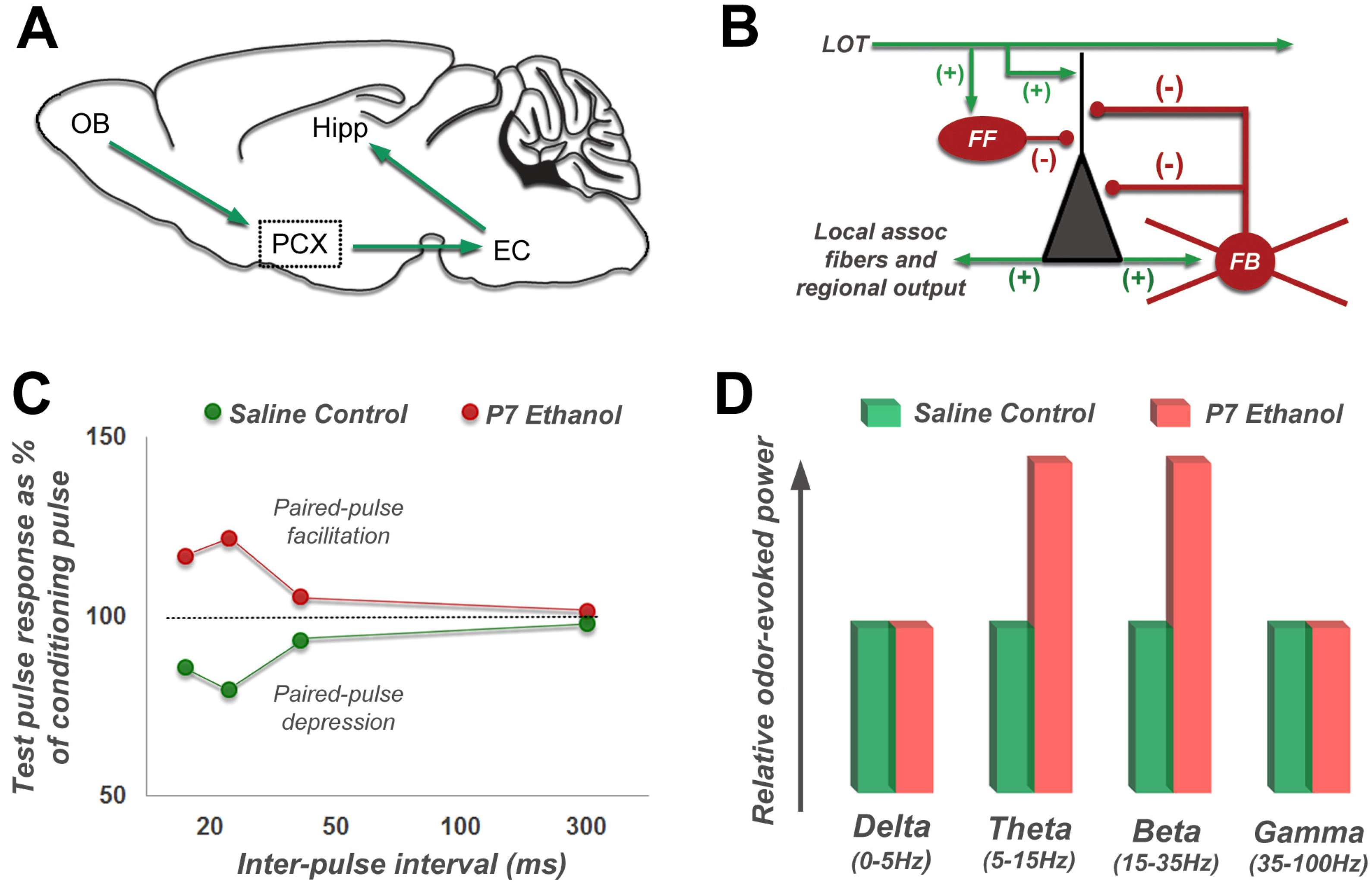
4.2. Long-Lasting Physiological Effects of Acute Developmental Ethanol Exposure
4.3. Long-Lasting Neurobehavioral Effects of Acute Developmental Ethanol Exposure
5. Relationship of FASD to Other Disorders
6. Summary
Acknowledgements
Conflict of Interest
References
- Chudley, A.E.; Kilgour, A.R.; Cranston, M.; Edwards, M. Challenges of diagnosis in fetal alcohol syndrome and fetal alcohol spectrum disorder in the adult. Am. J. Med. Genet. C Semin. Med. Genet. 2007, 145C, 261–272. [Google Scholar] [CrossRef]
- Carr, J.L.; Agnihotri, S.; Keightley, M. Sensory processing and adaptive behavior deficits of children across the fetal alcohol spectrum disorder continuum. Alcohol. Clin. Exp. Res. 2010, 34, 1022–1032. [Google Scholar]
- Gogolla, N.; Leblanc, J.J.; Quast, K.B.; Sudhof, T.C.; Fagiolini, M.; Hensch, T.K. Common circuit defect of excitatory-inhibitory balance in mouse models of autism. J. Neurodev. Disord. 2009, 1, 172–181. [Google Scholar]
- Riley, E.P.; McGee, C.L. Fetal alcohol spectrum disorders: An overview with emphasis on changes in brain and behavior. Exp. Biol. Med. (Maywood) 2005, 230, 357–365. [Google Scholar]
- Bell, S.H.; Stade, B.; Reynolds, J.N.; Rasmussen, C.; Andrew, G.; Hwang, P.A.; Carlen, P.L. The remarkably high prevalence of epilepsy and seizure history in fetal alcohol spectrum disorders. Alcohol. Clin. Exp. Res. 2010, 34, 1084–1089. [Google Scholar]
- May, P.A.; Gossage, J.P. Estimating the prevalence of fetal alcohol syndrome: A summary. Alcohol Res. Health. 2001, 25, 159–167. [Google Scholar]
- Abel, E.L. Fetal alcohol syndrome: The “American Paradox”. Alcohol Alcohol. 1998, 33, 195–201. [Google Scholar] [CrossRef]
- Abel, E.L.; Hannigan, J.H. Maternal risk factors in fetal alcohol syndrome: Provocative and permissive influences. Neurotoxicol. Teratol. 1995, 17, 445–462. [Google Scholar] [CrossRef]
- Sullivan, W. A note on the influence of maternal inebriety on the offspring. J. Ment. Sci. 1899, 45, 489–503. [Google Scholar]
- Jones, K.L.; Smith, D.W. Recognition of the fetal alcohol syndrome in early infancy. Lancet 1973, 302, 999–1001. [Google Scholar] [CrossRef]
- Abel, E.L. Prevention of alcohol abuse-related birth effects—II. Targeting and pricing. Alcohol Alcohol. 1998, 33, 417–420. [Google Scholar] [CrossRef]
- Ingersoll, K.S.; Ceperich, S.D.; Nettleman, M.D.; Karanda, K.; Brocksen, S.; Johnson, B.A. Reducing alcohol-exposed pregnancy risk in college women: Initial outcomes of a clinical trial of a motivational intervention. J. Subst. Abuse Treat. 2005, 29, 173–180. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention (CDC). Motivational intervention to reduce alcohol-exposed pregnancies—Florida, Texas, and Virginia, 1997–2001. MMWR Morb. Mortal. Wkly. Rep. 2003, 52, 441–444.
- Mwansa-Kambafwile, J.; Rendall-Mkosi, K.; Jacobs, R.; Nel, E.; London, L. Evaluation of a service provider short course for prevention of fetal alcohol syndrome. J. Stud. Alcohol Drugs 2011, 72, 530–535. [Google Scholar]
- Velasquez, M.M.; Ingersoll, K.S.; Sobell, M.B.; Floyd, R.L.; Sobell, L.C.; von Sternberg, K. A dual-focus motivational intervention to reduce the risk of alcohol-exposed pregnancy. Cogn. Behav. Pract. 2010, 17, 203–212. [Google Scholar]
- Burd, L.; Cotsonas-Hassler, T.M.; Martsolf, J.T.; Kerbeshian, J. Recognition and management of fetal alcohol syndrome. Neurotoxicol. Teratol. 2003, 25, 681–688. [Google Scholar]
- Lupton, C.; Burd, L.; Harwood, R. Cost of fetal alcohol spectrum disorders. Am. J. Med. Genet. C Semin. Med. Genet. 2004, 127C, 42–50. [Google Scholar] [CrossRef]
- Tanaka, H.; Suzuki, N.; Arima, M. Experimental studies on the influence of male alcoholism on fetal development. Brain Dev. 1982, 4, 1–6. [Google Scholar] [CrossRef]
- Ramsay, M. Genetic and epigenetic insights into fetal alcohol spectrum disorders. Genome Med. 2010, 2, 27. [Google Scholar] [CrossRef]
- Kim, J.S.; Shukla, S.D. Acute in vivo effect of ethanol (binge drinking) on histone H3 modifications in rat tissues. Alcohol Alcohol. 2006, 41, 126–132. [Google Scholar] [CrossRef]
- Govorko, D.; Bekdash, R.A.; Zhang, C.; Sarkar, D.K. Male germline transmits fetal alcohol adverse effect on hypothalamic proopiomelanocortin gene across generations. Biol. Psychiatry 2012, 72, 378–388. [Google Scholar] [CrossRef]
- Luo, J. Lithium-mediated protection against ethanol neurotoxicity. Front. Neurosci. 2010, 4, 41. [Google Scholar]
- Sadrian, B.; Subbanna, S.; Wilson, D.A.; Basavarajappa, B.S.; Saito, M. Lithium prevents long-term neural and behavioral pathology induced by early alcohol exposure. Neuroscience 2012, 206, 122–135. [Google Scholar] [CrossRef]
- Zhong, J.; Yang, X.; Yao, W.; Lee, W. Lithium protects ethanol-induced neuronal apoptosis. Biochem. Biophys. Res. Commun. 2006, 350, 905–910. [Google Scholar] [CrossRef]
- Chakraborty, G.; Saito, M.; Mao, R.F.; Wang, R.; Vadasz, C. Lithium blocks ethanol-induced modulation of protein kinases in the developing brain. Biochem. Biophys. Res. Commun. 2008, 367, 597–602. [Google Scholar] [CrossRef]
- Messiha, F.S. Lithium and the neonate: Developmental and metabolic aspects. Alcohol 1986, 3, 107–112. [Google Scholar] [CrossRef]
- Antonio, A.M.; Druse, M.J. Antioxidants prevent ethanol-associated apoptosis in fetal rhombencephalic neurons. Brain Res. 2008, 1204, 16–23. [Google Scholar] [CrossRef]
- Mitchell, J.J.; Paiva, M.; Heaton, M.B. The antioxidants vitamin E and beta-carotene protect against ethanol-induced neurotoxicity in embryonic rat hippocampal cultures. Alcohol 1999, 17, 163–168. [Google Scholar] [CrossRef]
- Ramachandran, V.; Watts, L.T.; Maffi, S.K.; Chen, J.; Schenker, S.; Henderson, G. Ethanol-induced oxidative stress precedes mitochondrially mediated apoptotic death of cultured fetal cortical neurons. J. Neurosci. Res. 2003, 74, 577–588. [Google Scholar] [CrossRef]
- Taranukhin, A.G.; Taranukhina, E.Y.; Saransaari, P.; Podkletnova, I.M.; Pelto-Huikko, M.; Oja, S.S. Neuroprotection by taurine in ethanol-induced apoptosis in the developing cerebellum. J. Biomed. Sci. 2010, 17 (Suppl. 1), S12. [Google Scholar]
- Ieraci, A.; Herrera, D.G. Nicotinamide protects against ethanol-induced apoptotic neurodegeneration in the developing mouse brain. PLoS Med. 2006, 3, e101. [Google Scholar] [CrossRef]
- Saito, M.; Mao, R.F.; Wang, R.; Vadasz, C.; Saito, M. Effects of gangliosides on ethanol-induced neurodegeneration in the developing mouse brain. Alcohol. Clin. Exp. Res. 2007, 31, 665–674. [Google Scholar]
- Kumar, A.; Singh, C.K.; Lavoie, H.A.; Dipette, D.J.; Singh, U.S. Resveratrol restores Nrf2 level and prevents ethanol-induced toxic effects in the cerebellum of a rodent model of fetal alcohol spectrum disorders. Mol. Pharmacol. 2011, 80, 446–457. [Google Scholar] [CrossRef]
- Paul, A.P.; Pohl-Guimaraes, F.; Krahe, T.E.; Filgueiras, C.C.; Lantz, C.L.; Colello, R.J.; Wang, W.; Medina, A.E. Overexpression of serum response factor restores ocular dominance plasticity in a model of fetal alcohol spectrum disorders. J. Neurosci. 2010, 30, 2513–2520. [Google Scholar]
- Wilson, D.A.; Peterson, J.; Basavaraj, B.S.; Saito, M. Local and regional network function in behaviorally relevant cortical circuits of adult mice following postnatal alcohol exposure. Alcohol. Clin. Exp. Res. 2011, 35, 1974–1984. [Google Scholar] [CrossRef]
- Criado, J.R.; Ehlers, C.L. Effects of adolescent ethanol exposure on event-related oscillations (EROs) in the hippocampus of adult rats. Behav. Brain Res. 2010, 210, 164–170. [Google Scholar] [CrossRef]
- Ehlers, C.L.; Criado, J.R. Adolescent ethanol exposure: Does it produce long-lasting electrophysiological effects? Alcohol 2010, 44, 27–37. [Google Scholar] [CrossRef]
- Lantz, C.L.; Wang, W.; Medina, A.E. Early alcohol exposure disrupts visual cortex plasticity in mice. Int. J. Dev. Neurosci 2012, 30, 351–357. [Google Scholar] [CrossRef]
- Norman, A.L.; O’Brien, J.W.; Spadoni, A.D.; Tapert, S.F.; Jones, K.L.; Riley, E.P.; Mattson, S.N. A functional magnetic resonance imaging study of spatial working memory in children with prenatal alcohol exposure: Contribution of familial history of alcohol use disorders. Alcohol. Clin. Exp. Res. 2012, 37, 132–140. [Google Scholar]
- Wozniak, J.R.; Mueller, B.A.; Muetzel, R.L.; Bell, C.J.; Hoecker, H.L.; Nelson, M.L.; Chang, P.N.; Lim, K.O. Inter-hemispheric functional connectivity disruption in children with prenatal alcohol exposure. Alcohol. Clin. Exp. Res. 2011, 35, 849–861. [Google Scholar]
- Chen, X.; Gabitto, M.; Peng, Y.; Ryba, N.J.; Zuker, C.S. A gustotopic map of taste qualities in the mammalian brain. Science 2011, 333, 1262–1266. [Google Scholar] [CrossRef]
- Buzsaki, G. Rhythms of the Brain; Oxford University Press: New York, NY, USA, 2006. [Google Scholar]
- Hebb, D.O. The Organization of Behavior: A Neuropsychological Theory; John Wiley: New York, NY, USA, 1949. [Google Scholar]
- Verret, L.; Mann, E.O.; Hang, G.B.; Barth, A.M.; Cobos, I.; Ho, K.; Devidze, N.; Masliah, E.; Kreitzer, A.C.; Mody, I.; Mucke, L.; Palop, J.J. Inhibitory interneuron deficit links altered network activity and cognitive dysfunction in Alzheimer model. Cell 2012, 149, 708–721. [Google Scholar] [CrossRef]
- Markram, K.; Markram, H. The intense world theory—a unifying theory of the neurobiology of autism. Front. Hum. Neurosci. 2010, 4, 224. [Google Scholar]
- Vattikuti, S.; Chow, C.C. A computational model for cerebral cortical dysfunction in autism spectrum disorders. Biol. Psychiatry 2010, 67, 672–678. [Google Scholar] [CrossRef]
- Kehrer, C.; Maziashvili, N.; Dugladze, T.; Gloveli, T. Altered excitatory-inhibitory balance in the NMDA-hypofunction model of schizophrenia. Front. Mol. Neurosci. 2008, 1, 6. [Google Scholar]
- Lisman, J. Excitation, inhibition, local oscillations, or large-scale loops: What causes the symptoms of schizophrenia? Cu. Opin. Neurobiol. 2012, 22, 537–544. [Google Scholar] [CrossRef]
- Won, H.; Mah, W.; Kim, E.; Kim, J.W.; Hahm, E.K.; Kim, M.H.; Cho, S.; Kim, J.; Jang, H.; Cho, S.C.; et al. GIT1 is associated with ADHD in humans and ADHD-like behaviors in mice. Nat. Med. 2011, 17, 566–572. [Google Scholar] [CrossRef]
- Hannigan, J.H. What research with animals is telling us about alcohol-related neurodevelopmental disorder. Pharmacol. Biochem. Behav. 1996, 55, 489–499. [Google Scholar] [CrossRef]
- Olney, J.W.; Tenkova, T.; Dikranian, K.; Qin, Y.Q.; Labruyere, J.; Ikonomidou, C. Ethanol-induced apoptotic neurodegeneration in the developing C57BL/6 mouse brain. Brain Res. Dev. Brain Res. 2002, 133, 115–126. [Google Scholar] [CrossRef]
- Youngentob, S.L.; Molina, J.C.; Spear, N.E.; Youngentob, L.M. The effect of gestational ethanol exposure on voluntary ethanol intake in early postnatal and adult rats. Behav. Neurosci. 2007, 121, 1306–1315. [Google Scholar] [CrossRef]
- Gil-Mohapel, J.; Boehme, F.; Kainer, L.; Christie, B.R. Hippocampal cell loss and neurogenesis after fetal alcohol exposure: Insights from different rodent models. Brain Res. Rev. 2010, 64, 283–303. [Google Scholar]
- Cudd, T.A. Animal model systems for the study of alcohol teratology. Exp. Biol. Med. (Maywood) 2005, 230, 389–393. [Google Scholar]
- Krulewitch, C.J. Alcohol consumption during pregnancy. Annu. Rev. Nurs. Res. 2005, 23, 101–134. [Google Scholar]
- Kleiber, M.L.; Wright, E.; Singh, S.M. Maternal voluntary drinking in C57BL/6J mice: Advancing a model for fetal alcohol spectrum disorders. Behav. Brain Res. 2011, 223, 376–387. [Google Scholar] [CrossRef]
- Rodrigo, R.; Vergara, L.; Oberhauser, E. Effect of chronic ethanol consumption on postnatal development of renal (Na + K)-ATPase in the rat. Cell Biochem. Funct. 1991, 9, 215–222. [Google Scholar] [CrossRef]
- Henderson, G.I.; Hoyumpa, A.M., Jr.; McClain, C.; Schenker, S. The effects of chronic and acute alcohol administration on fetal development in the rat. Alcohol. Clin. Exp. Res. 1979, 3, 99–106. [Google Scholar] [CrossRef]
- Abel, E. Fetal Alcohol Abuse Syndrome; Plenum Press: New York, NY, USA, 1998. [Google Scholar]
- Traves, C.; Lopez-Tejero, D. Ethanol elimination in alcohol-treated pregnant rats. Alcohol Alcohol. 1994, 29, 385–395. [Google Scholar]
- Maier, S.E.; Chen, W.J.; Miller, J.A.; West, J.R. Fetal alcohol exposure and temporal vulnerability regional differences in alcohol-induced microencephaly as a function of the timing of binge-like alcohol exposure during rat brain development. Alcohol. Clin. Exp. Res. 1997, 21, 1418–1428. [Google Scholar]
- Dobbing, J.; Sands, J. Comparative aspects of the brain growth spurt. Early Hum. Dev. 1979, 3, 79–83. [Google Scholar] [CrossRef]
- Young, C.; Olney, J.W. Neuroapoptosis in the infant mouse brain triggered by a transient small increase in blood alcohol concentration. Neurobiol. Dis. 2006, 22, 548–554. [Google Scholar] [CrossRef]
- Bonthius, D.J.; West, J.R. Blood alcohol concentration and microencephaly: A dose-response study in the neonatal rat. Teratology 1988, 37, 223–231. [Google Scholar] [CrossRef]
- Klintsova, A.Y.; Helfer, J.L.; Calizo, L.H.; Dong, W.K.; Goodlett, C.R.; Greenough, W.T. Persistent impairment of hippocampal neurogenesis in young adult rats following early postnatal alcohol exposure. Alcohol. Clin. Exp. Res. 2007, 31, 2073–2082. [Google Scholar] [CrossRef]
- Coleman, L.G., Jr.; Oguz, I.; Lee, J.; Styner, M.; Crews, F.T. Postnatal day 7 ethanol treatment causes persistent reductions in adult mouse brain volume and cortical neurons with sex specific effects on neurogenesis. Alcohol 2012, 46, 603–612. [Google Scholar] [CrossRef]
- Medina, A.E.; Krahe, T.E.; Ramoa, A.S. Early alcohol exposure induces persistent alteration of cortical columnar organization and reduced orientation selectivity in the visual cortex. J. Neurophysiol. 2005, 93, 1317–1325. [Google Scholar] [CrossRef]
- Slawecki, C.J.; Thomas, J.D.; Riley, E.P.; Ehlers, C.L. Neurophysiologic consequences of neonatal ethanol exposure in the rat. Alcohol 2004, 34, 187–196. [Google Scholar] [CrossRef]
- Wozniak, D.F.; Hartman, R.E.; Boyle, M.P.; Vogt, S.K.; Brooks, A.R.; Tenkova, T.; Young, C.; Olney, J.W.; Muglia, L.J. Apoptotic neurodegeneration induced by ethanol in neonatal mice is associated with profound learning/memory deficits in juveniles followed by progressive functional recovery in adults. Neurobiol. Dis. 2004, 17, 403–414. [Google Scholar] [CrossRef]
- Costa, E.T.; Savage, D.D.; Valenzuela, C.F. A review of the effects of prenatal or early postnatal ethanol exposure on brain ligand-gated ion channels. Alcohol. Clin. Exp. Res. 2000, 24, 706–715. [Google Scholar] [CrossRef]
- Izumi, Y.; Kitabayashi, R.; Funatsu, M.; Izumi, M.; Yuede, C.; Hartman, R.E.; Wozniak, D.F.; Zorumski, C.F. A single day of ethanol exposure during development has persistent effects on bi-directional plasticity, N-methyl-d-aspartate receptor function and ethanol sensitivity. Neuroscience 2005, 136, 269–279. [Google Scholar] [CrossRef]
- Puglia, M.P.; Valenzuela, C.F. Repeated third trimester-equivalent ethanol exposure inhibits long-term potentiation in the hippocampal CA1 region of neonatal rats. Alcohol 2010, 44, 283–290. [Google Scholar] [CrossRef]
- Berman, R.F.; Hannigan, J.H. Effects of prenatal alcohol exposure on the hippocampus: Spatial behavior, electrophysiology, and neuroanatomy. Hippocampus 2000, 10, 94–110. [Google Scholar] [CrossRef]
- Maier, S.E.; Miller, J.A.; West, J.R. Prenatal binge-like alcohol exposure in the rat results in region-specific deficits in brain growth. Neurotoxicol. Teratol. 1999, 21, 285–291. [Google Scholar] [CrossRef]
- Bonthius, D.J.; West, J.R. Alcohol-induced neuronal loss in developing rats: Increased brain damage with binge exposure. Alcohol. Clin. Exp. Res. 1990, 14, 107–118. [Google Scholar] [CrossRef]
- Lawrence, R.C.; Otero, N.K.; Kelly, S.J. Selective effects of perinatal ethanol exposure in medial prefrontal cortex and nucleus accumbens. Neurotoxicol. Teratol. 2012, 34, 128–135. [Google Scholar] [CrossRef]
- Uban, K.A.; Sliwowska, J.H.; Lieblich, S.; Ellis, L.A.; Yu, W.K.; Weinberg, J.; Galea, L.A. Prenatal alcohol exposure reduces the proportion of newly produced neurons and glia in the dentate gyrus of the hippocampus in female rats. Horm. Behav. 2010, 58, 835–843. [Google Scholar] [CrossRef]
- Ikonomidou, C.; Bittigau, P.; Ishimaru, M.J.; Wozniak, D.F.; Koch, C.; Genz, K.; Price, M.T.; Stefovska, V.; Horster, F.; Tenkova, T.; et al. Ethanol-induced apoptotic neurodegeneration and fetal alcohol syndrome. Science 2000, 287, 1056–1060. [Google Scholar] [CrossRef]
- Olney, J.W.; Tenkova, T.; Dikranian, K.; Muglia, L.J.; Jermakowicz, W.J.; D’Sa, C.; Roth, K.A. Ethanol-induced caspase-3 activation in the in vivo developing mouse brain. Neurobiol. Dis. 2002, 9, 205–219. [Google Scholar] [CrossRef]
- Saito, M.; Chakraborty, G.; Mao, R.F.; Paik, S.M.; Vadasz, C. Tau phosphorylation and cleavage in ethanol-induced neurodegeneration in the developing mouse brain. Neurochem. Res. 2010, 35, 651–659. [Google Scholar] [CrossRef]
- Olney, J.W. Fetal alcohol syndrome at the cellular level. Addict. Biol. 2004, 9, 137–149; discussion 151. [Google Scholar] [CrossRef]
- Goodlett, C.R.; Peterson, S.D.; Lundahl, K.R.; Pearlman, A.D. Binge-like alcohol exposure of neonatal rats via intragastric intubation induces both Purkinje cell loss and cortical astrogliosis. Alcohol. Clin. Exp. Res. 1997, 21, 1010–1017. [Google Scholar] [CrossRef]
- Guerri, C.; Pascual, M.; Renau-Piqueras, J. Glia and fetal alcohol syndrome. Neurotoxicology 2001, 22, 593–599. [Google Scholar] [CrossRef]
- Guerri, C.; Bazinet, A.; Riley, E.P. Foetal Alcohol Spectrum Disorders and alterations in brain and behaviour. Alcohol Alcohol. 2009, 44, 108–114. [Google Scholar]
- Hayes, D.M.; Deeny, M.A.; Shaner, C.A.; Nixon, K. Determining the threshold for alcohol-induced brain damage: New evidence with gliosis markers. Alcohol. Clin. Exp. Res. 2013, 37, 425–434. [Google Scholar] [CrossRef]
- Semmler, A.; Okulla, T.; Sastre, M.; Dumitrescu-Ozimek, L.; Heneka, M.T. Systemic inflammation induces apoptosis with variable vulnerability of different brain regions. J. Chem. Neuroanat. 2005, 30, 144–157. [Google Scholar] [CrossRef]
- Clarren, S.K.; Alvord, E.C., Jr.; Sumi, S.M.; Streissguth, A.P.; Smith, D.W. Brain malformations related to prenatal exposure to ethanol. J. Pediatr. 1978, 92, 64–67. [Google Scholar] [CrossRef]
- West, J.R.; Goodlett, C.R.; Bonthius, D.J.; Hamre, K.M.; Marcussen, B.L. Cell population depletion associated with fetal alcohol brain damage: Mechanisms of BAC-dependent cell loss. Alcohol. Clin. Exp. Res. 1990, 14, 813–818. [Google Scholar]
- Kirstein, C.L.; Philpot, R.M.; Dark, T. Fetal alcohol syndrome: Early olfactory learning as a model system to study neurobehavioral deficits. Int. J. Neurosci. 1997, 89, 119–132. [Google Scholar]
- O’Leary-Moore, S.K.; Parnell, S.E.; Lipinski, R.J.; Sulik, K.K. Magnetic resonance-based imaging in animal models of fetal alcohol spectrum disorder. Neuropsychol. Rev. 2011, 21, 167–185. [Google Scholar]
- Granato, A.; Van Pelt, J. Effects of early ethanol exposure on dendrite growth of cortical pyramidal neurons: Inferences from a computational model. Brain Res. Dev. Brain Res. 2003, 142, 223–227. [Google Scholar] [CrossRef]
- Hamilton, G.F.; Whitcher, L.T.; Klintsova, A.Y. Postnatal binge-like alcohol exposure decreases dendritic complexity while increasing the density of mature spines in mPFC Layer II/III pyramidal neurons. Synapse 2010, 64, 127–135. [Google Scholar]
- Wozniak, J.R.; Muetzel, R.L. What does diffusion tensor imaging reveal about the brain and cognition in fetal alcohol spectrum disorders? Neuropsychol. Rev. 2011, 21, 133–147. [Google Scholar] [CrossRef]
- Wozniak, J.R.; Mueller, B.A.; Chang, P.N.; Muetzel, R.L.; Caros, L.; Lim, K.O. Diffusion tensor imaging in children with fetal alcohol spectrum disorders. Alcohol. Clin. Exp. Res. 2006, 30, 1799–1806. [Google Scholar]
- De Giorgio, A.; Comparini, S.E.; Intra, F.S.; Granato, A. Long-term alterations of striatal parvalbumin interneurons in a rat model of early exposure to alcohol. J. Neurodev. Disord. 2012, 4, 18. [Google Scholar] [CrossRef]
- Moore, D.B.; Ruygrok, A.C.; Walker, D.W.; Heaton, M.B. Effects of prenatal ethanol exposure on parvalbumin-expressing GABAergic neurons in the adult rat medial septum. Alcohol. Clin. Exp. Res. 1997, 21, 849–856. [Google Scholar]
- Mitchell, J.J.; Paiva, M.; Heaton, M.B. Effect of neonatal ethanol exposure on parvalbumin-expressing GABAergic neurons of the rat medial septum and cingulate cortex. Alcohol 2000, 21, 49–57. [Google Scholar] [CrossRef]
- Bartos, M.; Vida, I.; Jonas, P. Synaptic mechanisms of synchronized gamma oscillations in inhibitory interneuron networks. Nat. Rev. Neurosci. 2007, 8, 45–56. [Google Scholar] [CrossRef]
- Lewis, D.A.; Curley, A.A.; Glausier, J.R.; Volk, D.W. Cortical parvalbumin interneurons and cognitive dysfunction in schizophrenia. Trends Neurosci. 2012, 35, 57–67. [Google Scholar] [CrossRef]
- Magloczky, Z.; Freund, T.F. Impaired and repaired inhibitory circuits in the epileptic human hippocampus. Trends Neurosci. 2005, 28, 334–340. [Google Scholar]
- Nusser, Z.; Kay, L.M.; Laurent, G.; Homanics, G.E.; Mody, I. Disruption of GABA(A) receptors on GABAergic interneurons leads to increased oscillatory power in the olfactory bulb network. J. Neurophysiol. 2001, 86, 2823–2833. [Google Scholar]
- Maier, S.E.; Miller, J.A.; Blackwell, J.M.; West, J.R. Fetal alcohol exposure and temporal vulnerability: Regional differences in cell loss as a function of the timing of binge-like alcohol exposure during brain development. Alcohol. Clin. Exp. Res. 1999, 23, 726–734. [Google Scholar]
- Sadrian, B.; Saito, M.; Wilson, D.A. Nathan Kline Institute: Orangeburg, NY, USA, Unpublished Work. 2013.
- Bavelier, D.; Levi, D.M.; Li, R.W.; Dan, Y.; Hensch, T.K. Removing brakes on adult brain plasticity: From molecular to behavioral interventions. J. Neurosci. 2010, 30, 14964–14971. [Google Scholar] [CrossRef]
- Sutherland, R.J.; McDonald, R.J.; Savage, D.D. Prenatal exposure to moderate levels of ethanol can have long-lasting effects on hippocampal synaptic plasticity in adult offspring. Hippocampus 1997, 7, 232–238. [Google Scholar] [CrossRef]
- Olney, J.W.; Wozniak, D.F.; Jevtovic-Todorovic, V.; Ikonomidou, C. Glutamate signaling and the fetal alcohol syndrome. Ment. Retard. Dev. Disabil. Res. Rev. 2001, 7, 267–275. [Google Scholar] [CrossRef]
- Lovinger, D.M.; White, G.; Weight, F.F. Ethanol inhibits NMDA-activated ion current in hippocampal neurons. Science 1989, 243, 1721–1724. [Google Scholar]
- Pian, J.P.; Criado, J.R.; Milner, R.; Ehlers, C.L. N-methyl-d-aspartate receptor subunit expression in adult and adolescent brain following chronic ethanol exposure. Neuroscience 2010, 170, 645–654. [Google Scholar] [CrossRef]
- Nixon, K.; Hughes, P.D.; Amsel, A.; Leslie, S.W. NMDA receptor subunit expression following early postnatal exposure to ethanol. Brain Res. Dev. Brain Res. 2002, 139, 295–299. [Google Scholar] [CrossRef]
- Takadera, T.; Suzuki, R.; Mohri, T. Protection by ethanol of cortical neurons from N-methyl-d-aspartate-induced neurotoxicity is associated with blocking calcium influx. Brain Res. 1990, 537, 109–114. [Google Scholar] [CrossRef]
- Lustig, H.S.; von Brauchitsch, K.L.; Chan, J.; Greenberg, D.A. Ethanol and excitotoxicity in cultured cortical neurons: Differential sensitivity of N-methyl-d-aspartate and sodium nitroprusside toxicity. J. Neurochem. 1992, 59, 2193–2200. [Google Scholar]
- Kelly, D.F. Alcohol and head injury: An issue revisited. J. Neurotrauma 1995, 12, 883–890. [Google Scholar] [CrossRef]
- Farber, N.B.; Heinkel, C.; Dribben, W.H.; Nemmers, B.; Jiang, X. In the adult CNS, ethanol prevents rather than produces NMDA antagonist-induced neurotoxicity. Brain Res. 2004, 1028, 66–74. [Google Scholar] [CrossRef]
- Scholz, J.; Broom, D.C.; Youn, D.H.; Mills, C.D.; Kohno, T.; Suter, M.R.; Moore, K.A.; Decosterd, I.; Coggeshall, R.E.; Woolf, C.J. Blocking caspase activity prevents transsynaptic neuronal apoptosis and the loss of inhibition in lamina II of the dorsal horn after peripheral nerve injury. J. Neurosci. 2005, 25, 7317–7323. [Google Scholar] [CrossRef]
- Slawecki, C.J.; Betancourt, M.; Cole, M.; Ehlers, C.L. Periadolescent alcohol exposure has lasting effects on adult neurophysiological function in rats. Brain Res. Dev. Brain Res. 2001, 128, 63–72. [Google Scholar] [CrossRef]
- Balaszczuk, V.; Bender, C.; Pereno, G.L.; Beltramino, C.A. Alcohol-induced neuronal death in central extended amygdala and pyriform cortex during the postnatal period of the rat. Int. J. Dev. Neurosci. 2011, 29, 733–742. [Google Scholar] [CrossRef]
- Sanderson, J.L.; Donald Partridge, L.; Valenzuela, C.F. Modulation of GABAergic and glutamatergic transmission by ethanol in the developing neocortex: An in vitro test of the excessive inhibition hypothesis of fetal alcohol spectrum disorder. Neuropharmacology 2009, 56, 541–555. [Google Scholar] [CrossRef]
- Gonzalez-Burgos, G.; Lewis, D.A. NMDA receptor hypofunction, parvalbumin-positive neurons, and cortical gamma oscillations in schizophrenia. Schizophr. Bull. 2012, 38, 950–957. [Google Scholar] [CrossRef]
- Lesh, T.A.; Niendam, T.A.; Minzenberg, M.J.; Carter, C.S. Cognitive control deficits in schizophrenia: Mechanisms and meaning. Neuropsychopharmacology 2011, 36, 316–338. [Google Scholar] [CrossRef]
- Knipper, M.; Rylett, R.J. A new twist in an old story: The role for crosstalk of neuronal and trophic activity. Neurochem. Int. 1997, 31, 659–676. [Google Scholar] [CrossRef]
- Megias, M.; Emri, Z.; Freund, T.F.; Gulyas, A.I. Total number and distribution of inhibitory and excitatory synapses on hippocampal CA1 pyramidal cells. Neuroscience 2001, 102, 527–540. [Google Scholar] [CrossRef]
- Akers, K.G.; Kushner, S.A.; Leslie, A.T.; Clarke, L.; van der Kooy, D.; Lerch, J.P.; Frankland, P.W. Fetal alcohol exposure leads to abnormal olfactory bulb development and impaired odor discrimination in adult mice. Mol. Brain 2011, 4, 29. [Google Scholar] [CrossRef]
- Bernstein, J.G.; Boyden, E.S. Optogenetic tools for analyzing the neural circuits of behavior. Trends Cogn. Sci. 2011, 15, 592–600. [Google Scholar] [CrossRef]
- Youngentob, S.L.; Glendinning, J.I. Fetal ethanol exposure increases ethanol intake by making it smell and taste better. Proc. Natl. Acad. Sci. USA 2009, 106, 5359–5364. [Google Scholar] [CrossRef]
- Kaneko, W.M.; Riley, E.P.; Ehlers, C.L. Electrophysiological and behavioral findings in rats prenatally exposed to alcohol. Alcohol 1993, 10, 169–178. [Google Scholar] [CrossRef]
- Baker, A.E.; Lane, A.; Angley, M.T.; Young, R.L. The relationship between sensory processing patterns and behavioural responsiveness in autistic disorder: A pilot study. J. Autism Dev. Disord. 2008, 38, 867–875. [Google Scholar] [CrossRef]
- Ganellen, R.J. Assessing normal and abnormal personality functioning: Strengths and weaknesses of self-report, observer, and performance-based methods. J. Pers. Assess. 2007, 89, 30–40. [Google Scholar] [CrossRef]
- Jirikowic, T.; Olson, H.C.; Kartin, D. Sensory processing, school performance, and adaptive behavior of young school-age children with fetal alcohol spectrum disorders. Phys. Occup. Ther. Pediatr. 2008, 28, 117–136. [Google Scholar] [CrossRef]
- Wengel, T.; Hanlon-Dearman, A.C.; Fjeldsted, B. Sleep and sensory characteristics in young children with fetal alcohol spectrum disorder. J. Dev. Behav. Pediatr. 2011, 32, 384–392. [Google Scholar] [CrossRef]
- Eichler, S.A.; Meier, J.C. E-I balance and human diseases—from molecules to networking. Front. Mol. Neurosci. 2008, 1, 2. [Google Scholar]
- Hensch, T.K. Critical period plasticity in local cortical circuits. Nat. Rev. Neurosci. 2005, 6, 877–888. [Google Scholar] [CrossRef]
- Ramamoorthi, K.; Lin, Y. The contribution of GABAergic dysfunction to neurodevelopmental disorders. Trends Mol. Med. 2011, 17, 452–462. [Google Scholar]
- Yizhar, O.; Fenno, L.E.; Prigge, M.; Schneider, F.; Davidson, T.J.; O’Shea, D.J.; Sohal, V.S.; Goshen, I.; Finkelstein, J.; Paz, J.T.; et al. Neocortical excitation/inhibition balance in information processing and social dysfunction. Nature 2011, 477, 171–178. [Google Scholar] [CrossRef]
- Fatemi, S.H.; Stary, J.M.; Earle, J.A.; Araghi-Niknam, M.; Eagan, E. GABAergic dysfunction in schizophrenia and mood disorders as reflected by decreased levels of glutamic acid decarboxylase 65 and 67 kDa and Reelin proteins in cerebellum. Schizophr. Res. 2005, 72, 109–122. [Google Scholar] [CrossRef]
- Busche, M.A.; Chen, X.; Henning, H.A.; Reichwald, J.; Staufenbiel, M.; Sakmann, B.; Konnerth, A. Critical role of soluble amyloid-beta for early hippocampal hyperactivity in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2012, 109, 8740–8745. [Google Scholar]
- Sanchez, P.E.; Zhu, L.; Verret, L.; Vossel, K.A.; Orr, A.G.; Cirrito, J.R.; Devidze, N.; Ho, K.; Yu, G.Q.; Palop, J.J.; Mucke, L. Levetiracetam suppresses neuronal network dysfunction and reverses synaptic and cognitive deficits in an Alzheimer’s disease model. Proc. Natl. Acad. Sci. USA 2012, 109, E2895–E2903. [Google Scholar]
- Wesson, D.W.; Borkowski, A.H.; Landreth, G.E.; Nixon, R.A.; Levy, E.; Wilson, D.A. Sensory network dysfunction, behavioral impairments, and their reversibility in an Alzheimer’s beta-amyloidosis mouse model. J. Neurosci. 2011, 31, 15962–15971. [Google Scholar] [CrossRef]
- Brambilla, P.; Perez, J.; Barale, F.; Schettini, G.; Soares, J.C. GABAergic dysfunction in mood disorders. Mol. Psychiatry 2003, 8, 721–737, 715. [Google Scholar]
- Naylor, D.E. Glutamate and GABA in the balance: Convergent pathways sustain seizures during status epilepticus. Epilepsia 2010, 51 (Suppl. 3), 106–109. [Google Scholar] [CrossRef]
- Disterhoft, J.F.; Wu, W.W.; Ohno, M. Biophysical alterations of hippocampal pyramidal neurons in learning, ageing and Alzheimer’s disease. Ageing Res. Rev. 2004, 3, 383–406. [Google Scholar] [CrossRef]
- Kaczorowski, C.C.; Sametsky, E.; Shah, S.; Vassar, R.; Disterhoft, J.F. Mechanisms underlying basal and learning-related intrinsic excitability in a mouse model of Alzheimer’s disease. Neurobiol. Aging 2011, 32, 1452–1465. [Google Scholar]
- Moriceau, S.; Sullivan, R.M. Unique neural circuitry for neonatal olfactory learning. J. Neurosci. 2004, 24, 1182–1189. [Google Scholar] [CrossRef]
- Roskam, S.; Koch, M. Effects of neonatal and peripubertal ethanol treatment on various aspects of adult rat behavior and brain anatomy. Int. J. Dev. Neurosci. 2009, 27, 249–256. [Google Scholar] [CrossRef]
- Bishop, S.; Gahagan, S.; Lord, C. Re-examining the core features of autism: A comparison of autism spectrum disorder and fetal alcohol spectrum disorder. J. Child Psychol. Psychiatry 2007, 48, 1111–1121. [Google Scholar] [CrossRef]
- Stevens, S.A.; Nash, K.; Koren, G.; Rovet, J. Autism characteristics in children with fetal alcohol spectrum disorders. Child Neuropsychol. 2012. [Google Scholar] [CrossRef]
- Han, S.; Tai, C.; Westenbroek, R.E.; Yu, F.H.; Cheah, C.S.; Potter, G.B.; Rubenstein, J.L.; Scheuer, T.; de la Iglesia, H.O.; Catterall, W.A. Autistic-like behaviour in Scn1a+/− mice and rescue by enhanced GABA-mediated neurotransmission. Nature 2012, 489, 385–390. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Sadrian, B.; Wilson, D.A.; Saito, M. Long-Lasting Neural Circuit Dysfunction Following Developmental Ethanol Exposure. Brain Sci. 2013, 3, 704-727. https://doi.org/10.3390/brainsci3020704
Sadrian B, Wilson DA, Saito M. Long-Lasting Neural Circuit Dysfunction Following Developmental Ethanol Exposure. Brain Sciences. 2013; 3(2):704-727. https://doi.org/10.3390/brainsci3020704
Chicago/Turabian StyleSadrian, Benjamin, Donald A. Wilson, and Mariko Saito. 2013. "Long-Lasting Neural Circuit Dysfunction Following Developmental Ethanol Exposure" Brain Sciences 3, no. 2: 704-727. https://doi.org/10.3390/brainsci3020704