
A Comprehensive Diverse ‘-omics’ Approach to Better Understanding the Molecular Pathomechanisms of Down Syndrome


Department of Pathological Biochemistry, Division of Pathological Science, Kyoto Pharmaceutical University, 5 Misasagi Nakauchi-cho, Ymashina-ku, Kyoto 607-8414, Japan
*
Author to whom correspondence should be addressed.
Brain Sci. 2017, 7(4), 44; https://doi.org/10.3390/brainsci7040044
Submission received: 28 March 2017
/
Revised: 17 April 2017
/
Accepted: 18 April 2017
/
Published: 21 April 2017
Abstract
:Diverse ‘-omics’ technologies permit the comprehensive quantitative profiling of a variety of biological molecules. Comparative ‘-omics’ analyses, such as transcriptomics and proteomics, are powerful and useful tools for unraveling the molecular pathomechanisms of various diseases. As enhanced oxidative stress has been demonstrated in humans and mice with Down syndrome (DS), a redox proteomic analysis is useful for understanding how enhanced oxidative stress aggravates the state of individuals with oxidative stress-related disorders. In this review, ‘-omics’ analyses in humans with DS and mouse models of DS are summarized, and the molecular dissection of this syndrome is discussed.
1. Introduction
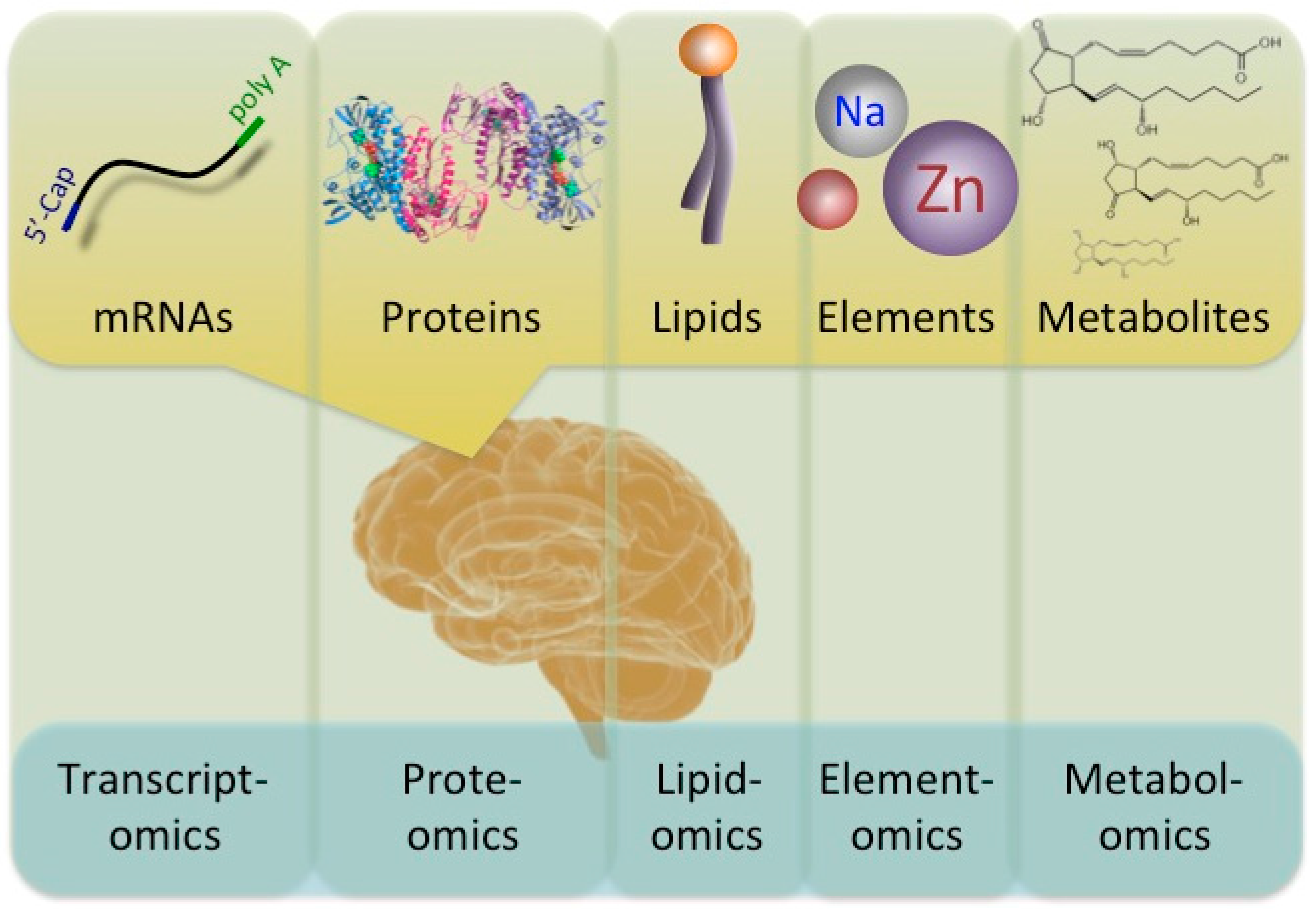
Understanding the changes in molecules provides important and valuable information for evaluating the molecular pathomechanisms of diseases. Diverse ‘-omics’ technologies permit the quantitative monitoring of various biological molecules in a comprehensive manner. Comprehensive expression profiles are designated by adding the suffix ‘-omics’ onto previously used terms, such as prote-omics (protein), transcript-omics (transcripts), lipid-omics (lipid), metabol-omics (metabolites), and so on (Figure 1). Comparative ‘-omics’ analyses comparing the profiles of healthy subjects and populations with some illness are a powerful and useful approach for better understanding molecular pathomechanisms. Indeed, a variety of disorders, including neurological disorders, have been subjected to ‘-omics’ analyses.
Although most ‘-omics’ analyses assess the levels of target molecules, protein, transcripts, fatty acids, and various metabolites in organs, redox proteomics and phospho-proteomics indicate the degree of oxidative stress and activation of signal transduction or transcriptional factors, respectively. Thus, proteomics can be used to determine not only the protein expression, but also the degree of post-translational modifications, such as phosphorylation and oxidation. However, no single ‘-omics’ analysis can fully unravel the complexities of some disorders. Therefore, the integration of multiple layers of information—the multi-’-omics’ approach—is required to acquire a precise picture of the pathophysiology of a given disease.
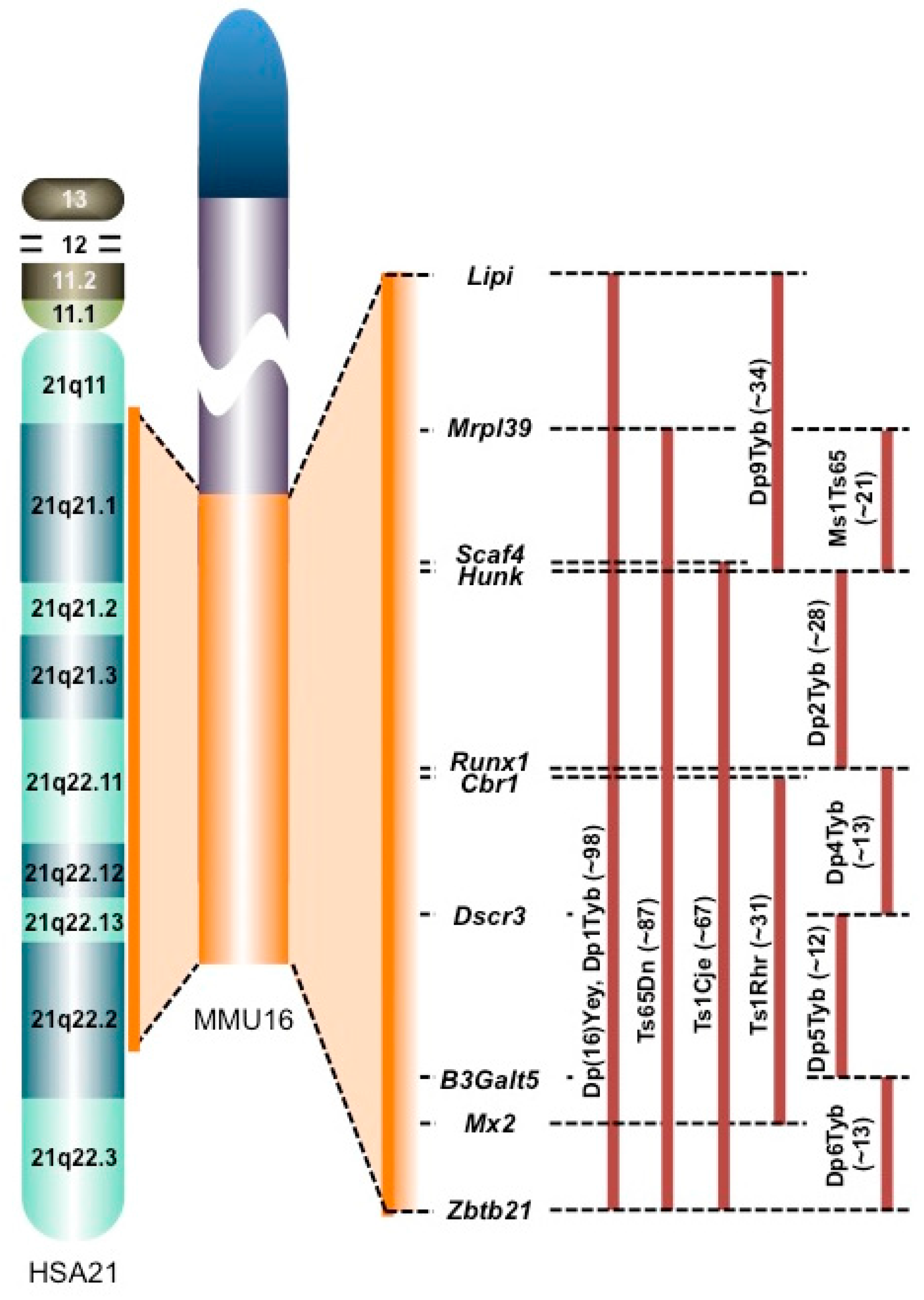
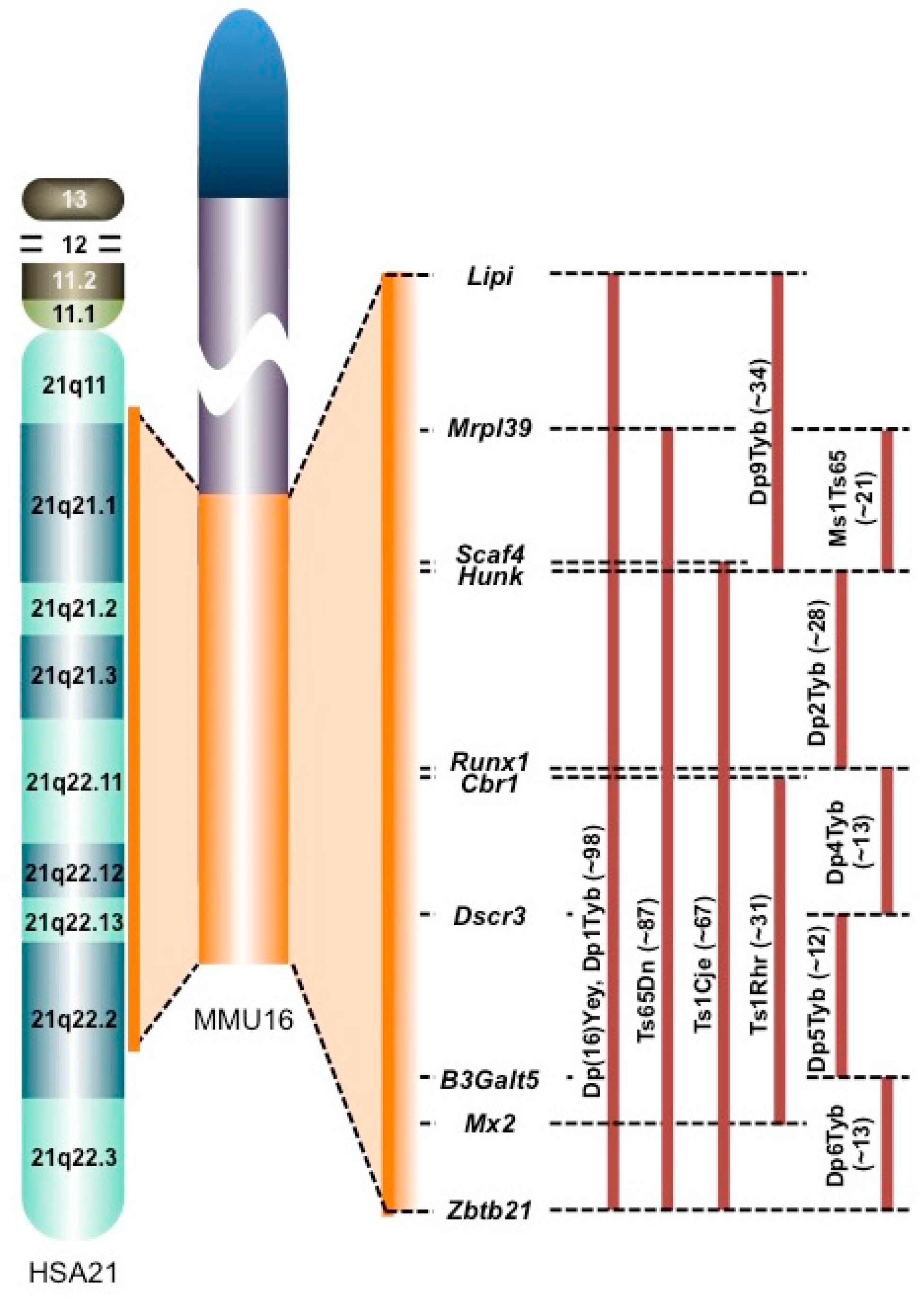
In this review, we discuss the changes in the molecules in brains from individuals with Down syndrome (DS) and DS mouse models according to ‘-omics’ analyses. Although DS is caused by trisomy of human chromosome 21 (HSA21), the distal end of mouse chromosome 16 (MMU16) and part of MMU10 and MMU17 share synteny with human chromosome 21 (HSA21) [1] (Figure 2). Therefore, several mouse models carrying an extra partial MMU16, MMU10, or MMU17 have been established as mouse models for DS (Figure 2). In particular, MMU16 has synteny with a large portion of HSA21, which has led to the establishment of several mouse models of DS: Dp(16)Yey [2], Ts65Dn [3], Ts1Cje [4], Ms1Ts65 [5], and Ts1Rhr mice [6] (Figure 2). Furthermore, seven mouse models of DS carrying three copies of various segments of MMU16 syntenic to HSA21 have been recently established: Dp1Tyb, Dp2Tyb, Dp3Tyb, Dp4Tyb, Dp5Tyb, Dp6Tyb, and Dp9Tyb [7] (Figure 2). The Ts65Dn model is the most intensively studied model of DS to date and carries a 13.2 Mb segmental trisomy of MMU16, and the Ts1Cje model carrying a shorter segmental trisomy (7.6 Mb) is also widely used. Both models display DS-like phenotypes, including learning and memory deficits and/or an abnormal neuron morphology and function [3,4,8,9,10].
2. Comparative Transcriptomics in the DS Brain
The analysis of the expression of transcripts in humans with DS and mouse models of DS provides valuable information for understanding the pathophysiological mechanisms underlying this disease. However, whether or not the expression of trisomic genes is upregulated in a gene-dose-dependent manner and the effect of trisomic gene expression on the expression of transcripts from euploid genes remain unclear.
A number of comprehensive gene expression analyses using tissue of humans with DS and mouse models of DS have been performed. The findings from studies using brain tissue are summarized in Table 1. Although relatively few studies have been performed with human DS samples, two included in Table 1 indicate that a gene-dose-dependent increase in transcription was detected in the brain both before and after birth [11,12].
A number of comparative transcriptomic analyses in brains of mouse models of DS have been performed (Table 1). The precise brain region and the age of the samples analyzed must be considered for an accurate interpretation of these comparative transcriptomics data. However, a variety of samples have been used, and most (but not all) transcriptomic analyses comparing trisomic and euploid tissues support the hypothesis of increased mRNA levels in a gene-dose-dependent manner. In addition, a number of reports have shown that the mRNA expression of some euploid genes is disturbed in the brain of DS model mice.
Although transcriptomic analyses using DS brains in humans and mice provide important information, as described above, the interpretation of the data is very complex. RNA sequencing is a powerful analytical tool that is expected to replace microarrays for transcriptomics profiling [25]. RNA sequencing has several advantages over microarrays, such as the avoidance cross-hybridization, the limited detection range of individual probes, and non-specific hybridization. RNA sequencing could, therefore, provide new information on the gene expression in brains with DS.
3. Comparative Proteomics in the DS Brain
The protein expression does not always correlate with the expression of mRNA, so a comprehensive analysis of the protein expression in brains with DS is very important for understanding the pathomechanisms of the disease. A number of comparative proteomics analyses using the brains from patients and mouse models have been performed. The reports including such analyses for understanding changes in the protein expression in the brains with DS and neuronal cells derived from embryonic stem (ES) cells with an extra HSA21 are listed in Table 2. In contrast to the findings on gene expression analyses, a number of proteomics analyses failed to detect any increased expression of trisomic genes at the protein level (Table 2). This might be due to the sensitivity of the proteomic analysis, especially two-dimensional (2D)-electrophoresis, since antibody arrays successfully detected an increase in the protein expression of trisomic genes [26]. Recently-developed chemical labeling techniques using isobaric tags (i.e., tags with the same molecular weight) for the relative and absolute quantification (iTRAQ) [27] and tandem mass tags (TMT) [28] are expected to resolve the issue of the low sensitivity of separation on 2D-electrophoresis.
A number of proteins associated with proteolysis, energy metabolism, the cytoskeleton, and cell proliferation that are coded in disomic genes were found to be dysregulated in humans and mice with DS (Table 2) [29,30,31], although proteins, which were differentially expressed in both human and mouse brain with DS, have not yet been identified in these proteomic analysis. For instance, ubiquitin carboxyl-terminal hydrolase isozyme L1 (Uchl1) coded on an euploid gene showed a decreased expression in the brain of a 141G6 mouse carrying a yeast artificial chromosome (YAC), which contains HSA21 genes, Down syndrome critical region gene 3 (Dscr3), phosphatidylinositol glycan anchor biosynthesis, class P (Pigp), ripply transcriptional repressor 3 (Ripply3), and tetratricopeptide repeat domain 3 (Ttc3), as well as neurons differentiated from ES cells carrying an extra HSA21 [32,31]. In addition, proteolysis and energy metabolism-related proteins, such as Uchl1 and α-enolase, show not only differential expression, but also oxidative modification under DS conditions, as will be explain in the next section.
4. Redox Proteomics in the DS Brain
Since proteins are major targets of reactive oxygen, nitrogen species and aldehyde products of lipid peroxidation (LPO), comparative analysis of these protein modifications reveals the degree of oxidative stress, and the certain target molecules of enhanced oxidative stress. Therefore, redox proteomics is a useful tool to decipher the effect of oxidative stress in the brain with DS. As shown in Table 3, redox proteomics detecting carbomylated- or 4-hydroxynonenal (4-HNE) adducted-proteins revealed that the carbonylated UchL1 level was increased in the human DS brain [33]. Furthermore, although the levels of LPO-related modified proteins, such as 13(S)-hydroperoxyoctadeca-9Z,11E-dienoic acid (13-HPODE)-adducted α-enolase, glycolytic enzyme, and 4-HNE-adducted neuron specific enolase, are increased in Ts1Cje mice [34], the expression of α-enolase and neuron-specific enolase decreased in the human DS cortex and the hippocampus of 141G6 mice [29,32]. The impairment of the energy metabolism is suggested to be closely related to several age-related neurodegenerative disorders, such as Alzheimer’s disease and mild cognitive impairment (MCI) [35]. Thus, the oxidative stress-mediated decline in certain protein functions may be associated with abnormalities of the DS brain.
5. Other ‘-omics’ Analyses
Other ‘-omics’ analyses, such as metabolomics including lipidomics and elementomics, including metallomics, are expected to prove useful in understanding the pathophysiological state of DS. As no proteins were found to be differentially expressed in the Ts1Cje mouse brain during neonatal and postnatal life by 2D-electrophoresis-based proteomics [30], these other ‘-omics’ approaches are thought to be particularly useful.
A metabolomics approach can digest a very large volume of information regarding the levels of the complete set of low-molecular-weight molecules or metabolites synthesized by a cell. The alteration of biogenic metabolites, such as neurotransmitters and prostaglandins, in abundance provides a unique chemical fingerprint that specific cellular processes leave behind. Although there have been a number of metabolomics studies on the accumulation of such elements in the brains of Alzheimer’s disease patients (see [44]), no metabolomics studies with DS subjects have yet been reported.
Elementomics, including metallomics, is another new ‘-omics’ approach using inductively-coupled plasma-mass spectrometry (ICP-MS) [45]. Elementomics comprehensively elucidate the levels of the most common elements, including biogenic trace metals, in the tissues and cells. This approach has only been applied to study Alzheimer’s disease, with no reports in DS subjects available at present [46,47]. The dyshomeostasis of intrinsic metals, such as zinc, copper, and iron, may play a role in cognitive impairment in individuals with DS, as intersectin 1 coded in the trisomic region in DS is suggested to be involved in iron internalization. Indeed, increases in the levels of non-protein-bound iron in serum and iron-binding protein lactotransferrin in the brain have been demonstrated in individuals with DS [48,49]. Other ‘-omics’ analyses, such as metabolomic and elementomics analyses, for DS samples are expected to help unravel the pathophysiological mechanisms underlying abnormalities in the DS brain.
Acknowledgments
This work is supported, in part, by KAKENHI (#20790089 and #25460077 to K.I.), the Takeda Science Foundation (to K.I.), the Shimizu Foundation for Immunology and Neuroscience Grant for 2014 (to K.I.), Kyoto Pharmaceutical University Fund for the Promotion of Scientific Research (to K.I.), and the Strategic Research Foundation Grant-aided Project for Private Universities from the Ministry of Education, Culture, Sport, Science, and Technology, Japan (MEXT), 2012–2016 (S1201008).
Author Contributions
K.I. wrote the manuscript, S.A. edited the paper.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Davisson, M.T.; Bechtel, L.J.; Akeson, E.C.; Fortna, A.; Slavov, D.; Gardiner, K. Evolutionary breakpoints on human chromosome 21. Genomics 2001, 78, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Yu, T.; Morishima, M.; Pao, A.; LaDuca, J.; Conroy, J.; Nowak, N.; Matsui, S.; Shiraishi, I.; Yu, Y.E. Duplication of the entire 22.9 Mb human chromosome 21 syntenic region on mouse chromosome 16 causes cardiovascular and gastrointestinal abnormalities. Hum. Mol. Genet. 2007, 16, 1359–1366. [Google Scholar] [CrossRef] [PubMed]
- Davisson, M.T.; Schmidt, C.; Akeson, E.C. Segmental trisomy of murine chromosome 16: A new model system for studying Down syndrome. Prog. Clin. Biol. Res. 1990, 360, 263–280. [Google Scholar] [PubMed]
- Sago, H.; Carlson, E.J.; Smith, D.J.; Kilbridge, J.; Rubin, E.M.; Mobley, W.C.; Epstein, C.J.; Huang, T.T. Ts1Cje, a partial trisomy 16 mousemodel for Downsyndrome, exhibits learning and behavioral abnormalities. Proc. Natl. Acad. Sci. USA 1998, 95, 6256–6261. [Google Scholar] [CrossRef] [PubMed]
- Sago, H.; Carlson, E.J.; Smith, D.J.; Rubin, E.M.; Crnic, L.S.; Huang, T.T.; Epstein, C.J. Genetic dissection of region associated with behavioral abnormalities in mouse models for Down syndrome. Pediatr. Res. 2000, 48, 606–613. [Google Scholar] [CrossRef] [PubMed]
- Olson, L.E.; Richtsmeier, J.T.; Leszl, J.; Reeves, R.H. A chromosome 21 critical region does not cause specific Down syndrome phenotypes. Science 2004, 306, 687–690. [Google Scholar] [CrossRef] [PubMed]
- Lana-Elola, E.; Watson-Scales, S.; Slender, A.; Gibbins, D.; Martineau, A.; Douglas, C.; Mohun, T.; Fisher, E.M.; Tybulewicz, V.L. Genetic dissection of Down syndrome-associated congenital heart defects using a new mouse mapping panel. Elife 2016, 14, e11614. [Google Scholar] [CrossRef] [PubMed]
- Siarey, R.J.; Villar, A.J.; Epstein, C.J.; Galdzicki, Z. Abnormal synaptic plasticity in the Ts1Cje segmental trisomy 16 mouse model of Down syndrome. Neuropharmacology 2005, 49, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Belichenko, P.V.; Kleschevnikov, A.M.; Salehi, A.; Epstein, C.J.; Mobley, W.C. Synaptic and cognitive abnormalities in mouse models of Down syndrome: Exploring genotype-phenotype relationships. J. Comp. Neurol. 2007, 504, 329–345. [Google Scholar] [CrossRef] [PubMed]
- Belichenko, N.P.; Belichenko, P.V.; Kleschevnikov, A.M.; Salehi, A.; Reeves, R.H.; Mobley, W.C. The “Down syndrome critical region” is sufficient in the mouse model to confer behavioral, neurophysiological, and synaptic phenotypes characteristic of Down syndrome. J. Neurosci. 2009, 29, 5938–5948. [Google Scholar] [CrossRef] [PubMed]
- Mao, R.; Wang, X.; Spitznagel, E.L., Jr.; Frelin, L.P.; Ting, J.C.; Ding, H.; Kim, J.W.; Ruczinski, I.; Downey, T.J.; Pevsner, J. Primary and secondary transcriptional effects in the developing human Down syndrome brain and heart. Genome Biol. 2005, 6, R107. [Google Scholar] [CrossRef] [PubMed]
- Lockstone, H.E.; Harris, L.W.; Swatton, J.E.; Wayland, M.T.; Holland, A.J.; Bahn, S. Gene expression profiling in the adult Down syndrome brain. Genomics 2007, 90, 647–660. [Google Scholar] [CrossRef] [PubMed]
- Chrast, R.; Scott, H.S.; Papasavvas, M.P.; Rossier, C.; Antonarakis, E.S.; Barras, C.; Davisson, M.T.; Schmidt, C.; Estivill, X.; Dierssen, M.; et al. The mouse brain transcriptome by SAGE: Differences in gene expression between P30 brains of the partial trisomy 16 mouse model of Down syndrome (Ts65Dn) and normals. Genome Res. 2000, 10, 2006–2021. [Google Scholar] [CrossRef] [PubMed]
- Sultan, M.; Piccini, I.; Balzereit, D.; Herwig, R.; Saran, N.G.; Lehrach, H.; Reeves, R.H.; Yaspo, M.L. Gene expression variation in Down’s syndrome mice allows prioritization of candidate genes. Genome Biol. 2007, 8, R91. [Google Scholar] [CrossRef] [PubMed]
- Alldred, M.J.; Lee, S.H.; Petkova, E.; Ginsberg, S.D. Expression profile analysis of vulnerable CA1 pyramidal neurons in young-Middle-Aged Ts65Dn mice. J. Comp. Neurol. 2015, 523, 61–74. [Google Scholar] [CrossRef] [PubMed]
- Walus, M.; Kida, E.; Rabe, A.; Albertini, G.; Golabek, A.A. Widespread cerebellar transcriptome changes in Ts65Dn Down syndrome mouse model after lifelong running. Behav. Brain Res. 2016, 296, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Alldred, M.J.; Lee, S.H.; Petkova, E.; Ginsberg, S.D. Expression profile analysis of hippocampal CA1 pyramidal neurons in aged Ts65Dn mice, a model of Down syndrome (DS) and Alzheimer’s disease (AD). Brain Struct. Funct. 2015, 220, 2983–2996. [Google Scholar] [CrossRef] [PubMed]
- Guedj, F.; Pennings, J.L.; Ferres, M.A.; Graham, L.C.; Wick, H.C.; Miczek, K.A.; Slonim, D.K.; Bianchi, D.W. The fetal brain transcriptome and neonatal behavioral phenotype in the Ts1Cje mouse model of Down syndrome. Am. J. Med. Genet. A. 2015, 167, 1993–2008. [Google Scholar] [CrossRef] [PubMed]
- Amano, K.; Sago, H.; Uchikawa, C.; Suzuki, T.; Kotliarova, S.E.; Nukina, N.; Epstein, C.J.; Yamakawa, K. Dosage-dependent over-expression of genes in the trisomic region of Ts1Cje mouse model for Down syndrome. Hum. Mol. Genet. 2004, 13, 1333–1340. [Google Scholar] [CrossRef] [PubMed]
- Dauphinot, L.; Lyle, R.; Rivals, I.; Dang, M.T.; Moldrich, R.X.; Golfier, G.; Ettwiller, L.; Toyama, K.; Rossier, J.; Personnaz, L.; et al. The cerebellar transcriptome during postnatal development of the Ts1Cje mouse, a segmental trisomy model for Down syndrome. Hum. Mol. Genet. 2005, 14, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Laffaire, J.; Rivals, I.; Dauphinot, L.; Pasteau, F.; Wehrle, R.; Larrat, B.; Vitalis, T.; Moldrich, R.X.; Rossier, J.; Sinkus, R.; et al. Gene expression signature of cerebellar hypoplasia in a mouse model of Down syndrome during postnatal development. BMC Genom. 2009, 30, 138. [Google Scholar] [CrossRef] [PubMed]
- Ling, K.H.; Hewitt, C.A.; Tan, K.L.; Cheah, P.S.; Vidyadaran, S.; Lai, M.I.; Lee, H.C.; Simpson, K.; Hyde, L.; Pritchard, M.A.; et al. Functional transcriptome analysis of the postnatal brain of the Ts1Cje mouse model for Down syndrome reveals global disruption of interferon-related molecular networks. BMC Genom. 2014, 22, 624. [Google Scholar] [CrossRef] [PubMed]
- Guedj, F.; Pennings, J.L.; Wick, H.C.; Bianchi, D.W. Analysis of adult cerebral cortex and hippocampus transcriptomes reveals unique molecular changes in the Ts1Cje mouse model of down syndrome. Brain Pathol. 2015, 25, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Moldrich, R.X.; Dauphinot, L.; Laffaire, J.; Vitalis, T.; Hérault, Y.; Beart, P.M.; Rossier, J.; Vivien, D.; Gehrig, C.; Antonarakis, S.E.; et al. Proliferation deficits and gene expression dysregulation in Down’s syndrome (Ts1Cje) neural progenitor cells cultured from neurospheres. J. Neurosci. Res. 2009, 87, 3143–3152. [Google Scholar] [CrossRef] [PubMed]
- Mutz, K.O.; Heilkenbrinker, A.; Lönne, M.; Walter, J.G.; Stahl, F. Transcriptome analysis using next-generation sequencing. Curr. Opin. Biotechnol. 2013, 24, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.M.; Dhanasekaran, A.R.; Block, A.; Tong, S.; Costa, A.C.; Stasko, M.; Gardiner, K.J. Protein dynamics associated with failed and rescued learning in the Ts65Dn mouse model of Down syndrome. PLoS ONE 2015, 10, e0119491. [Google Scholar] [CrossRef] [PubMed]
- Ross, P.L.; Huang, Y.N.; Marchese, J.N.; Williamson, B.; Parker, K.; Hattan, S.; Khainovski, N.; Pillai, S.; Dey, S.; Daniels, S.; et al. Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol. Cell Proteom. 2004, 3, 1154–1169. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.; Schäfer, J.; Kuhn, K.; Kienle, S.; Schwarz, J.; Schmidt, G.; Neumann, T.; Johnstone, R.; Mohammed, A.K.; Hamon, C. Tandem mass tags: A novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Anal. Chem. 2003, 75, 1895–1904. [Google Scholar] [CrossRef] [PubMed]
- Cenini, G.; Fiorini, A.; Sultana, R.; Perluigi, M.; Cai, J.; Klein, J.B.; Head, E.; Butterfield, D.A. An investigation of the molecular mechanisms engaged before and after the development of Alzheimer disease neuropathology in Down syndrome: A proteomics approach. Free Radic. Biol. Med. 2014, 76, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, K.; Kanai, S.; Sago, H.; Yamakawa, K.; Akiba, S. Comparative proteomic profiling reveals aberrant cell proliferation in the brain of embryonic Ts1Cje, a mouse model of Down syndrome. Neuroscience 2014, 281, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Kadota, M.; Nishigaki, R.; Wang, C.C.; Toda, T.; Shirayoshi, Y.; Inoue, T.; Gojobori, T.; Ikeo, K.; Rogers, M.S.; Oshimura, M. Proteomic signatures and aberrations of mouse embryonic stem cells containing a single human chromosome 21 in neuronal differentiation: An in vitro model of Down syndrome. Neuroscience 2004, 129, 325–335. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.H.; Gulesserian, T.; Verger, E.; Delabar, J.M.; Lubec, G. Protein dysregulation in mouse hippocampus polytransgenic for chromosome 21 structures in the Down Syndrome Critical Region. J. Proteome Res. 2006, 5, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Di Domenico, F.; Coccia, R.; Cocciolo, A.; Murphy, M.P.; Cenini, G.; Head, E.; Butterfield, D.A.; Giorgi, A.; Schinina, M.E.; Mancuso, C.; et al. Impairment of proteostasis network in Down syndrome prior to the development of Alzheimer’s disease neuropathology: Redox proteomics analysis of human brain. Biochim. Biophys. Acta 2013, 1832, 1249–1259. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, K.; Amano, K.; Takaki, E.; Ebrahim, A.S.; Shimohata, A.; Shibazaki, N.; Inoue, I.; Takaki, M.; Ueda, Y.; Sago, H.; et al. Increased lipid peroxidation in Down’s syndrome mouse models. J. Neurochem. 2009, 110, 1965–1976. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Poon, H.F.; St Clair, D.; Keller, J.N.; Pierce, W.M.; Klein, J.B.; Markesbery, W.R. Redox proteomics identification of oxidatively modified hippocampal proteins in mild cognitive impairment: Insights into the development of Alzheimer’s disease. Neurobiol. Dis. 2006, 22, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Peyrl, A.; Weitzdoerfer, R.; Gulesserian, T.; Fountoulakis, M.; Lubec, G. Aberrant expression of signaling-related proteins 14-3-3 gamma and RACK1 in fetal Down syndrome brain (trisomy 21). Electrophoresis 2002, 23, 152–157. [Google Scholar] [CrossRef]
- Engidawork, E.; Gulesserian, T.; Fountoulakis, M.; Lubec, G. Aberrant protein expression in cerebral cortex of fetus with Down syndrome. Neuroscience 2003, 122, 145–154. [Google Scholar] [CrossRef]
- Kim, S.H.; Krapfenbauer, K.; Cheon, M.S.; Fountoulakis, M.; Cairns, N.J.; Lubec, G. Human brain cytosolic histamine-N-methyltransferase is decreased in Down syndrome and increased in Pick’s disease. Neurosci. Lett. 2002, 321, 169–172. [Google Scholar] [CrossRef]
- Ahmed, M.M.; Sturgeon, X.; Ellison, M.; Davisson, M.T.; Gardiner, K.J. Loss of correlations among proteins in brains of the Ts65Dn mouse model of Down syndrome. J. Proteome Res. 2012, 11, 1251–1263. [Google Scholar] [CrossRef] [PubMed]
- Palminiello, S.; Kida, E.; Kaur, K.; Walus, M.; Wisniewski, K.E.; Wierzba-Bobrowicz, T.; Rabe, A.; Albertini, G.; Golabek, A.A. Increased levels of carbonic anhydrase II in the developing Down syndrome brain. Brain Res. 2008, 1190, 193–205. [Google Scholar] [CrossRef] [PubMed]
- Block, A.; Ahmed, M.M.; Dhanasekaran, A.R.; Tong, S.; Gardiner, K.J. Sex differences in protein expression in the mouse brain and their perturbations in a model of Down syndrome. Biol. Sex Differ. 2015, 9, 24. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.H.; Guedj, F.; Delabar, J.M.; Lubec, G. Dysregulation of growth factor receptor-bound protein 2 and fascin in hippocampus of mice polytransgenic for chromosome 21 structures. Hippocampus 2007, 17, 1180–1192. [Google Scholar] [CrossRef] [PubMed]
- Di Domenico, F.; Pupo, G.; Tramutola, A.; Giorgi, A.; Schininà, M.E.; Coccia, R.; Head, E.; Butterfield, D.A.; Perluigi, M. Redox proteomics analysis of HNE-modified proteins in Down syndrome brain: Clues for understanding the development of Alzheimer disease. Free Radic. Biol. Med. 2014, 71, 270–280. [Google Scholar] [CrossRef] [PubMed]
- González-Domínguez, R.; Sayago, A.; Fernández-Recamales, Á. Metabolomics in Alzheimer’s disease: The need of complementary analytical platforms for the identification of biomarkers to unravel the underlying pathology. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2017, in press. [Google Scholar]
- Li, Y.F.; Chen, C.Y.; Qu, Y.; Gao, Y.X.; Li, B.; Zhao, Y.L.; Chai, Z.F. Metallomics, elementomics, and analytical techniques. Pure Appl. Chem. 2008, 80, 2577–2594. [Google Scholar] [CrossRef]
- Braidy, N.; Poljak, A.; Marjo, C.; Rutlidge, H.; Rich, A.; Jayasena, T.; Inestrosa, N.C.; Sachdev, P. Metal and complementary molecular bioimaging in Alzheimer's disease. Front. Aging Neurosci. 2014, 15, 138. [Google Scholar] [CrossRef] [PubMed]
- González-Domínguez, R.; García-Barrera, T.; Gómez-Ariza, J.L. Characterization of metal profiles in serum during the progression of Alzheimer’s disease. Metallomics 2014, 6, 292–300. [Google Scholar] [CrossRef] [PubMed]
- Manna, C.; Officioso, A.; Trojsi, F.; Tedeschi, G.; Leoncini, S.; Signorini, C.; Ciccoli, L.; De Felice, C. Increased non-protein bound iron in Down syndrome: Contribution to lipid peroxidation and cognitive decline. Free Radic. Res. 2016, 50, 1422–1431. [Google Scholar] [CrossRef] [PubMed]
- Leveugle, B.; Spik, G.; Perl, D.P.; Bouras, C.; Fillit, H.M.; Hof, P.R. The iron-binding protein lactotransferrin is present in pathologic lesions in a variety of neurodegenerative disorders: A comparative immunohistochemical analysis. Brain Res. 1994, 650, 20–31. [Google Scholar] [CrossRef]
Figure 1.
A diagram of the interaction between ‘-omics’ and the flow of biological molecules. A number of ‘-omics’ analyses have been developed. Transcriptomics, proteomics, lipidomics, elementomics, and metabolomics analyses describe the levels of mRNA, proteins, lipids, such as phospholipids, elements, such as trace biometals, and various metabolites, such as prostaglandins, respectively.
Figure 1.
A diagram of the interaction between ‘-omics’ and the flow of biological molecules. A number of ‘-omics’ analyses have been developed. Transcriptomics, proteomics, lipidomics, elementomics, and metabolomics analyses describe the levels of mRNA, proteins, lipids, such as phospholipids, elements, such as trace biometals, and various metabolites, such as prostaglandins, respectively.
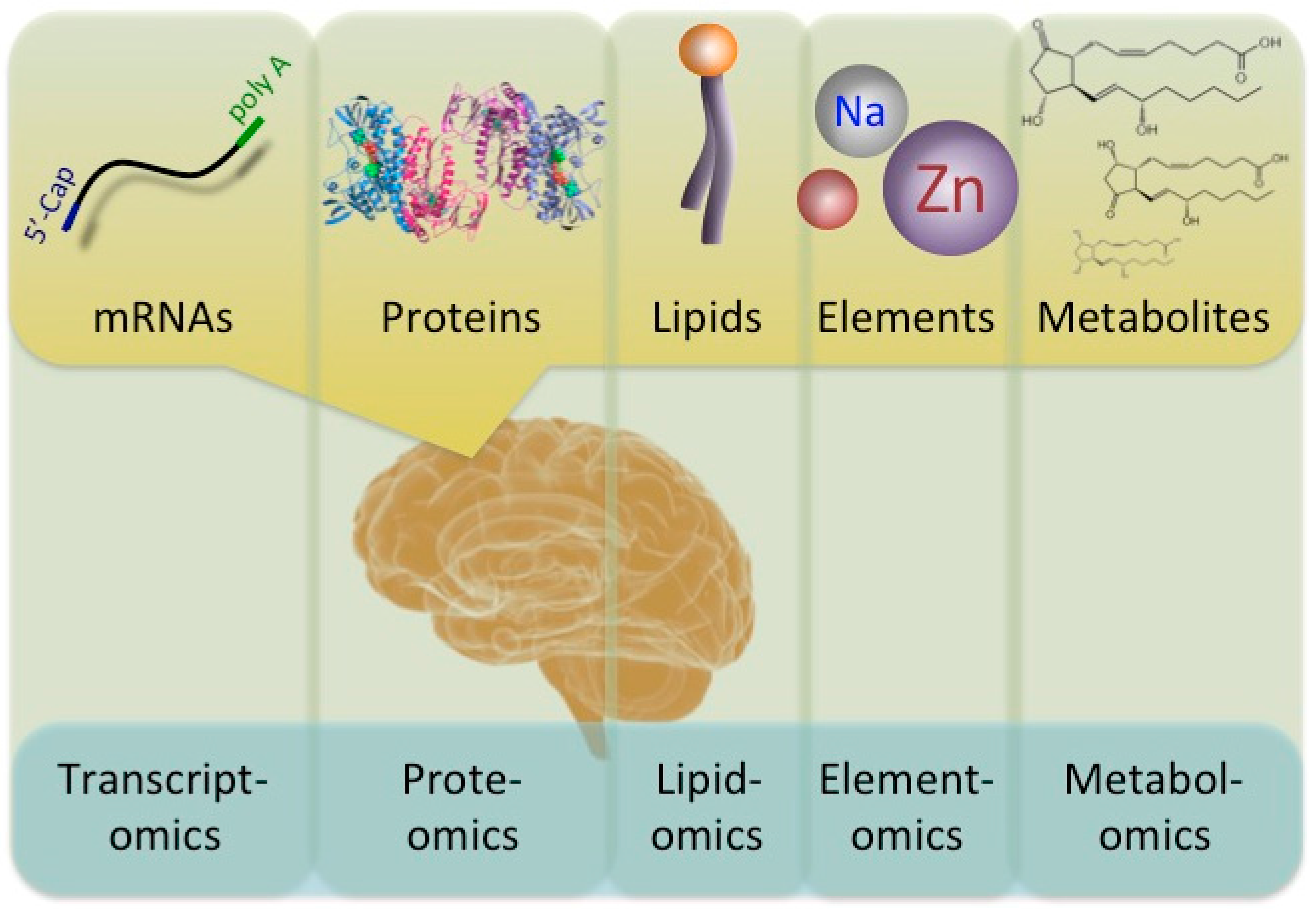
Figure 2.
Segmentally trisomic regions of DS mouse models. Most of HSA21 is syntenic to the distal end of MMU16. The trisomic regions in several mouse models of DS are compared on the right of the MMU16 (orange bars). The numbers in brackets represent the number of protein-coding genes within each MMU16 region according to Ensembl release 87. Lipi: lipase, member I, Mrpl39: mitochondrial ribosomal protein L39, Scaf4: SR-related CTD-associated factor 4, Hunk: hormonally upregulated Neu-associated kinase, Runx1: runt-related transcription factor 1, Cbr1: carbonyl reductase 1, Dscr3: Down syndrome critical region gene 3, B3Galt5: UDP-Gal:betaGlcNAc beta 1,3-galactosyltransferase, polypeptide 5, Mx2: MX dynamin-like GTPase 2, and Zbtb21: zinc finger and BTB domain containing 21.
Figure 2.
Segmentally trisomic regions of DS mouse models. Most of HSA21 is syntenic to the distal end of MMU16. The trisomic regions in several mouse models of DS are compared on the right of the MMU16 (orange bars). The numbers in brackets represent the number of protein-coding genes within each MMU16 region according to Ensembl release 87. Lipi: lipase, member I, Mrpl39: mitochondrial ribosomal protein L39, Scaf4: SR-related CTD-associated factor 4, Hunk: hormonally upregulated Neu-associated kinase, Runx1: runt-related transcription factor 1, Cbr1: carbonyl reductase 1, Dscr3: Down syndrome critical region gene 3, B3Galt5: UDP-Gal:betaGlcNAc beta 1,3-galactosyltransferase, polypeptide 5, Mx2: MX dynamin-like GTPase 2, and Zbtb21: zinc finger and BTB domain containing 21.

{kind=link}
{kind=link}
Table 1.
A comparative transcriptomic analysis of the brain with DS.
| Sample (Genetic Background) | Tissue | Number of Samples | Stage | Method | Transcription of Trisomic Genes | Remarks | Reference |
|---|---|---|---|---|---|---|---|
| Human | Cerebellum | Control: n = 3; DS: n = 3 | 18–20 weeks of gestation | Affymetrix U133A GeneChip | A gene dosage-dependent increase in transcription was detected in the cerebellum of individuals with DS. | [11] | |
| Human | Dorsolateral prefrontal cortex | Control: n = 8; DS: n = 7 | Adult | Affymetrix human genome HG-U133A GeneChip | More than 25% of genes on HSA21 were differentially expressed, versus a median of 4.4% for all chromosomes. | Dysregulated genes are classified into development (notably Notch and Dlx family genes), lipid transport, and cellular proliferation. | [12] |
| Ts65Dn mice (B6Ei/C3) | Whole brain | Control: n = 7, 4 males, 3 males Ts65Dn: n = 3, 3 males | Postnatal day 30 | SAGE analysis with library (28,531 tags) | The expression of most known genes from the trisomic region of mouse MMU16 in Ts65Dn is too low. Only three genes (Ifnar2, Ufngr2, and Cbr) are overexpressed in Ts65Dn males compared to control males. | It has been suggested that abnormal ribosomal biogenesis may be involved in the development and maintenance of DS phenotypes. | [13] |
| Ts65Dn mice (B6Ei/C3) | Cerebellum, cortex, midbrain | Control: n = 8 Ts65Dn: n = 8 | Males at 1–4 months old | Quantitative real-time RT-PCR | A trend toward 1.5-fold over-expression for the trisomic genes was detected. The global over-expression level of trisomic genes in Ts65Dn was 1.44-fold in the cerebellum, 1.37-fold in the cortex, and 1.39-fold in the midbrain. | [14] | |
| Ts65Dn mice (B6Ei/C3) | CA1 pyramidal cells | Control: n = 7; Ts65Dn: n = 9 | 4–9 months old | Custom-designed array (576 cDNA/ESRs) | N/A | Downregulation of neutrophins and their cognate neutrophin receptors. | [15] |
| Ts65Dn mice (B6Ei/C3) | Cerebellum | Control (sedentary): n = 4 Control (running): n = 4 Ts65Dn (sedentary) n = 4 Ts65Dn (running): n = 4 | Females at 9–13 months old | Agilent oligonucleotide microarray (SurePrint G3 Mouse Gene Expression 8 × 60 K Microarray) | Forty tested trisomic genes showed higher expression in Ts65Dn mice than in euploid mice, with an average ratio of Ts65Dn/WT 1.47. | [16] | |
| Ts65Dn mice (B6Ei/C3) | CA1 pyramidal cells | Control: n = 12 Ts65Dn: n = 13 | 10–24 months old | Custom-designed array (576 cDNA/ESRs) | N/A | Dysregulation of excitatory and inhibitory neurotransmission receptor families and neurotrophins, including brain-derived neurotrophic factor, as well as several cognate neurotrophin receptors. | [17] |
| Ts1Cje mice (C57BL/6J) | Fetal brain | Control: n = 5 Ts1Cje: n = 5 | Embryonic day 15.5 | Affymetrix mouse gene 1.0 ST arrays | About half of the trisomic genes were significantly upregulated in the embryonic brain of Ts1Cje mice. | [18] | |
| Ts1Cje mice (C57BL/6J) | Whole brain | Control: n = 6 Ts1Cje: n = 6 | Males at postnatal day 0 | Affymetrix murine genome U74A and U74B microarrays | The expression of most genes in the trisomic region was increased approximately 1.5-fold, and the top 24 most consistently over-expressed genes in Ts1Cje mice were all located in the trisomic region. | The transcripts of trisomic genes were mainly overexpressed in a gene-dose-dependent manner. | [19] |
| Ts1Cje mice (C57BL/6) | Cerebellum | Control: n = 2 for each stage Ts1Cje: n = 2 for each stage | Postnatal day 0, 15, and 30 | Affymetrix murine genome U74A version | The mean expression ratios of trisomic genes between Ts1Cje and controls were 1.66, 1.32, and 1.32 at P0, P15, and P30, respectively, whereas with euploid genes, the ratios were 1.08, 1.12, and 1.02 at P0, P15, and P30, respectively. | In the cerebellum of Ts1Cje mice, six homeobox genes and two genes belonging to the Notch pathway showed severely decreased expression | [20] |
| Ts1Cje mice (B6C3SnF1/ Orl) | Cerebellum | Control: n = 2 for each stage Ts1Cje: n = 2 for each stage | Postnatal day 0, 3, 7, and 10 | Agilent RNA 6000 | A prevailing gene dosage effect of trisomy and a limited secondary effect on postnatal development were noted. Approximately 80% of gene expression differences were attributed to dosage imbalance, suggesting that the trisomic genes are likely to be directly responsible for the phenotype present in cerebellum of Ts1Cje mice. | [21] | |
| Ts1Cje mice (C57BL/6) | Cerebral cortexcerebellum hippocampus | Control: n = 3 for each stage Ts1Cje: n = 3 for each stage | Postnatal day 1, 15, 30, and 84 | Affymetrix murine genome U74A version 3 microarray | A gene dosage-dependent increase in transcription was detected in the cerebellum of individuals with DS. | The Jak-Stat pathway may be overstimulated in the brain of Ts1Cje mice. | [22] |
| Ts1Cje mice (C57BL/6J) | Cerebral cortex hippocampus | Control: n = 5 Ts1Cje: n = 6 | Females at 2–2.5 months old | Affymetrix mouse gene 1.0 ST arrays | Of the 77 genes present in the trisomic region of Ts1Cje mice, 22 (28.6%) were differentially regulated in either the cortex or hippocampus, while the expression of the remaining 46 (71.4%) was not affected. | Dysregulation of NFAT signaling, and G-protein signaling (e.g., olfactory perception) | [23] |
| Ts1Cje mice (B6C3SnF1/Orl) | Neural progenitor cells | Control: n = 3 Ts1Cje: n = 3 | Neurospheres were derived from E14.5 cortex | DNA microarrays (RNG-MRC_MM25k_EVRY) | The expression ratios of 54% of trisomic genes (Ts1Cje/WT) were significantly higher than the expected diploid gene ratio of 1.0. | Ts1Cje neural progenitors proliferated at a slower rate. Some euploid genes involved in proliferation, differentiation, and the glial function were dysregulated. | [24] |
Table 2.
A comparative proteomic analysis of the brain with DS.
| Sample | Tissue | Sample Information | Method | Differentially Expressed Proteins | Remarks | Reference | |
|---|---|---|---|---|---|---|---|
| Human | fetal cortex | controls (18.8 ± 2.2 weeks of gestation) n = 7 (one female, six males) DS (19.6 ± 2.0 weeks of gestation) n = 9 (two females, seven males) | 2D-PAGE/MALDI- TOF-MS | 14-3-3γ (↓), Receptor of activated protein C kinase 1 (RACK1) (↓) | [36] | ||
| Human | fetal cortex | controls (18.8 ± 2.2 weeks of gestation) n = 7 (one female, six males) DS (19.6 ± 2.0 weeks of gestation) n = 9 (two females, seven males) | 2D-PAGE/MALDI- TOF-MS | Double-strand-break repair protein rad 21 (Rad21) (↑), Eukaryotic initiation factor 3 (eIF3) p47 subunit 5 (↑), heat shock protein (Hsp) 75 (↑), septin 7 (↓), β- amyloid precursor-like protein 1 (↓), β-tubulin (↓) | [37] | ||
| Human | frontal cortex | Young controls (13.1 ± 15.3 years old) n = 6 Old controls (53.0 ± 8.5 years old ) n = 6 DS (11.01 ± 10.9 years old) n = 6 | 2D-PAGE/nano-LC-MS | DS vs. young controls Ras-related protein Rab3a (↓), Guanine nucleotide-binding protein 1 (Bnb1) (↓), Apolipoprotein E (↓), Transitional endoplasmic reticulum ATPase (Vcp) (↓), pyridoxal phosphate phosphatase (↓), Malate Dehydrogenase 2 (Mdh2) (↑), α-enolase (↓) | Overlapping and independent molecular pathways, such as energy metabolism, oxidative damage, protein synthesis, and autophagy, are suggested to be involved in DS, aging, and DA/AD. | [29] | |
| Human | frontal cortex, cerebellum | frontal cortex controls (62.80 ± 8.61 years old) n = 5; AD (60.80 ±103.62 years old) n = 5 DS (56.00 ± 10.49 years old) n = 5 | cerebellum controls (68.50 ± 6.25 years old) n = 4 AD (59.86 ± 6.47 years old) n = 7 DS (55.43 ±8.62 years old) n = 7 | 2D-PAGE/ MALDI-TOF-MS | frontal cortex Histamine-N-methyltransferase (↓) | [38] | |
| Ts65Dn mice | cerebellum, cerebral cortex | Control males (4.4–7.8 months old) n = 6; Ts65Dn males (4.4–7.8 months old) n = 5 | Protein arrays (64 proteins/protein modifications) | Only a small number of trisomic proteins were increased in a gene-dose-dependent manner. | Ts65Dn mice have lost the correlations seen in control mice among levels of functionally related proteins, including the components of the MAP kinase pathway and subunits of the NMDA receptor. | [39] | |
| Ts65Dn mice | cerebellum | Control and Ts65Dn (postnatal day 0, 16, and 21 and 3, 4–6, 8, 12, and 14–21 months old) n = 52 (total) | 2D-PAGE/MS | Carbonic anhydrase II (↑) | Increased levels of carbonic anhydrase II in the developing brain with DS | [40] | |
| Ts65Dn mice | hippocampus | Wild-type/context-shock/saline (males at 3–4 months old) n = 10; wild-type/shock-context/saline (males at 3–4 months old) n = 10; wild-type/context-shock/memantine (males at 3–4 months old) n = 10; wild-type/shock-context/memantine (males at 3–4 months old) n = 10; Ts65Dn/context-shock/saline (males at 3–4 months old) n = 9; Ts65Dn/shock-context/saline (males at 3-4 m-old) n = 10; Ts65Dn/context-shock/memantine (males at 3–4 months old) n = 10; Ts65Dn/shock-context/memantine (males at 3–4 months old) n = 10 | Reverse phase protein arrays (85 proteins/protein modifications) | (i) the dynamic responses seen in control mice in normal learning, >40% also occur in Ts65Dn in failed learning or are compensated by baseline abnormalities, and thus are considered necessary but not sufficient for successful learning, and (ii) treatment with memantine does not in general normalize the initial protein levels but instead induces direct and indirect responses in approximately half the proteins measured and results in normalization of the endpoint protein levels. | [26] | ||
| Ts1Cje mice | Whole brain | Control males (E14.5) n = 5 Ts1Cje males (E14.5) n = 5 control males (P0) n = 5 Ts1Cje males (P0) n = 5 control males (3 m-old) n = 5 Ts1Cje (three months old) n = 5 | 2D-PAGE/MALDI-TOF- MS | Calcyclin-binding protein (↑), transketolase (↑), pyruvate kinase (↑), 60S acidic ribosomal protein P0 (↑), nucleoside diphosphate kinase-B (↓) | The epression of several proteins were dysregulated in the brain of Ts1Cje mice at E14.5, but not at postnatal day 0 and 90. | [30] | |
| Dp(10)1Yey mice * | hippocampus, cerebellum, cerebral cortex | Control females (7–9 months old) n = 10 Dp10 females (7–9 months old) n = 7 Control males (7–9 months old) n = 9 Dp10 males (7–9 months old) n = 10 | Protein arrays (approximately 100 proteins/ protein modifications) | S100B (trisomic) (↑), App, Itsn, Rcan1, Pknox (in hippocampus) (↑) | The gender-specific abnormalities in the Dp10 suggest the possibility of gender-specific phenotypes in DS. | [41] | |
| 141G6 mice ** (YAC Tg) | hippocampus | Wild-type males (three months old) n = 10 141G6 males (three months old) n = 10 | 2D-PAGE/MALDI- TOF-MS | Electron-transfer flavoprotein α, mitochondrial (↓), NADH dehydrogenase Fe-S protein 3 (↓), NG, NG- dimethylargine dimethylaminhydrolase (↓), Flotillin-1 (↓), Profilin II (↓), Tubulin α6 (↓), Tubulin β3/4 (↑), Vimentin (↓), Hsp60 (↓), Hsp90β (↓), Peptidyl-prolyl cis-trans isomerase A (↓), 3-phosphoglycerate dehydrogenase (↑), ATP synthase α, mitochondrial (↓), Creatine kinase (↓), Fructose-bisphophate dehydrogenase (↓), Neuron specific enolase (↓), Glycerol-3-phosphate dehydrogenase (↑), Glyoxylate reductase/hydroxypyruvate reductase (↓), Guanylate kinase (↓), Isovaleryl coenzyme A dehydrogenase (↓), Phosphoglycerate kinase 1 (↓), Pyruvate kinase M2 (↓), UMP-CMP kinase (↓), Astrocytic phosphoprotein PEA-15 (↓), Dihydropyrimidinase related protein-1 (↑) -4 (↓), ES1 protein homolog, mitochondrial (↑), Protein CGI-51 homolog (↓), Heterogeneous nuclear ribonucleoprotein A2/B1/K (↓), Lamin receptor 1 (↓), ubiquitin carboxyl-terminal hydrolase isozyme L1 (UchL1) (↓), CamK2α (↓), EF-hand domain- containing protein 2 (↑), Voltage-dependent anion-selective channel protein 2 (↓) | A number of proteins were identified as molecules with altered expression in the hippocampus of 141G6 mice. In particular, a decreased level of calcium/calmodulin- dependent protein kinase type II alpha chain was identified as a candidate for cognitive impairment in DS. | [32] | |
| 152F7 mice *** (YAC Tg) | hippocampus | Wild-type males (three months old) n = 9; 152F7 males (three months old) n = 10 | 2D-PAGE/MALDI- TOF-MS | Fascin actin-bundling protein 1 (↑), growth factor receptor-bound protein 2 (Grb2) (↓) | Decreased Grb2 levels in the hippocampus of 152F7 mice may contribute to impaired cytoskeleton functions, and fascin dysregulation is involved in actin bundling for vesicle trafficking and may represent or lead to impaired neurotransmission. | [42] | |
| TT2F/hChr21 mice | Neurons differentiated from ES cells | TT2F controls (in vitro differentiation day 0, 3, 6, and 10) n = 2; TT2F/hChr21 (in vitro differentiation day 0, 3, TOF-MS 6 and 10) n = 2 | Calponin 3 (↓), eukaryotic translation elongation factor 1D (↓), heterogeneous nuclear ribonucleoprotein C (↓) , Hsp70 (↓), Hsp84 (↓), Hsp86-1 (↓), microtubule associated protein RP/EB family member 2 (↓), UCHL1 (↓), ubiquitin-specific-processing protease OTUB1 (↓) Annexin A4 (↑), ATPase H+ transporting V1 subunit A1 (↑), ATPase H+ transporting V1 subunit B2 (↑), Keratin 2-8 (↑), Plastin 3 (↑), Ezrin (↑) | HSA21 gene-dosage effects or chromosomal imbalance may affect the expression of cytoskeleton proteins, chaperon proteins, translation regulators, energy metabolism. | [31] | ||
Enumerated molecules with changes of ≥25% in expression shown. * Dp(10)1Yey mice have a duplication of the Hsa21 homologous region on Mmu10, from Prmt2 to Pdxk; ** 141G6: YAC encompasses four genes: Dscr3, Pigp, Ripply3, and Ttc3; *** 152F7: YAC encompasses four genes: Dscr3, Dscr5, Ttc3, and dual-specificity tyrosine-(Y)-phosphorylation-regulated kinase 1a; LC, liquid chromatography; ESI, electrospray ionization; MALDI, matrix-assisted laser desorption/ionization; TOF-MS, time-of-flight mass spectrometer.
Table 3.
A comparative redox proteomic analysis of the brain with DS.
| Sample | Tissue | Sample Information | Method | Differentially Expressed Proteins | Remarks | Reference |
|---|---|---|---|---|---|---|
| Human | frontal cortex | Young controls (12.1 ± 4.7 years old) n = 8 (four females, four males) | Redox proteomics (2D-PAGE/Oxyblot/MALDI-TOF-MS, Ion Trap-OrbitrapMS) | pTau(Ser404) (↑) carbonylated proteins UchL1 (↑), cathepsin D (↑), 78-kDa glucose- regulated protein (↑), V0-type proton ATPase subunit B, brain isoform (↑), glial fibrillary acidic protein (GFAP) (↑), succinyl-CoA:3-ketoacid- coenzyme A transferase 1, mitochondrial (↑) | Impairment of the proteostasis network and autophagic pathway | [33] |
| Human | frontal cortex | Young controls (24.9 ± 9.95 years old) n = 6 (two females, four males) DS (26.9 ± 17.04 years old) n = 6 (two females, four males) Old controls (59.2 ±7.48 years old) n = 6 (two females, four males) DS/AD 59.3 ± 3.44 years old) n = 6 (four females, two males) | Redox proteomics (2D-PAGE/4-HNE immunoblot/MALD-TOF-MS) | Protein-bound-4-hydroxy-2-nonenal (4-HNE) DS vs young control cytochrome b-c1 complex Rieske subunit, mitochondrial (↑), GFAP (↑), glutamate dehydrogenase 1, mitochondrial (↑), peroxiredoxin-2 (↑), myelin basic protein (↑), UchL1 (↑), fructose-bisphosphate aldolase-A and -C (↑), α-internexin (↑), PK isozymes M1/M2 (↑) | Impairment of several processes, including the neuronal integrity, axonal transport, synapse connections, degenerative systems, energy production, and antioxidant defense, was noted in the brains of DS and DS/AD subjects. | [43] |
| Ts1Cje mice | whole brain (excluding cerebellum) | Control males (3 m-old) n = 3 Ts1Cje males (3 m-old) n = 3 | Redox proteomics (2D-PAGE/4-HNE and HEL immunoblot/LC-MS) | 13-HPODE-bound protein ATP synthase, β chain (↑), Neuron specific enolase (↑), α-enolase (↑), Peroxiredoxin 6 (↑), Triosephosphate isomerase 1 (↑) 4-HNE-bound protein Neuron specific enolase (↑), Peroxiredoxin 6(↑), Neurofilament, light polypeptide (↑), α-internexin (↑), Phosphoglycerate kinase 1 (↑), Triosephosphate isomerase 1 (↑) | A redox proteomics approach revealed that the proteins modified with 13-HPODE and/or 4-HNE are involved in either ATP generation, the neuronal cytoskeleton, or antioxidant activity. | [34] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Ishihara, K.; Akiba, S. A Comprehensive Diverse ‘-omics’ Approach to Better Understanding the Molecular Pathomechanisms of Down Syndrome. Brain Sci. 2017, 7, 44. https://doi.org/10.3390/brainsci7040044
AMA Style
Ishihara K, Akiba S. A Comprehensive Diverse ‘-omics’ Approach to Better Understanding the Molecular Pathomechanisms of Down Syndrome. Brain Sciences. 2017; 7(4):44. https://doi.org/10.3390/brainsci7040044
Chicago/Turabian StyleIshihara, Keiichi, and Satoshi Akiba. 2017. "A Comprehensive Diverse ‘-omics’ Approach to Better Understanding the Molecular Pathomechanisms of Down Syndrome" Brain Sciences 7, no. 4: 44. https://doi.org/10.3390/brainsci7040044
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.