Disturbed Glucose Metabolism in Rat Neurons Exposed to Cerebrospinal Fluid Obtained from Multiple Sclerosis Subjects

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Approval
2.2. Patient Cohort
2.2.1. Patient Population
2.2.2. Patient Characteristics
2.3. Cerebrospinal Fluid Samples of Patients
2.4. Animals
2.5. Primary Culture of CGNs
2.6. Confocal Microscopy
2.7. CSF Treatment and RNA Extraction
2.8. Gene Microarray and Data Normalization
2.9. Statistical Analysis
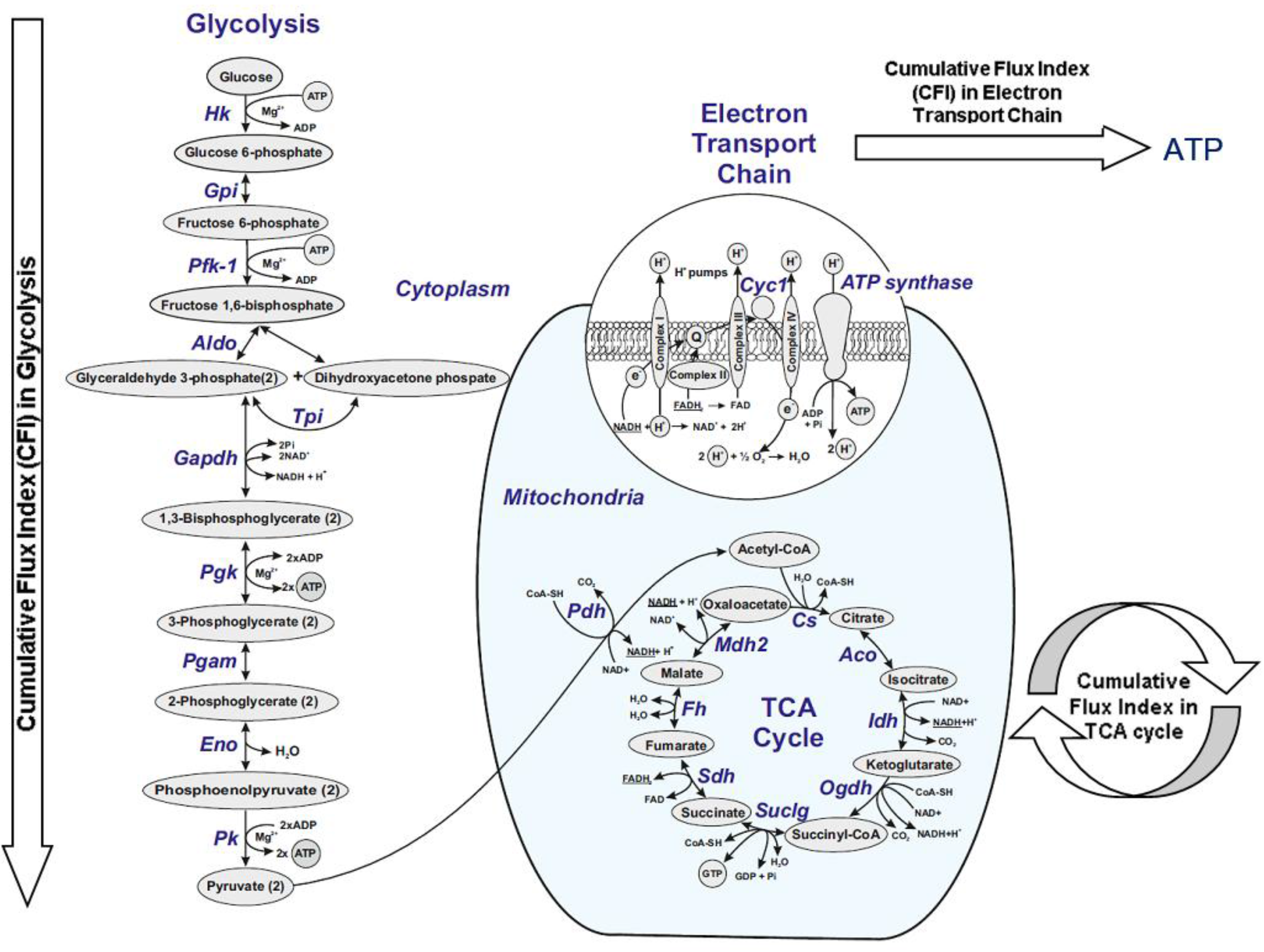
2.10. Analysis of Gene-Gene Interaction Networks Using String v10 Software
3. Results
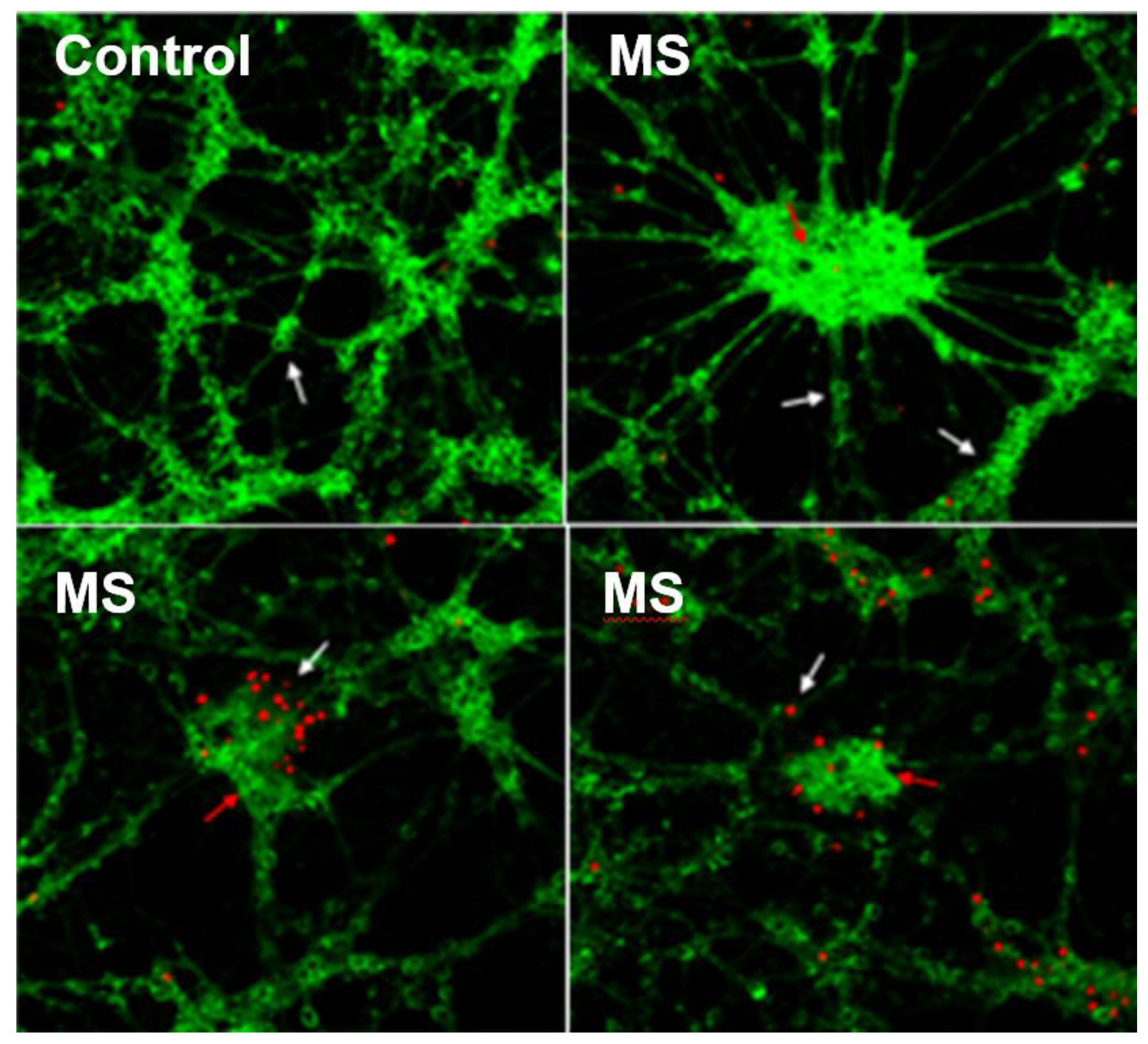
3.1. CSF Exposure Caused Neuronal Cell Death
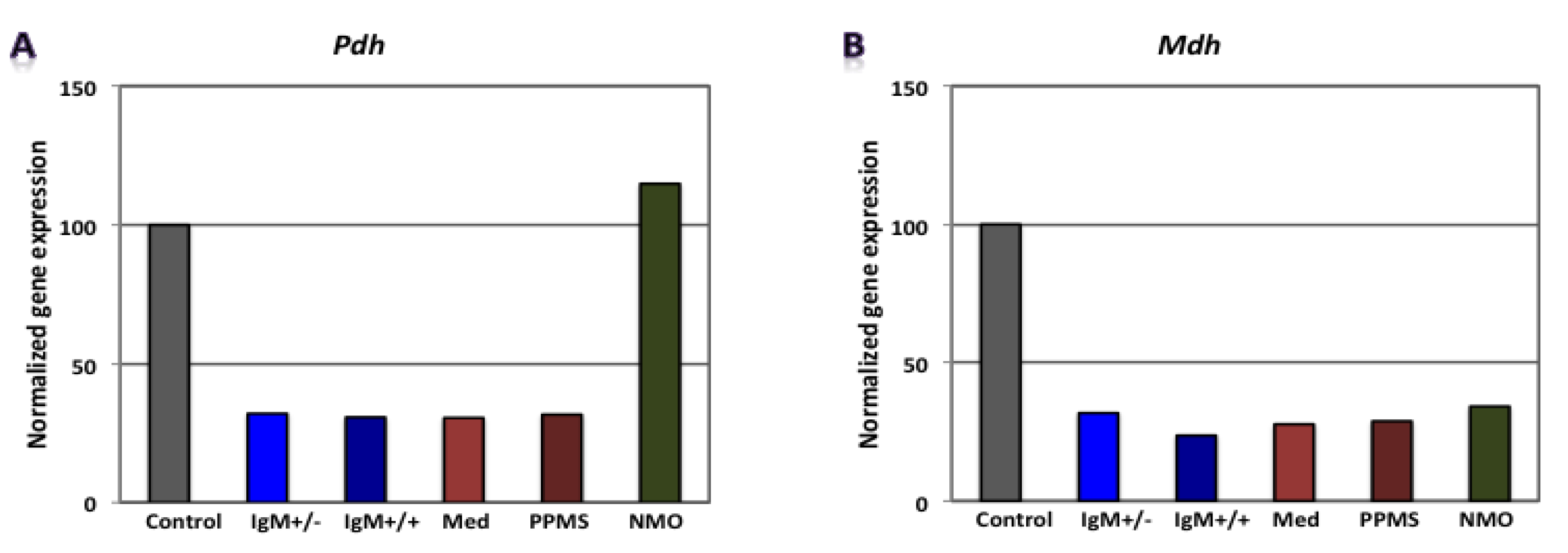
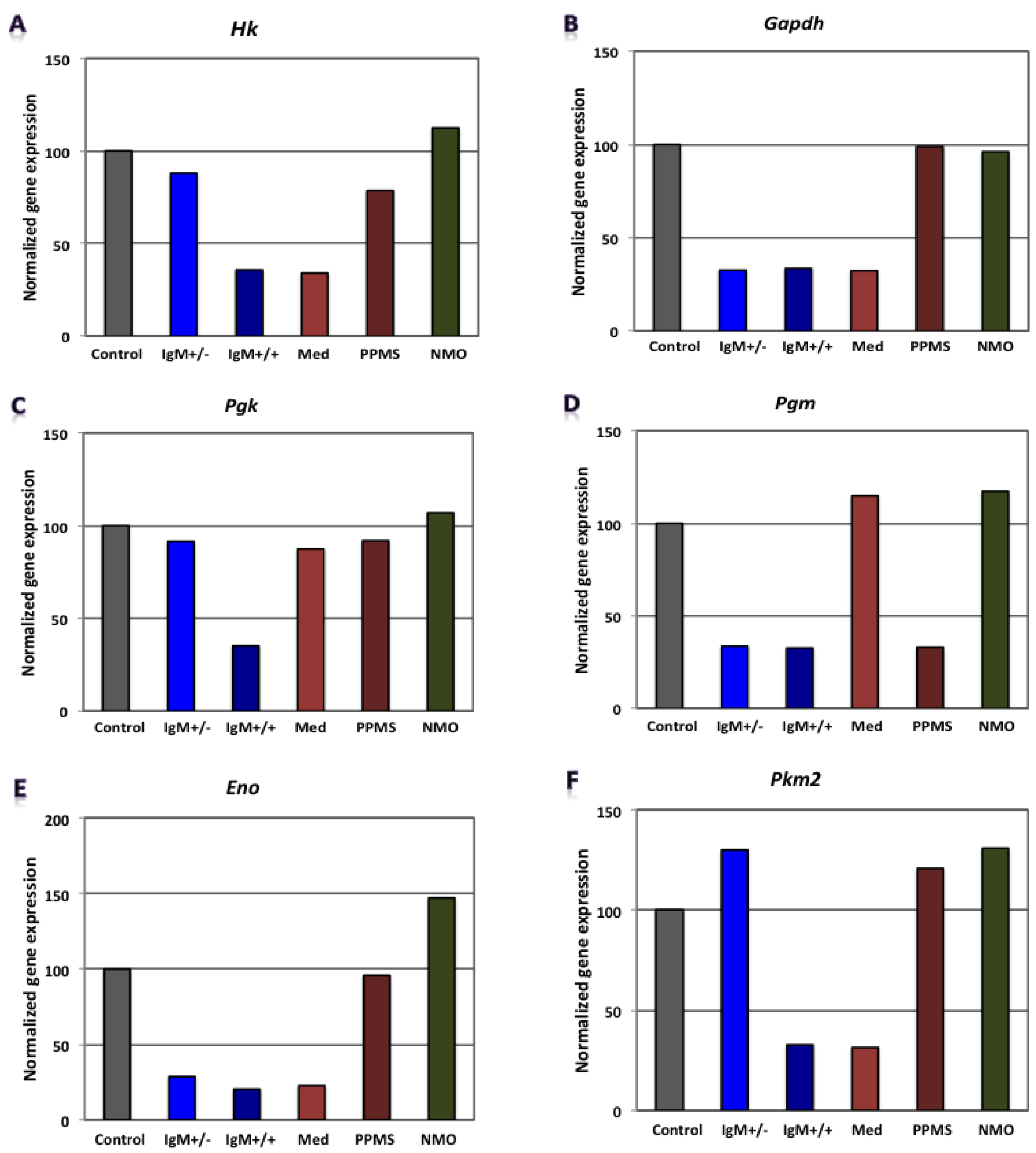
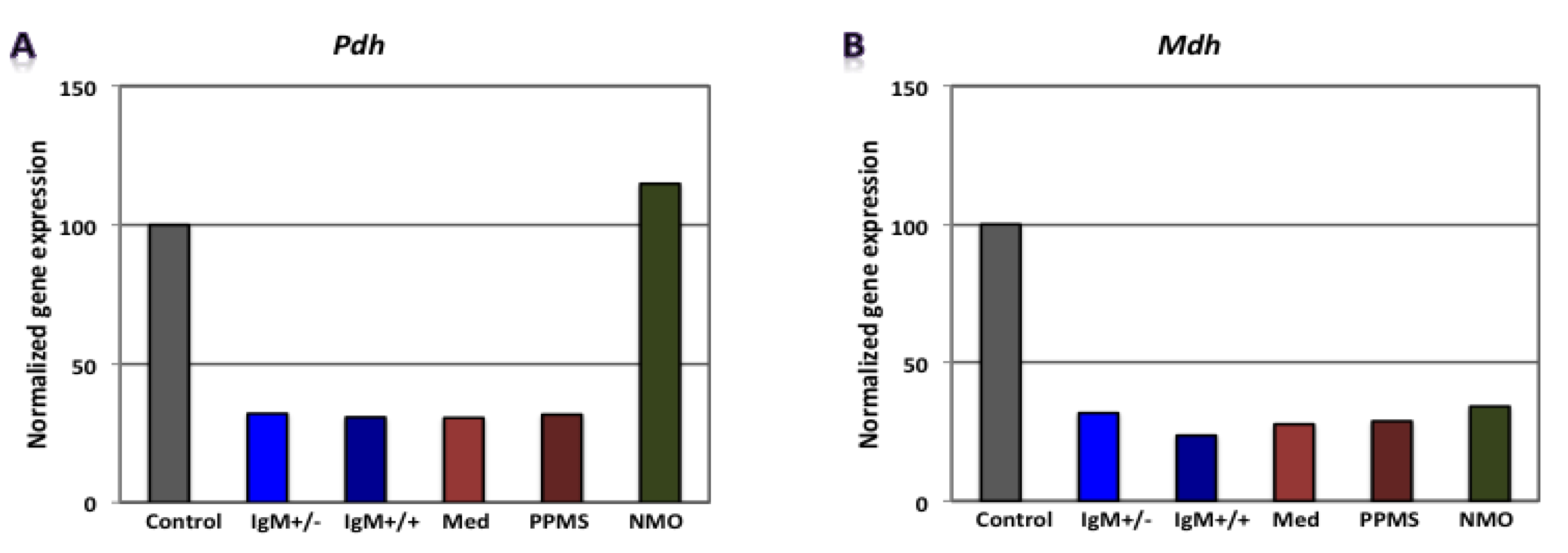
3.2. Differential Expression of Genes Involved in Glucose Metabolism in CGNs by Microarray Gene Expression Profiling
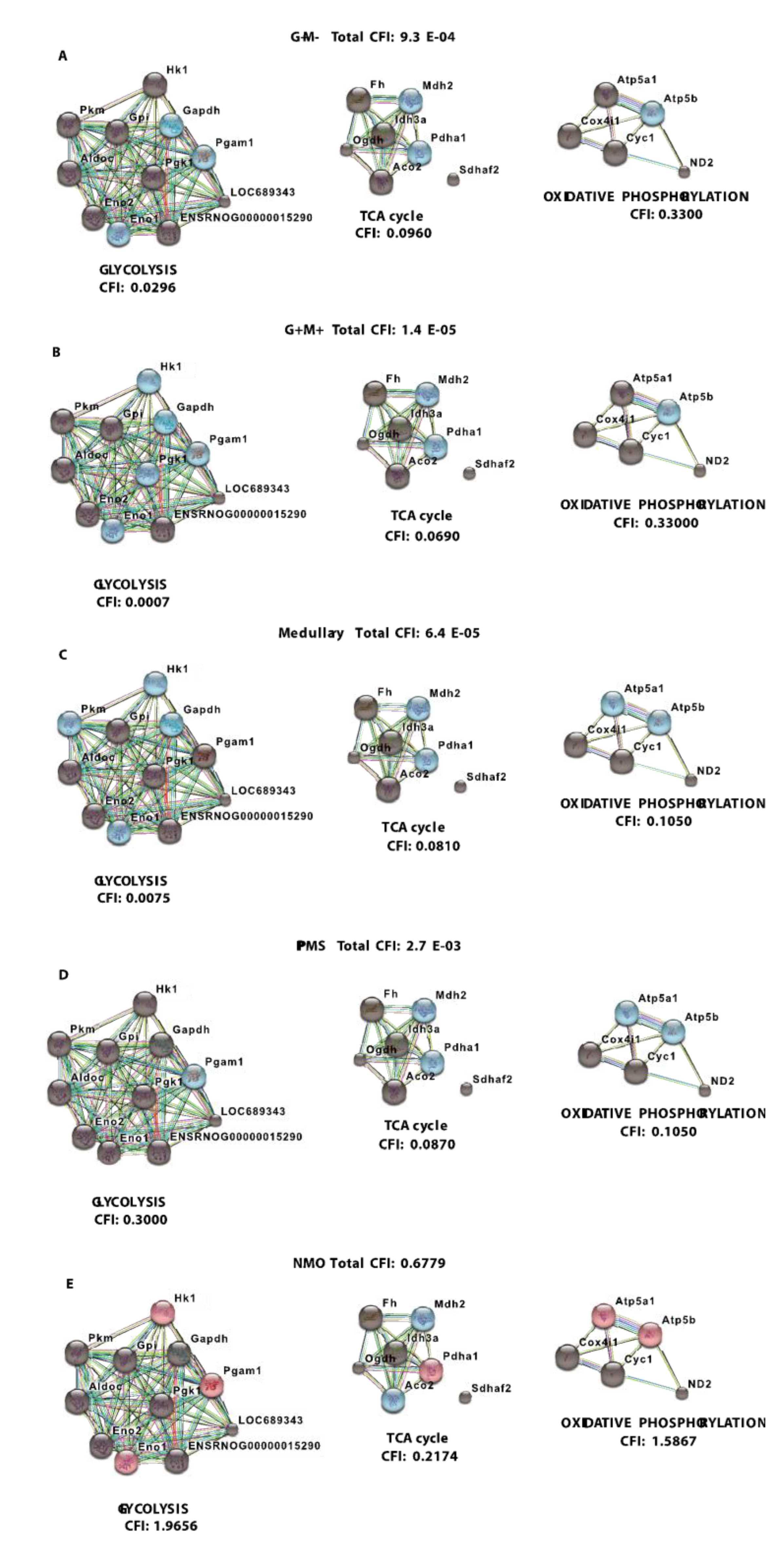
3.3. Analysis of Gene-Gene Interaction Networks Using String v10 Software
4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Xiao, B.G.; Zhang, G.X.; Ma, C.G.; Link, H. The cerebrospinal fluid from patients with multiple sclerosis promotes neuronal and oligodendrocyte damage by delayed production of nitric oxide in vitro. J. Neurol. Sci. 1996, 142, 114–120. [Google Scholar] [CrossRef]
- Alcázar, A.; Regidor, I.; Masjuan, J.; Salinas, M.; Álvarez-Cermeño, J.C. Axonal damage induced by cerebrospinal fluid from patients with relapsing-remitting multiple sclerosis. J. Neuroimmunol. 2000, 104, 58–67. [Google Scholar] [CrossRef]
- Vidaurre, O.G.; Haines, J.D.; Sand, I.K.; Adula, K.P.; Huynh, J.L.; McGraw, C.A.; Zhang, F.; Varghese, M.; Sotirchos, E.; Bhargava, P.; et al. Cerebrospinal fluid ceramides from patients with multiple sclerosis impair neuronal bioenergetics. Brain 2014, 137, 2271–2286. [Google Scholar] [CrossRef] [PubMed]
- Waxman, S.G. Ions, energy and axonal injury: Towards a molecular neurology of multiple sclerosis. Trends Mol. Med. 2006, 12, 192–195. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Morrison, B.M.; Li, Y.; Lengacher, S.; Farah, M.H.; Hoffman, P.N.; Liu, Y.; Tsingalia, A.; Jin, L.; Zhang, P.; et al. Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature 2012, 487, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Mazzola, J.L.; Sirover, M.A. Reduction of glyceraldehyde-3-phosphate dehydrogenase activity in Alzheimer’s disease and in Huntington’s disease fibroblasts. J. Neurochem. 2001, 76, 442–449. [Google Scholar] [CrossRef] [PubMed]
- Senatorov, V.V.; Charles, V.; Reddy, P.; Tagle, D.A.; Chuang, D.-M. Overexpression and nuclear accumulation of glyceraldehyde-3-phosphate dehydrogenase in a transgenic mouse model of Huntington’s disease. Mol. Cell. Neurosci. 2003, 22, 285–297. [Google Scholar] [CrossRef]
- Soucek, T.; Cumming, R.; Dargusch, R.; Maher, P.; Schubert, D. The Regulation of Glucose Metabolism by HIF-1 Mediates a Neuroprotective Response to Amyloid Beta Peptide. Neuron 2003, 39, 43–56. [Google Scholar] [CrossRef]
- Blalock, E.M.; Geddes, J.W.; Chen, K.C.; Porter, N.M.; Markesbery, W.R.; Landfield, P.W. Incipient Alzheimer’s disease: Microarray correlation analyses reveal major transcriptional and tumor suppressor responses. Proc. Natl. Acad. Sci. USA 2004, 101, 2173–2178. [Google Scholar] [CrossRef] [PubMed]
- Brooks, W.M.; Lynch, P.J.; Ingle, C.C.; Hatton, A.; Emson, P.C.; Faull, R.L.; Starkey, M.P. Gene expression profiles of metabolic enzyme transcripts in Alzheimer’s disease. Brain. Res. 2007, 1127, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Diamanduros, A.; Chowdhury, S.A.; Scelsa, S.; Latov, N.; Sadiq, S.A. Specific electron transport chain abnormalities in amyotrophic lateral sclerosis. J. Neurol. 2009, 256, 774–782. [Google Scholar] [CrossRef] [PubMed]
- Jones, H.H.; Jones, H.H., Jr.; Bunch, L.D. Biochemical studies in multiple sclerosis. Ann. Int. Med. 1950, 33, 831–840. [Google Scholar] [PubMed]
- McArdle, B.; Mackenzie, I.C.K.; Webster, G.R. Studies on intermediate carbohydrate metabolism in Multiple Sclerosis. J. Neurol. Neurosurg. Psychiatry 1960, 23, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Royds, J.A.; Timperley, W.R.; Taylor, C.B. Levels of enolase and other enzymes in the cerebrospinal fluid as indices of pathological change. J. Neurol. Neurosurg. Psychiatry 1981, 44, 1129–1135. [Google Scholar] [CrossRef] [PubMed]
- Kolln, J.; Zhang, Y.; Thai, G.; Demetriou, M.; Hermanowicz, N.; Duquette, P.; van den Noort, S.; Qin, Y. Inhibition of Glyceraldehyde-3-Phosphate Dehydrogenase Activity by Antibodies Present in the Cerebrospinal Fluid of Patients with Multiple Sclerosis. J. Immunol. 2010, 185, 1968–1975. [Google Scholar] [CrossRef] [PubMed]
- Henneman, D.H.; Altschule, M.D.; Goncz, R.M.; Alexander, L. Title of the article. A.M.A. Arch. Neurol. Psychiatry 1954, 72, 688. [Google Scholar] [CrossRef]
- Iñarrea, P.; Alarcia, R.; Alava, M.A.; Capablo, J.L.; Casanova, A.; Iñiguez, C.; Iturralde, M.; Larrodé, P.; Martín, J.; Mostacero, E.; et al. Mitochondrial Complex Enzyme Activities and Cytochrome c Expression Changes in Multiple Sclerosis. Mol. Neurobiol. 2013, 49, 1–9. [Google Scholar] [CrossRef]
- Safavizadeh, N.; Rahmani, S.A.; Zaefizadeh, M. Investigation of cytochrome c oxidase gene subunits expression on the Multiple sclerosis. Indian J. Hum. Genet. 2013, 19, 18–25. [Google Scholar] [PubMed]
- Mahad, D.; Ziabreva, I.; Lassmann, H.; Turnbull, D. Mitochondrial defects in acute multiple sclerosis lesions. Brain 2008, 131, 1722–1735. [Google Scholar] [CrossRef] [PubMed]
- Broadwater, L.; Pandit, A.; Azzam, S.; Clements, R.; Vadnal, J.; Sulak, M.; Yong, V.W.; Freeman, E.J.; Gregory, R.B.; McDonough, J. Analysis of the Mitochondrial Proteome in Multiple Sclerosis Cortex. Biochim. Biophys. Acta 2011, 1812, 630–641. [Google Scholar] [CrossRef] [PubMed]
- Mathur, D.; LÃ3pez-Rodas, G.; Casanova, B.; Marti, M.B. Perturbed Glucose Metabolism: Insights into Multiple Sclerosis Pathogenesis. Front. Neurol. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Afifi, A.K.; Aleu, F.P.; Goodgold, J.; MacKay, B. Ultrastructure of atrophic muscle in amyotrophic lateral sclerosis. Neurology 1966, 16, 475–481. [Google Scholar] [CrossRef] [PubMed]
- Atsumi, T. The ultrastructure of intramuscular nerves in amyotrophic lateral sclerosis. Acta Neuropathol. 1981, 55, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, S.; Iwata, M. Ultrastructural study of synapses in the anterior horn neurons of patients with amyotrophic lateral sclerosis. Neurosci. Lett. 1996, 204, 53–56. [Google Scholar] [CrossRef]
- Siklos, L.; Engelhardt, J.; Harati, Y.; Smith, R.G.; Joó, F.; Appel, S.H. Ultrastructural evidence for altered calcium in motor nerve terminals in amyotropic lateral sclerosis. Ann. Neurol. 1996, 39, 203–216. [Google Scholar] [CrossRef] [PubMed]
- Dal Canto, M.C.; Gurney, M.E. Neuropathological changes in two lines of mice carrying a transgene for mutant human Cu, Zn SOD and in mice overexpressing wild type human SOD: A model of familial amyotrophic lateral sclerosis (FALS). Brain Res. 1995, 676, 25–40. [Google Scholar] [CrossRef]
- Kong, J.; Xu, Z. Massive mitochondrial degeneration in motor neurons triggers the onset of amyotrophic lateral sclerosis in mice expressing a mutant SOD1. J. Neurosci. 1998, 18, 3241–3250. [Google Scholar] [PubMed]
- Wong, P.C.; Pardo, C.A.; Borchelt, D.R.; Lee, M.K.; Copeland, N.G.; Jenkins, N.A.; Sisodia, S.S.; Cleveland, D.W.; Price, D.L. An adverse property of a familial ALS-linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron 1995, 14, 1105–1116. [Google Scholar] [CrossRef]
- Jung, C.; Higgins, C.M.J.; Xu, Z. Mitochondrial electron transport chain complex dysfunction in a transgenic mouse model for amyotrophic lateral sclerosis. J. Neurochem. 2002, 83, 535–545. [Google Scholar] [CrossRef] [PubMed]
- Mattiazzi, M.; D’Aurelio, M.; Gajewski, C.D.; Martushova, K.; Kiaei, M.; Beal, M.F.; Manfredi, G. Mutated human SOD1 causes dysfunction of oxidative phosphorylation in mitochondria of transgenic mice. J. Biol. Chem. 2002, 277, 29626–29633. [Google Scholar] [CrossRef] [PubMed]
- Browne, S.E.; Yang, L.; DiMauro, J.-P.; Fuller, S.W.; Licata, S.C.; Beal, M.F. Bioenergetic abnormalities in discrete cerebral motor pathways presage spinal cord pathology in the G93A SOD1 mouse model of ALS. Neurobiol. Dis. 2006, 22, 599–610. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Perry, G.; Moreira, P.I.; Aliev, G.; Cash, A.D.; Hirai, K.; Smith, M.A. Mitochondrial abnormalities and oxidative imbalance in Alzheimer disease. J. Alzheimers Dis. 2006, 9, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Der, S.D.; Zhou, A.; Williams, B.R.G.; Silverman, R.H. Identification of genes differentially regulated by interferon α, β, or γ using the oligonucleotide arrays. Proc. Natl. Acad. Sci. USA 1998, 95, 15623–15628. [Google Scholar] [CrossRef] [PubMed]
- Wandinger, K.-P.; Stürzebecher, C.-S.; Bielekova, B.; Detore, G.; Rosenwald, A.; Staudt, L.M.; McFarland, H.F.; Martin, R. Complex immunomodulatory effects of interferon-β in multiple sclerosis include the upregulation of T helper 1-associated marker genes. Ann. Neurol. 2001, 50, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Bomprezzi, R. Gene expression profile in multiple sclerosis patients and healthy controls: Identifying pathways relevant to disease. Hum. Mol. Genet. 2003, 12, 2191–2199. [Google Scholar] [CrossRef] [PubMed]
- Koike, F.; Satoh, J.; Miyake, S.; Yamamoto, T.; Kawai, M.; Kikuchi, S.; Nomura, K.; Yokoyama, K.; Ota, K.; Kanda, T.; et al. Microarray analysis identifies interferon beta-regulated genes in multiple sclerosis. J. Neuroimmunol. 2003, 139, 109–118. [Google Scholar] [CrossRef]
- Hong, J.; Zang, Y.C.; Hutton, G.; Rivera, V.M.; Zhang, J.Z. Gene expression profiling of relevant biomarkers for treatment evaluation in multiple sclerosis. J. Neuroimmunol. 2004, 152, 126–139. [Google Scholar] [CrossRef] [PubMed]
- Becker, K.G.; Mattson, D.H.; Powers, J.M.; Gado, A.M.; Biddison, W.E. Analysis of a sequenced cDNA library from multiple sclerosis lesions. J. Neuroimmunol. 1997, 77, 27–38. [Google Scholar] [CrossRef]
- Whitney, L.W.; Becker, K.G.; Tresser, N.J.; Caballero-Ramos, C.I.; Munson, P.J.; Prabhu, V.V.; Trent, J.M.; McFarland, H.F.; Biddison, W.E. Analysis of gene expression in mutiple sclerosis lesions using cDNA microarrays. Ann. Neurol. 1999, 46, 425–428. [Google Scholar] [CrossRef]
- Chabas, D.; Baranzini, S.E.; Mitchell, D.; Bernard, C.C.; Rittling, S.R.; Denhardt, D.T.; Sobel, R.A.; Lock, C.; Karpuj, M.; Pedotti, R.; et al. The influence of the proinflammatory cytokine, osteopontin, on autoimmune demyelinating disease. Science 2001, 294, 1731–1735. [Google Scholar] [CrossRef] [PubMed]
- Brynedal, B.; Khademi, M.; Wallström, E.; Hillert, J.; Olsson, T.; Duvefelt, K. Gene expression profiling in multiple sclerosis: A disease of the central nervous system but with relapses triggered in the periphery? Neurobiol. Dis. 2010, 37, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Mathur, D.; Urena-Peralta, J.R.; Lopez-Rodas, G.; Casanova, B.; Coret-Ferrer, F.; Burgal-Marti, M. Bypassing hazard of housekeeping genes: Their evaluation in rat granule neurons treated with cerebrospinal fluid of multiple sclerosis subjects. Front. Cell. Neurosci. 2015, 9. [Google Scholar] [CrossRef] [PubMed]
- Mathur, D.; Riffo-Campos, A.L.; Castillo, J.; Haines, J.D.; Vidaurre, O.G.; Zhang, F.; Coret-Ferrer, F.; Casaccia, P.; Casanova, B.; Lopez-Rodas, G. Bioenergetic Failure in Rat Oligodendrocyte Progenitor Cells Treated with Cerebrospinal Fluid Derived from Multiple Sclerosis Patients. Front. Cell. Neurosci. 2017, 11, 209. [Google Scholar] [CrossRef] [PubMed]
- Lennon, V.A.; Wingerchuk, D.M.; Kryzer, T.J.; Pittock, S.J.; Lucchinetti, C.F.; Fujihara, K.; Nakashima, I.; Weinshenker, B.G. A serum autoantibody marker of neuromyelitis optica: Distinction from multiple sclerosis. Lancet 2004, 364, 2106–2112. [Google Scholar] [CrossRef]
- Lennon, V.A.; Kryzer, T.J.; Pittock, S.J.; Verkman, A.S.; Hinson, S.R. IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. J. Exp. Med. 2005, 202, 473–477. [Google Scholar] [CrossRef] [PubMed]
- Agre, P.; Kozono, D. Aquaporin water channels: Molecular mechanisms for human disease. FEBS Lett. 2003, 555, 72–78. [Google Scholar] [CrossRef]
- Nielsen, S.; Nagelhus, E.A.; Amiry-Moghaddam, M.; Bourque, C.; Agre, P.; Ottersen, O.P. Specialised membrane domains for water transport in glial cells: High-resolution immunogold cytochemistry of aquaporin-4 in rat brain. J. Neurosci. 1997, 17, 171–180. [Google Scholar] [PubMed]
- Lublin, F.D.; Reingold, S.C. Defining the clinical course of multiple sclerosis: Results of an international survey. Neurology 1996, 46, 907–911. [Google Scholar] [CrossRef] [PubMed]
- Wingerchuk, D.M.; Lennon, V.A.; Pittock, S.J.; Lucchinetti, C.F.; Weinshenker, B.G. Revised diagnostic criteria for neuromyelitis optica. Neurology 2006, 66, 1485–1489. [Google Scholar] [CrossRef] [PubMed]
- Kostulas, V.K.; Link, H.; Lefvert, A.K. Oligoclonal IgG bands in cerebrospinal fluid. Principles for demonstration and interpretation based on findings in 1114 neurological patients. Arch. Neurol. 1987, 44, 1041–1044. [Google Scholar] [CrossRef] [PubMed]
- Sharief, M.K.; Thompson, E.J. Intrathecal immunoglobulin M synthesis in multiple sclerosis. Brain 1991, 114, 181–195. [Google Scholar] [PubMed]
- Kearney, H.; Miller, D.H.; Ciccarelli, O. Spinal cord MRI in multiple sclerosis—Diagnostic, prognostic and clinical value. Nat Rev Neurol. 2015, 11, 327–338. [Google Scholar] [CrossRef] [PubMed]
- Miñana, M.-D.; Montoliu, C.; Llansola, M.; Grisolı́a, S.; Felipo, V. Nicotine prevents glutamate-induced proteolysis of the microtubule-associated protein MAP-2 and glutamate neurotoxicity in primary cultures of cerebellar neurons. Neuropharmacology 1998, 37, 847–857. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING v10: Protein–protein interaction networks, integrated over the tree of life. Nucleic Acids Res 2015, 43, D447–D452. [Google Scholar] [CrossRef] [PubMed]
- Smith, K.J.; Lassmann, H. The role of nitric oxide in multiple sclerosis. Lancet Neurol. 2002, 1, 232–241. [Google Scholar] [CrossRef]
- Kim, J.Y.; Shen, S.; Dietz, K.; He, Y.; Howell, O.; Reynolds, R.; Casaccia, P. HDAC1 nuclear export induced by pathological conditions is essential for the onset of axonal damage. Nat. Neurosci. 2009, 13, 180–189. [Google Scholar] [CrossRef] [PubMed]
- Nikic, I.; Merkler, D.; Sorbara, C.; Brinkoetter, M.; Kreutzfeldt, M.; Bareyre, F.M.; Brück, W.; Bishop, D.; Misgeld, T.; Kerschensteiner, M. A reversible form of axon damage in experimental autoimmune encephalomyelitis and multiple sclerosis. Nat. Med. 2011, 17, 495–499. [Google Scholar] [CrossRef] [PubMed]
- Dutta, R.; McDonough, J.; Yin, X.; Peterson, J.; Chang, A.; Torres, T.; Gudz, T.; Macklin, W.B.; Lewis, D.A.; Fox, R.J.; et al. Mitochondrial dysfunction as a cause of axonal degeneration in multiple sclerosis patients. Ann. Neurol. 2006, 59, 478–489. [Google Scholar] [CrossRef] [PubMed]
- Lu, F.; Selak, M.; O’Connor, J.; Croul, S.; Lorenzana, C.; Butunoi, C.; Kalman, B. Oxidative damage to mitochondrial DNA and activity of mitochondrial enzymes in chronic active lesions of multiple sclerosis. J. Neurol. Sci. 2000, 177, 95–103. [Google Scholar] [CrossRef]
- Kolln, J.; Ren, H.-M.; Da, R.-R.; Zhang, Y.; Spillner, E.; Olek, M.; Hermanowicz, N.; Hilgenberg, L.G.; Smith, M.A.; van den Noort, S.; et al. Triosephosphate Isomerase- and Glyceraldehyde-3-Phosphate Dehydrogenase-Reactive Autoantibodies in the Cerebrospinal Fluid of Patients with Multiple Sclerosis. J. Immunol. 2006, 177, 5652–5658. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Case | G | Age | Working Clinical Form | Clinical Form | P.T. | Actual EDSS | OCGB IgG | OCMB IgM | Ac-AQ4 | T |
|---|---|---|---|---|---|---|---|---|---|---|
| Multiple Sclerosis | ||||||||||
| 1 | F | 23 | RMS (G+/M−) | RRMS | 5 | 1.50 | G+ | M- | N.A. | IFN, FGM |
| 2 | F | 21 | RMS (G+/M−) | SPMS | 18 | 4.00 | G+ | M- | N.A. | IFN, MTZ |
| 3 | F | 36 | RMS (G+/M−) | CIS | 4 | 1.50 | G+ | M- | N.A. | N.T. |
| 4 | M | 22 | RMS (G+/M−) | RRMS | 6 | 1.50 | G+ | M- | N.A. | IFN, CPX, NTZ, FGM |
| 5 | F | 21 | RMS (G+/M−) | RRMS | 3 | 3.00 | G+ | M- | N.A. | IFN |
| 6 | F | 30 | RMS (G+/M−) | RRMS | 22 | 4.00 | G+ | M- | N.A. | IFN, NTZ, FGM |
| 7 | F | 29 | RMS (G+/M−) | RRMS | 10 | 1.50 | G+ | M- | N.A. | N.T. |
| 8 | F | 29 | RMS (G+/M−) | RRMS | 7 | 1.50 | G+ | M- | N.A. | IFN, NTZ |
| 9 | F | 28 | RMS (G+/M−) | RRMS | 10 | 5.50 | G+ | M- | N.A. | MTZ, IFN |
| 10 | F | 28 | RMS (G+/M−) | RRMS | 4 | 1.00 | G+ | M- | N.A. | N.T. |
| 11 | F | 37 | RMS (G+/M+) | RRMS | 7 | 3.50 | G++ | M+ | N.A. | IFN, NTZ, CPX |
| 12 | M | 32 | RMS (G+/M+) | RRMS | 4 | 1.00 | G+ | M+ | N.A. | IV-IgG |
| 13 | F | 44 | RMS (G+/M+) | RRMS | 5 | 2.00 | G+ | M+ | N.A. | N.T. |
| 14 | F | 26 | RMS (G+/M+) | RRMS | 5 | 2.00 | G++ | M++ | N.A. | U. |
| 15 | F | 14 | RMS (G+/M+) | RRMS | 18 | 3.50 | G+ | M+ | N.A. | PE, IFN, NTZ, ASCT |
| 16 | M | 25 | RMS (G+/M+) | RRMS | 11 | 2.00 | G+ | M+ | N.A. | IFN, FGM |
| 17 | F | 21 | RMS (G+/M+) | SPMS | 25 | 8.50 | G++ | M+ | N.A. | IFN, AZA, MTZ, IV-IgG |
| 18 | F | 17 | RMS (G+/M+) | RRMS | 16 | 2.00 | G+ | M+ | N.A. | U. |
| 19 | F | 23 | RMS (G+/M+) | SPMS | 18 | 6.50 | G+ | M+ | N.A. | IFN, IV-IgG, Cy |
| 20 | F | 22 | RMS (G+/M+) | SPMS | 5 | 4.00 | G+ | M+ | N.A. | IFN, FGM, CPX |
| 21 | F | 29 | RMS (G+/M+) | RRMS | 5 | 2.50 | G+ | M+ | N.A. | IFN, FGM, CPX |
| 22 | M | 39 | Spinal MS | SPMS | 10 | 4.50 | G+ | M+ | N.A. | IFN, Cy, FGM, NTZ |
| 23 | F | 25 | Spinal MS | SPMS | 6 | 7.00 | G+ | M- | N.A. | IFN, MTZ, RTX |
| 24 | F | 25 | Spinal MS | SPMS | 14 | 8.00 | G+ | M+ | N.A. | IFN, Cy, RTX |
| 25 | M | 34 | Spinal MS | SPMS | 9 | 6.00 | G+ | M- | N.A. | IFN, Cy |
| 26 | M | 34 | Spinal MS | SPMS | 6 | 6.50 | G+ | N.A. | N.A. | IFN, MTZ, IV-IgG |
| 27 | F | 23 | Spinal MS | RRMS | 5 | 4.00 | G- | M- | N.A. | IFN, NTZ, FGM, RTX |
| 28 | F | 40 | Spinal MS | SPMS | 10 | 7.50 | G- | M+ | N.A. | AZA, IV-IgG, Cy, RTX |
| 29 | F | 23 | Spinal MS | SPMS | 28 | 6.50 | G+ | M- | N.A. | IFN, MTZ, RTX |
| 30 | F | 54 | PPMS | PPMS | 12 | 7.00 | G+ | M- | N.A. | N.T. |
| 31 | M | 40 | PPMS | PPMS | 23 | 6.00 | G+ | M- | N.A. | MTZ, Cy, RTX |
| 32 | F | 52 | PPMS | PPMS | 14 | 5.50 | G++ | M- | N.A. | AZA |
| 33 | F | 38 | PPMS | PPMS | 11 | 5.50 | G+ | M- | N.A. | N.T. |
| 34 | M | 31 | PPMS | PPMS | 24 | 6.00 | G+ | M- | N.A. | N.T. |
| 35 | F | 47 | PPMS | PPMS | 14 | 5.50 | G++ | M- | N.A. | N.T. |
| 36 | M | 49 | PPMS | PPMS | 11 | 6.00 | G- | M- | N.A. | FGM |
| 37 | F | 26 | PPMS | PPMS | 13 | 6.50 | G++ | M- | N.A. | N.T. |
| 38 | F | 34 | PPMS | PPMS | 6 | 5.00 | G++ | M- | N.A. | N.T. |
| 39 | F | 39 | PPMS | PPMS | 8 | 8.50 | G- | M- | N.A. | Cy |
| 40 | M | 18 | PPMS | PPMS | 15 | 8.00 | U. | U. | N.A. | U. |
| Neuromyelitis Optica Patients | ||||||||||
| 41 | F | 39 | NMO | NMO | 5 | 9.00 | G- | M- | P. | IFN, MTZ, Cy, RTX |
| 42 | F | 50 | NMO | NMO | 4 | 7.00 | G- | M- | P. | IFN, NTZ, RTX |
| 43 | M | 15 | NMO | NMO | 17 | 4.00 | G+ | M+ | N. | IFN, IV-IgG |
| 44 | M | 42 | NMO | NMO | 5 | 3.50 | N.A. | N.A. | P. | IV-IgG |
| 45 | F | 22 | NMO | NMO | 5 | 2.50 | G+ | M+ | P. | IV-IgG, IFN, CPX |
| 46 | F | 27 | NMO | NMO | 5 | 2.00 | G+ | M- | N. | IFN, AZA |
| 47 | M | 9 | NMO | NMO | 14 | 1.00 | G- | M- | N. | IV-IgG, IFN, CPX |
| 48 | F | 8 | NMO | NMO | 32 | 4.00 | G+ | M+ | N. | IFN, IV-IgG |
| 49 | M | 19 | NMO | NMO | 20 | 8.50 | G- | M- | N. | IFN, MTZ, Cy, RTX |
| Non-Inflammatory Neurological Diseases (Control) | ||||||||||
| 50 | M | 23 | CONTROL | Non-inflammatory motor neuron neurological disease | N.A. | N.A. | G- | M- | N.A. | N.T. |
| 51 | F | 77 | CONTROL | Glioblastoma | N.A. | N.A. | G- | M- | N.A. | N.T. |
| 52 | F | 33 | CONTROL | Central Pontine myelinolisis | N.A. | N.A. | G- | M- | N.A. | N.T. |
| 53 | F | 32 | CONTROL | Central Pontine myelinolisis | N.A. | N.A. | G- | M- | N.A. | N.T. |
| 54 | M | 59 | CONTROL | Classic migraine | N.A. | N.A. | G- | M- | N.A. | N.T. |
| 55 | F | 36 | CONTROL | Headache in which subarachnoid hemorrhage was suspected | N.A. | N.A. | G- | M- | N.A. | N.T. |
| 56 | F | 57 | CONTROL | Headache in which subarachnoid hemorrhage was suspected | N.A. | N.A. | G- | M- | N.A. | N.T. |
| 57 | M | 37 | CONTROL | Headache in which subarachnoid hemorrhage was suspected | N.A. | N.A. | G- | M- | N.A. | N.T. |
| 58 | F | 21 | CONTROL | Benign intracranial hypertension | N.A. | N.A. | G- | M- | N.A. | N.T. |
| 59 | M | 13 | CONTROL | Chronic axonal polyneuropathy | N.A. | N.A. | G- | M- | N.A. | N.T. |
| Controls (n = 10) | MS Patients (n = 40) | NMO Patients (n = 9) | p | |
|---|---|---|---|---|
| % females (n) | 60.0 (6) | 75.0 (30) | 55.6 (5) | 0.40 (χ2) |
| Age (mean, SD) | 40.3 (19.5) | 30.7 (9.7) | 25.6 (15.0) | 0.04 (ANOVA test) |
| EDSS | NA | 4.5 (2.3) | 4.6 (2.8) | 0.94 (t-test) |
| Progression Time | NA | 11.1 (6.6) | 11.8 (9.7) | 0.79 (t-test) |
| RMS (n = 18) | SPMS (n = 11) | PPMS (n = 11) | p | |
|---|---|---|---|---|
| % females (n) | 83.3 (15) | 72.7 (8) | 63.6 (7) | 0.48 (χ2) |
| Age (mean, SD) | 27.3 (7.2) | 27.9 (7.3) | 38.9 (11.2) | 0.003 (ANOVA test) |
| EDSS | 2.4 (1.2) | 6.2 (1.5) | 6.3 (1.1) | <0.001 (ANOVA test) |
| Progression Time | 8.1 (5.4) | 13.5 (7.8) | 13.8 (5.7) | 0.043 (ANOVA test) |
| Relapsing MS (n = 21) G+/M− (n = 10) G+/M+ (n = 11) | Spinal MS (n = 8) | PPMS (n = 11) | p | ||
|---|---|---|---|---|---|
| % females (n) | 90 (9) | 81.8 (9) | 62.5 (5) | 63.6 (7) | 0.40 (χ2) |
| Age (mean, SD) | 26.7 (4.8) | 26.3 (8.7) | 31.4 (7.0) | 38.9 (11.2) | 0.005 |
| EDSS | 2.5 (1.5) | 3.4 (2.2) | 6.2 (1.4) | 6.3 (1.1) | <0.001 |
| MSSS * | 3.2 (1.9) | 4.5 (2.5) | 8.2 (1.6) | 6.9 (1.6) | <0.001 |
| Progression Time | 8.9 (6.3) | 10.8 (5.9) | 8.5 (3.1) | 13.8 (5.7) | 0.154 |
| Clinical Form | Genes Upregulated | Genes Downregulated | CFI | Total CFI | Prognosis | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Glycolysis | TCA Cycle | ETC | Glycolysis | TCA Cycle | ETC | Glycolysis | TCA Cycle | ETC | |||
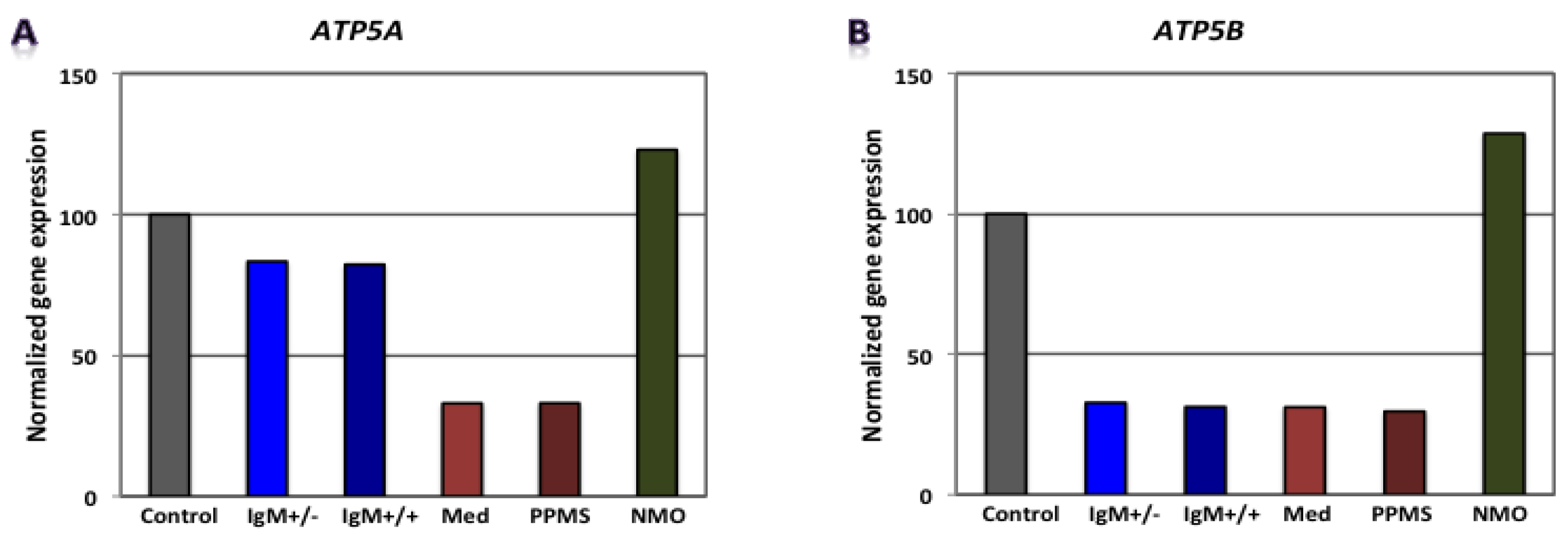
| RMS (G+/M−) | - | - | - | Gapdh, Pgam1, Eno1 | Mdh2, Pdha1 | Atp5b | 0.0296 | 0.0960 | 0.3300 | 9.3 × 10−4 | Poor prognosis |
| RMS (G+/M+) | - | - | - | Hk1, Gapdh, Pgam1, Pgk1, Eno1 | Mdh2, Pdha1 | Atp5b | 0.0007 | 0.0690 | 0.33000 | 1.4 × 10−5 | Worse prognosis |
| Spinal MS | - | - | - | Hk1, Gapdh, Eno1, Pk1 | Mdh2, Pdha1 | Atp5a1, Atp5b | 0.0075 | 0.0810 | 0.1050 | 6.4 × 10−5 | Worst prognosis |
| PPMS | - | - | - | Pgam1 | Mdh2, Pdha1 | Atp5a1, Atp5b | 0.3000 | 0.0870 | 0.1050 | 2.7 × 10−3 | - |
| NMO | Hk1, Pgam1, Eno1 | Pdha1 | Atp5a1, Atp5b | - | Mdh2, Aco2 | - | 1.9656 | 0.2174 | 1.5867 | 0.6779 | - |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mathur, D.; María-Lafuente, E.; Ureña-Peralta, J.R.; Sorribes, L.; Hernández, A.; Casanova, B.; López-Rodas, G.; Coret-Ferrer, F.; Burgal-Marti, M. Disturbed Glucose Metabolism in Rat Neurons Exposed to Cerebrospinal Fluid Obtained from Multiple Sclerosis Subjects. Brain Sci. 2018, 8, 1. https://doi.org/10.3390/brainsci8010001
Mathur D, María-Lafuente E, Ureña-Peralta JR, Sorribes L, Hernández A, Casanova B, López-Rodas G, Coret-Ferrer F, Burgal-Marti M. Disturbed Glucose Metabolism in Rat Neurons Exposed to Cerebrospinal Fluid Obtained from Multiple Sclerosis Subjects. Brain Sciences. 2018; 8(1):1. https://doi.org/10.3390/brainsci8010001
Chicago/Turabian StyleMathur, Deepali, Eva María-Lafuente, Juan R. Ureña-Peralta, Lucas Sorribes, Alberto Hernández, Bonaventura Casanova, Gerardo López-Rodas, Francisco Coret-Ferrer, and Maria Burgal-Marti. 2018. "Disturbed Glucose Metabolism in Rat Neurons Exposed to Cerebrospinal Fluid Obtained from Multiple Sclerosis Subjects" Brain Sciences 8, no. 1: 1. https://doi.org/10.3390/brainsci8010001