Particle-Based Microarrays of Oligonucleotides and Oligopeptides

Abstract
:1. Introduction
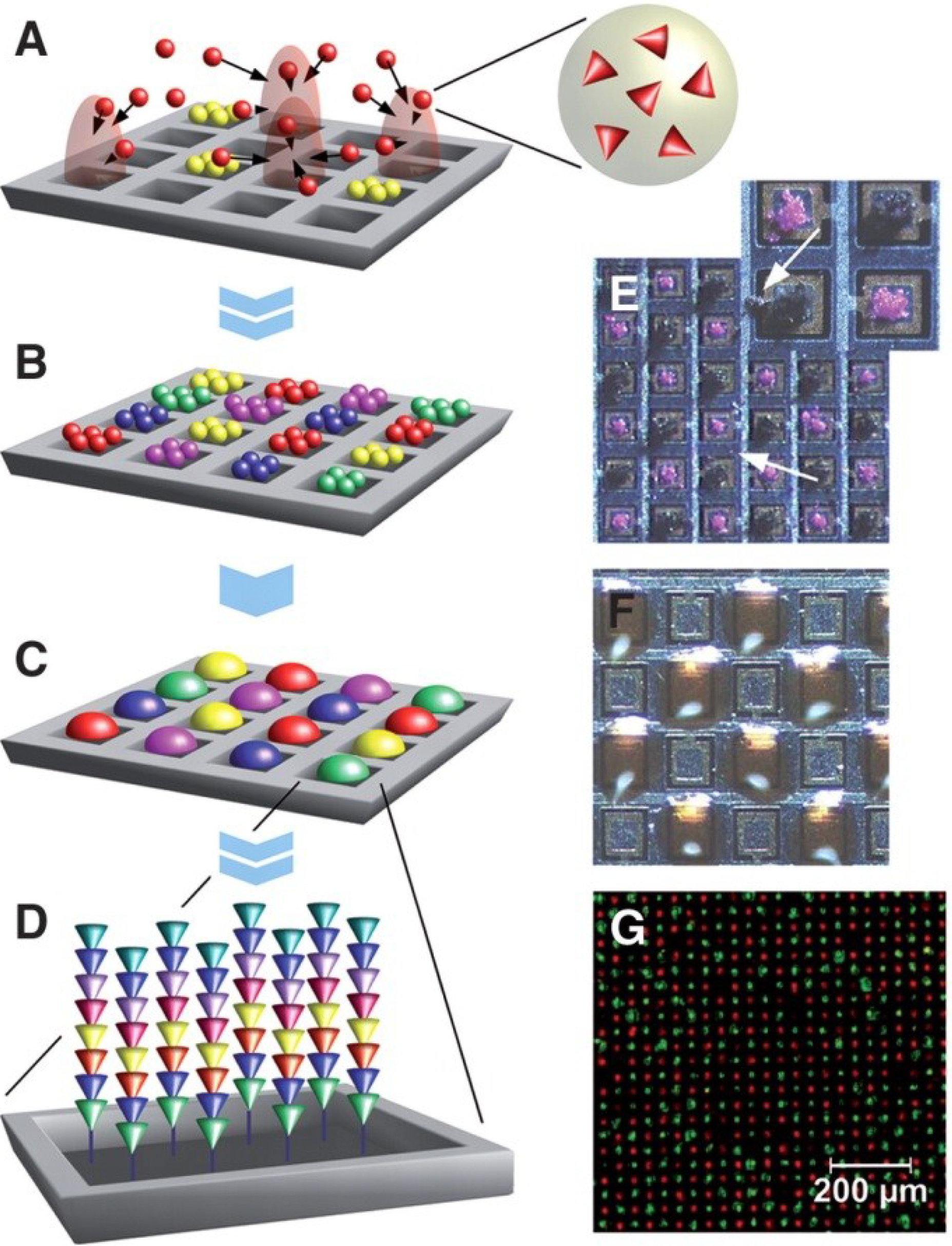
2. Particle-Based Oligonucleotide Arrays
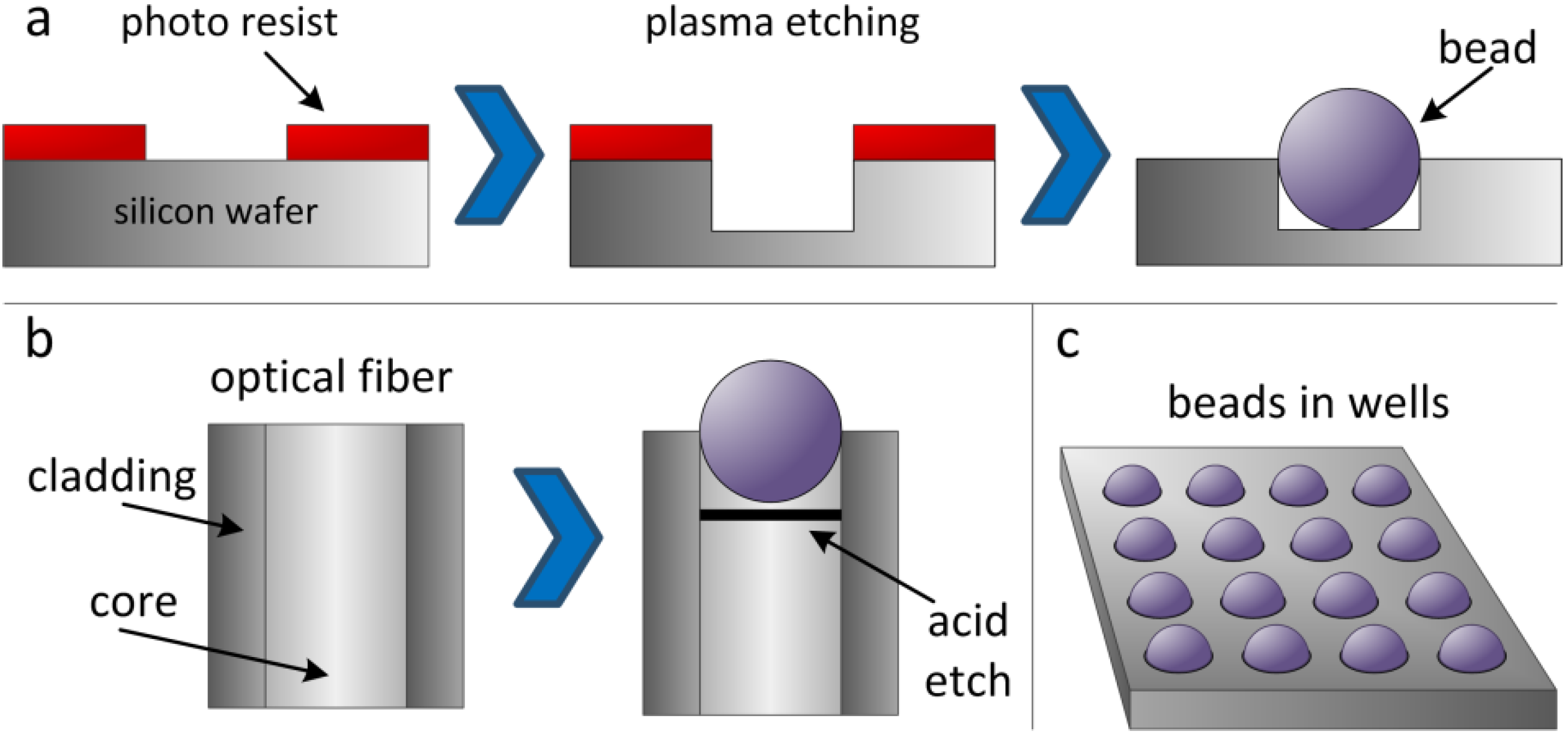
2.1. Illumina Arrays

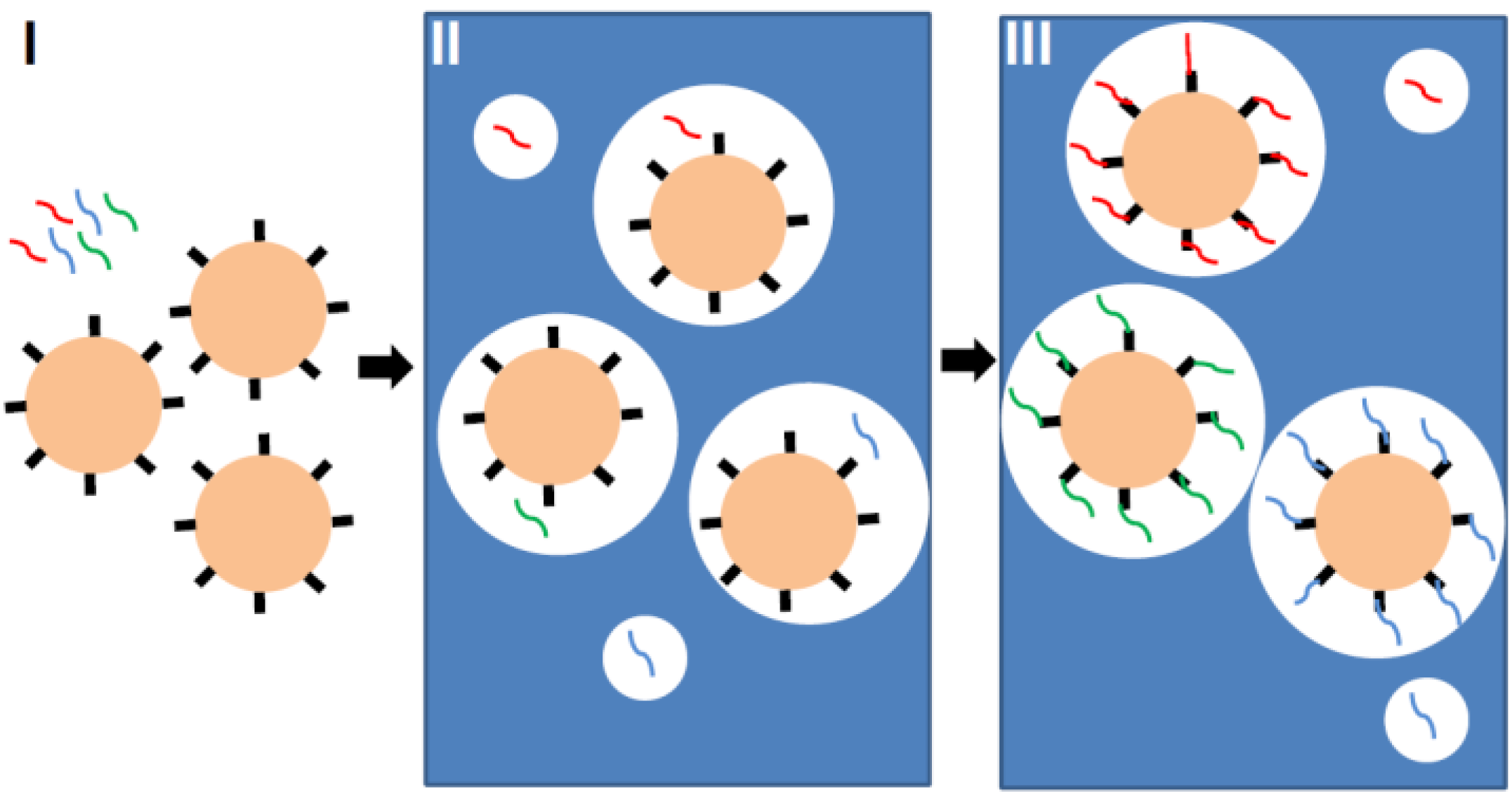
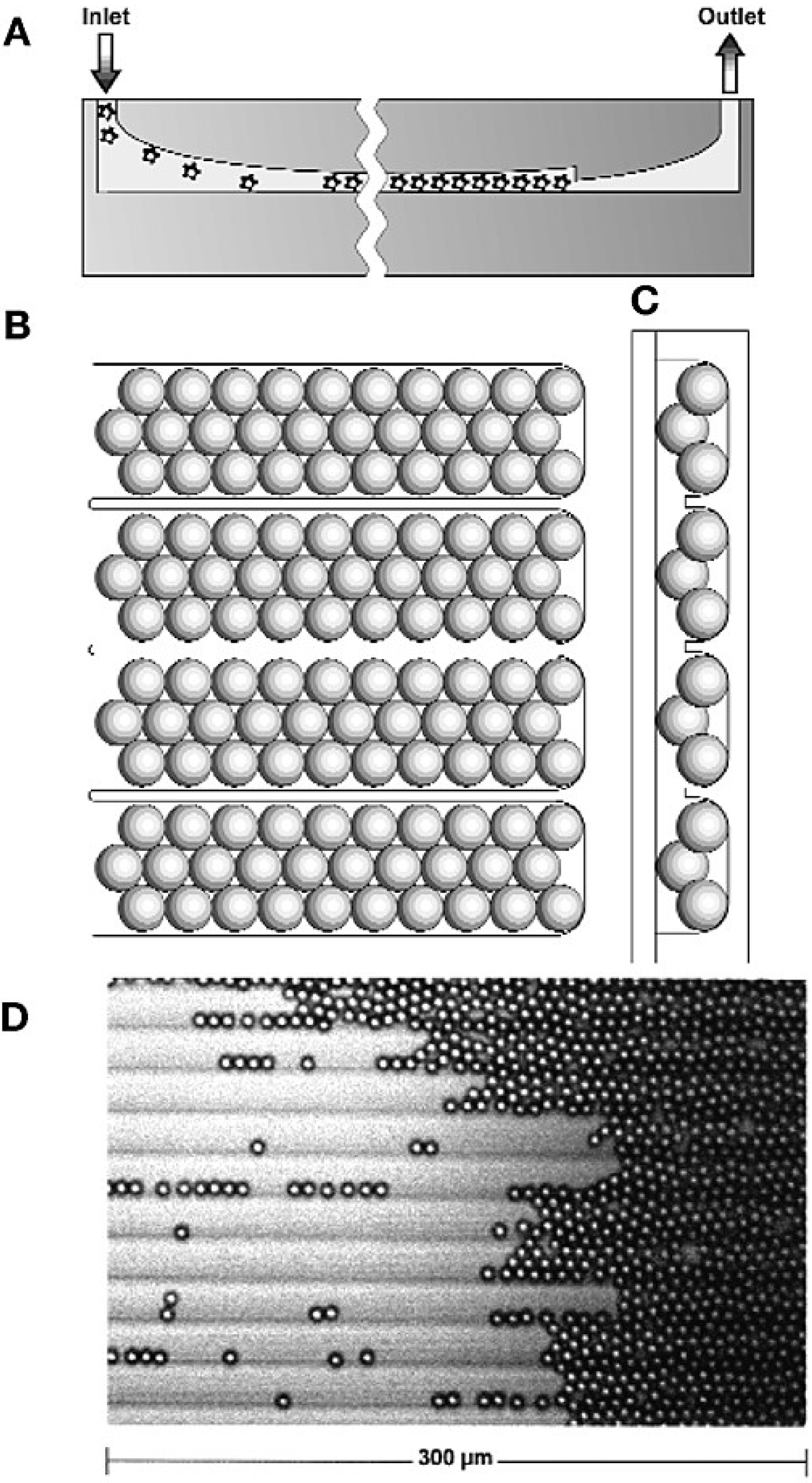
2.2. Particle-Based Emulsion PCR


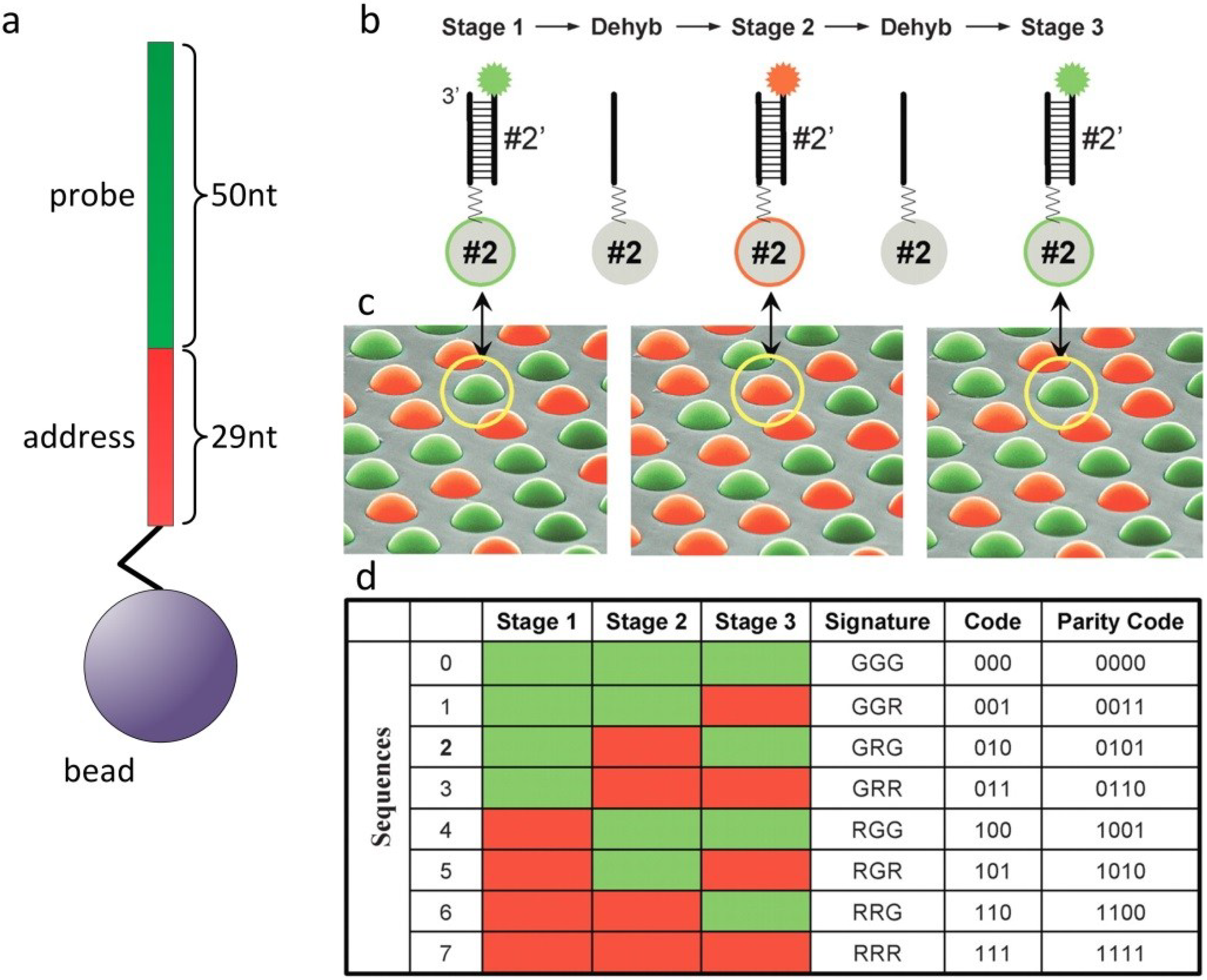
2.3. Particle-Based Arrays for Sequencing



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | Throughput Mb/day | Read Length bp | Quality % | Costs $/Mb |
|---|---|---|---|---|
| 454/Roche | 750 | ~400 | 99.9 | ~20 |
| Illumina/Solexa | 5,000 | ~100 | 98 | ~0.5 |
3. Particle-Based Peptide Arrays
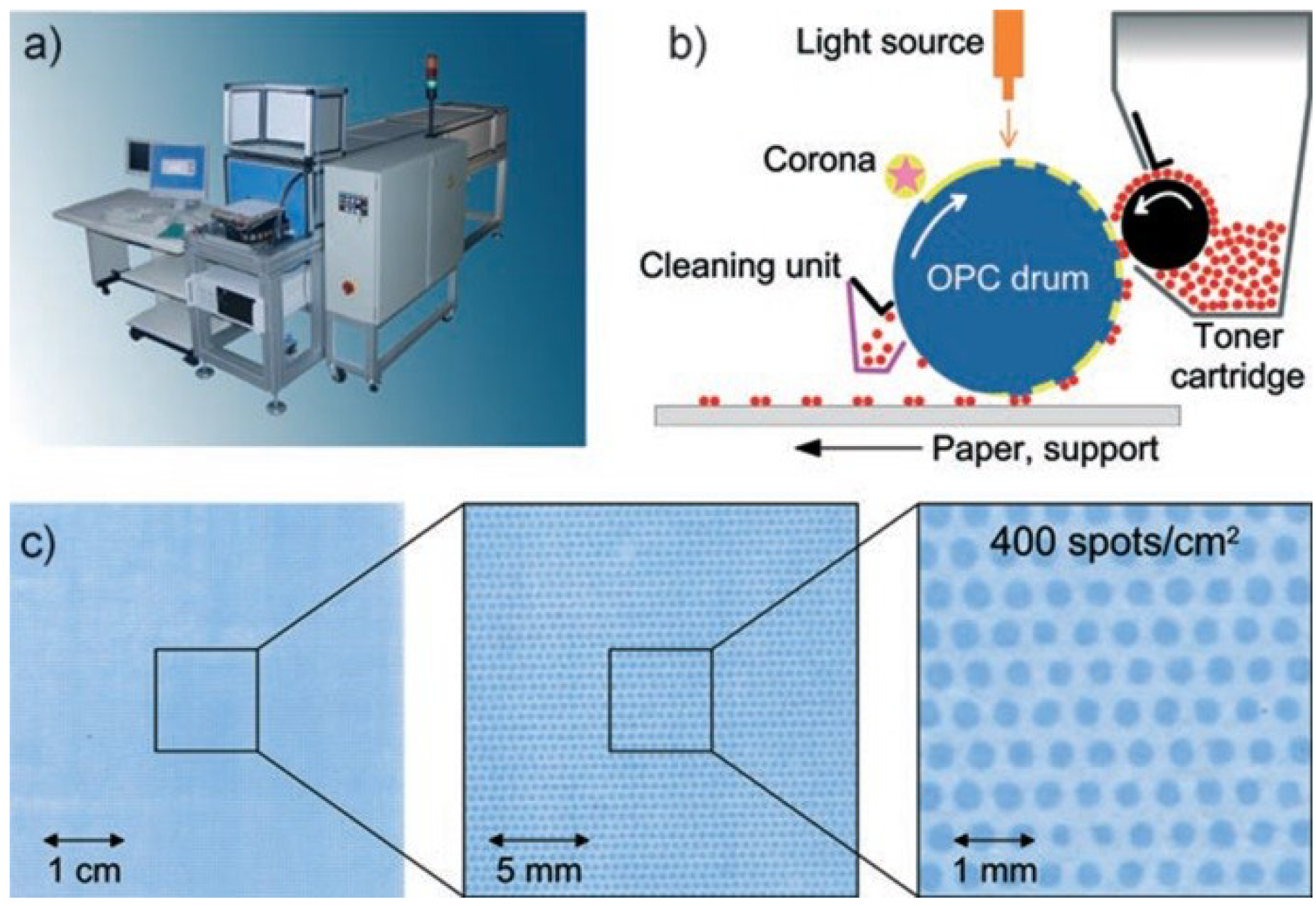
3.1. Amino Acid Particles and Xerographic Methods


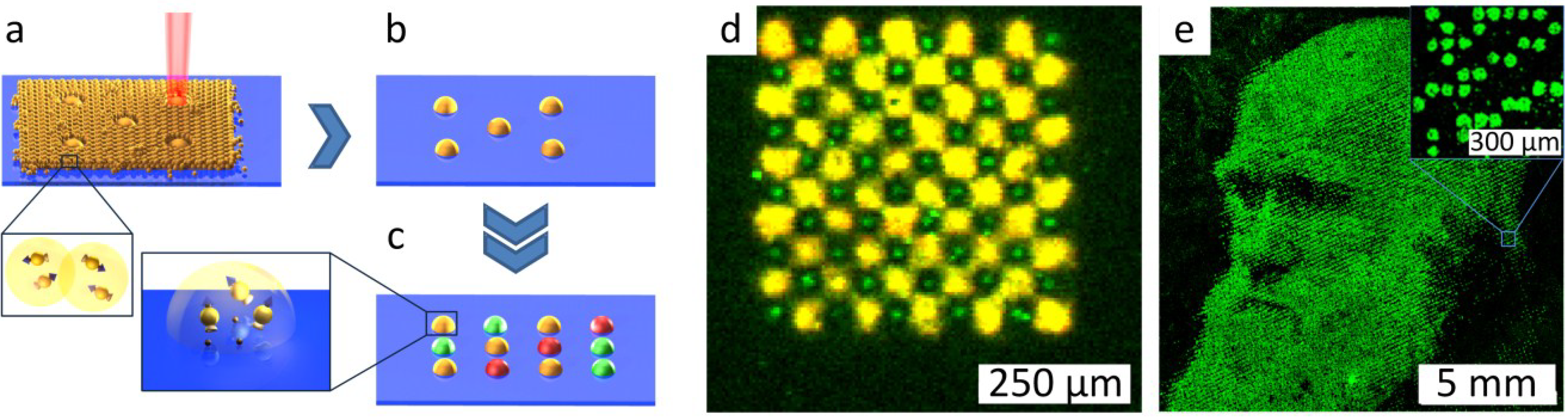
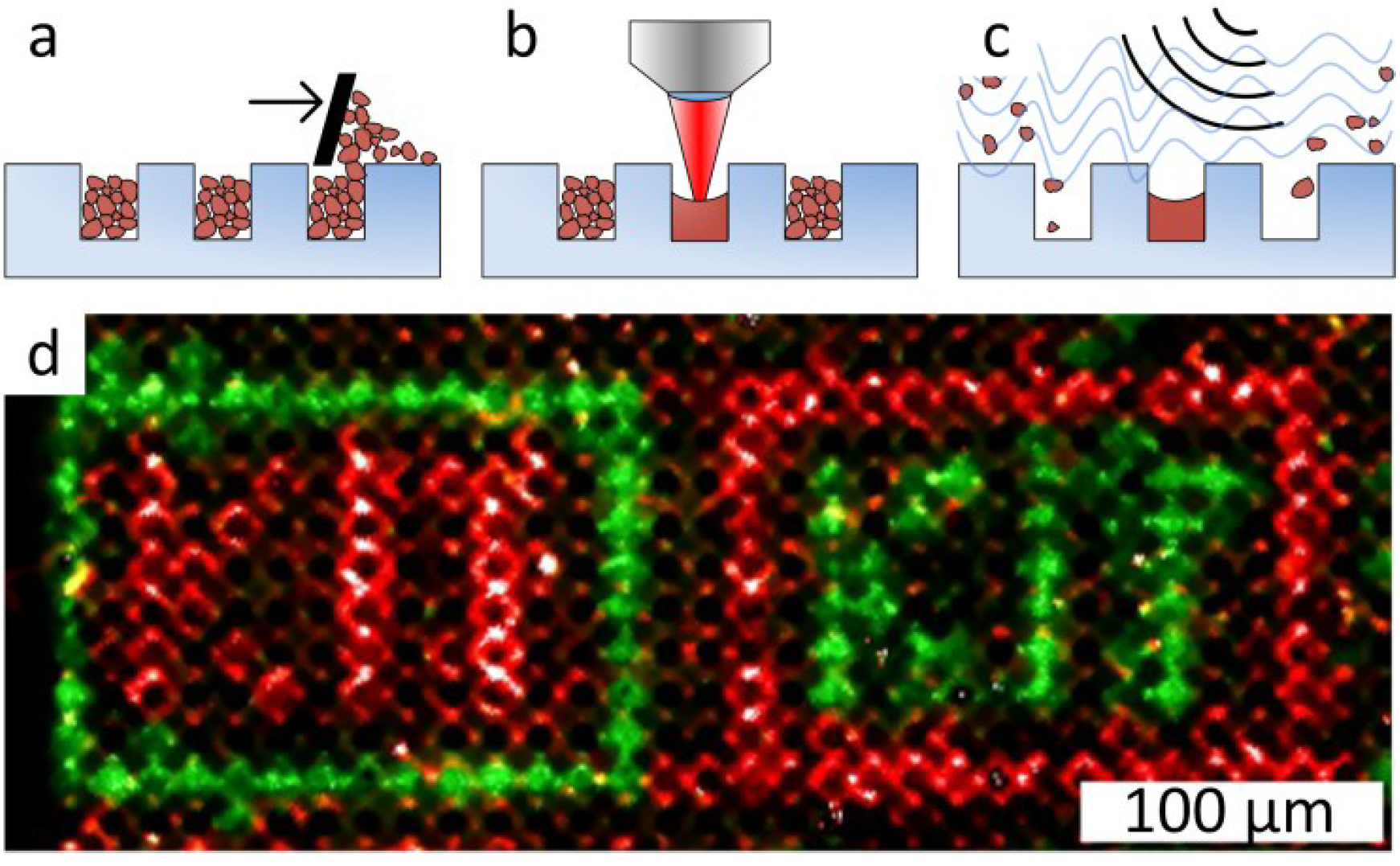
3.2. Combinatorial Laser Fusing of Amino Acid Particles


4. Conclusions
Acknowledgments
Conflicts of Interest
References and Notes
- Frank, R. Spot-synthesis—An easy technique for the positionally addressable, parallel chemical synthesis on a membrane support. Tetrahedron 1992, 48, 9217–9232. [Google Scholar] [CrossRef]
- Frank, R. The SPOT synthesis technique—Synthetic peptide arrays on membrane supports—Principles and applications. J. Immunol. Methods 2002, 267, 13–26. [Google Scholar] [CrossRef] [PubMed]
- JPT. Available online: http://www.jpt.com/ (accessed on 1 September 2014).
- Fodor, S.P.A.; Read, J.L.; Pirrung, M.C.; Stryer, L.; Lu, A.T.; Solas, D. Light-directed, spatially addressable parallel chemical synthesis. Science 1991, 251, 767–773. [Google Scholar] [CrossRef]
- Affimetryx. Available online: http://www.affymetrix.com/estore/ (accessed on 1 September 2014).
- Furka, A.; Sebestyen, F.; Asgedom, M.; Dibo, G. General-method for rapid synthesis of multicomponent peptide mixtures. Int. J. Pept. Protein Res. 1991, 37, 487–493. [Google Scholar] [CrossRef] [PubMed]
- Lam, K.S.; Salmon, S.E.; Hersh, E.M.; Hruby, V.J.; Kazmierski, W.M.; Knapp, R.J. A new type of synthetic peptide library for identifying ligand-binding activity. Nature 1991, 354, 82–84. [Google Scholar] [CrossRef] [PubMed]
- Merrifield, R.B. Solid phase peptide synthesis.1. Synthesis of a tetrapeptide. J. Am. Chem. Soc. 1963, 85, 2149–2154. [Google Scholar]
- Merrifield, R.B. Solid-phase synthesis (Nobel Lecture). Angew. Chem. Int. Ed. Engl. 1985, 24, 799–810. [Google Scholar] [CrossRef]
- Katz, C.; Levy-Beladev, L.; Rotem-Bamberger, S.; Rito, T.; Rudiger, S.G.D.; Friedler, A. Studying protein-protein interactions using peptide arrays. Chem. Soc. Rev. 2011, 40, 2131–2145. [Google Scholar] [CrossRef] [PubMed]
- Dressman, D.; Yan, H.; Traverso, G.; Kinzler, K.W.; Vogelstein, B. Transforming single DNA molecules into fluorescent magnetic particles for detection and enumeration of genetic variations. Proc. Natl. Acad. Sci. USA 2003, 100, 8817–8822. [Google Scholar] [CrossRef]
- Illumina. Available online: http://www.illumina.com/ (accessed on 1 September 2014).
- Steinberg, G.; Stromsborg, K.; Thomas, L.; Barker, D.; Zhao, C.F. Strategies for covalent attachment of DNA to beads. Biopolymers 2004, 73, 597–605. [Google Scholar] [CrossRef]
- Steemers, F.J.; Ferguson, J.A.; Walt, D.R. Screening unlabeled DNA targets with randomly ordered fiber-optic gene arrays. Nat. Biotechnol. 2000, 18, 91–94. [Google Scholar] [CrossRef] [PubMed]
- Walt, D.R. Molecular biology—Bead-based fiber-optic arrays. Science 2000, 287, 451–452. [Google Scholar] [CrossRef] [PubMed]
- Taylor, L.C.; Walt, D.R. Application of high-density optical microwell arrays in a live-cell biosensing system. Anal. Biochem. 2000, 278, 132–142. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, J.A.; Steemers, F.J.; Walt, D.R. High-density fiber-optic DNA random microsphere array. Anal. Chem. 2000, 72, 5618–5624. [Google Scholar] [CrossRef] [PubMed]
- Gunderson, K.L.; Kruglyak, S.; Graige, M.S.; Garcia, F.; Kermani, B.G.; Zhao, C.F.; Che, D.P.; Dickinson, T.; Wickham, E.; Bierle, J.; et al. Decoding randomly ordered DNA arrays. Genome Res. 2004, 14, 870–877. [Google Scholar] [CrossRef] [PubMed]
- Epstein, J.R.; Ferguson, J.A.; Lee, K.H.; Walt, D.R. Combinatorial decoding: An approach for universal DNA array fabrication. J. Am. Chem. Soc. 2003, 125, 13753–13759. [Google Scholar] [CrossRef]
- Brenner, S.; Johnson, M.; Bridgham, J.; Golda, G.; Lloyd, D.H.; Johnson, D.; Luo, S.J.; McCurdy, S.; Foy, M.; Ewan, M.; et al. Gene expression analysis by massively parallel signature sequencing (MPSS) on microbead arrays. Nat. Biotechnol. 2000, 18, 630–634. [Google Scholar] [CrossRef]
- Shendure, J.; Porreca, G.J.; Reppas, N.B.; Lin, X.X.; McCutcheon, J.P.; Rosenbaum, A.M.; Wang, M.D.; Zhang, K.; Mitra, R.D.; Church, G.M. Accurate multiplex polony sequencing of an evolved bacterial genome. Science 2005, 309, 1728–1732. [Google Scholar] [CrossRef]
- Kim, J.B.; Porreca, G.J.; Song, L.; Greenway, S.C.; Gorham, J.M.; Church, G.M.; Seidman, C.E.; Seidman, J.G. Polony multiplex analysis of gene expression (PMAGE) in mouse hypertrophic cardiomyopathy. Science 2007, 316, 1481–1484. [Google Scholar] [CrossRef]
- Leamon, J.H.; Lee, W.L.; Tartaro, K.R.; Lanza, J.R.; Sarkis, G.J.; deWinter, A.D.; Berka, J.; Lohman, K.L. A massively parallel PicoTiterPlate (TM) based platform for discrete picoliter-scale polymerase chain reactions. Electrophoresis 2003, 24, 3769–3777. [Google Scholar] [CrossRef]
- Metzker, M.L. Applications of next-generation sequencing, sequencing technologies—The next generation. Nat. Rev. Genet. 2010, 11, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Michael, K.L.; Taylor, L.C.; Schultz, S.L.; Walt, D.R. Randomly ordered addressable high-density optical sensor arrays. Anal. Chem. 1998, 70, 1242–1248. [Google Scholar] [CrossRef]
- Margulies, M.; Egholm, M.; Altman, W.E.; Attiya, S.; Bader, J.S.; Bemben, L.A.; Berka, J.; Braverman, M.S.; Chen, Y.J.; Chen, Z.T.; et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature 2005, 437, 376–380. [Google Scholar]
- Nyren, P. The history of pyrosequencing. Methods Mol Biol. 2007, 373, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Bentley, D.R.; Balasubramanian, S.; Swerdlow, H.P.; Smith, G.P.; Milton, J.; Brown, C.G.; Hall, K.P.; Evers, D.J.; Barnes, C.L.; Bignell, H.R.; et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature 2008, 456, 53–59. [Google Scholar] [CrossRef]
- Eid, J.; Fehr, A.; Gray, J.; Luong, K.; Lyle, J.; Otto, G.; Peluso, P.; Rank, D.; Baybayan, P.; Bettman, B.; et al. Real-time DNA sequencing from single polymerase molecules. Science 2009, 323, 133–138. [Google Scholar] [CrossRef]
- Kircher, M.; Kelso, J. High-throughput DNA sequencing—Concepts and limitations. Bioessays 2010, 32, 524–536. [Google Scholar] [CrossRef] [PubMed]
- Fedurco, M.; Romieu, A.; Williams, S.; Lawrence, I.; Turcatti, G. BTA, a novel reagent for DNA attachment on glass and efficient generation of solid-phase amplified DNA colonies. Nucl. Acids Res. 2006, 34, e22. [Google Scholar] [CrossRef]
- Next generation sequencing market expected to grow to $2.7 billion by 2017. Pharma Letter. 4 July 2013. Available online: http://www.thepharmaletter.com/ (accessed on 4 July 2013).
- Pareek, C.S.; Smoczynski, R.; Tretyn, A. Sequencing technologies and genome sequencing. J. Appl. Genet. 2011, 52, 413–435. [Google Scholar] [CrossRef] [PubMed]
- Chapin, S.C.; Appleyard, D.C.; Pregibon, D.C.; Doyle, P.S. Rapid microRNA profiling on encoded gel microparticles. Angew. Chem. Int. Ed. 2011, 50, 2289–2293. [Google Scholar] [CrossRef]
- Nolan, J.P.; Sklar, L.A. Suspension array technology: Evolution of the flat-array paradigm. Trends Biotechnol. 2002, 20, 9–12. [Google Scholar] [CrossRef] [PubMed]
- Akhras, M.S.; Pettersson, E.; Diamond, L.; Unemo, M.; Okamoto, J.; Davis, R.W.; Pourmand, N. The sequencing bead array (SBA), a next-generation digital suspension array. PLoS One 2013, 8, e76696. [Google Scholar] [CrossRef] [PubMed]
- Pregibon, D.C.; Toner, M.; Doyle, P.S. Multifunctional encoded particles for high-throughput biomolecule analysis. Science 2007, 315, 1393–1396. [Google Scholar] [CrossRef] [PubMed]
- Beyer, M.; Nesterov, A.; Block, I.; Konig, K.; Felgenhauer, T.; Fernandez, S.; Leibe, K.; Torralba, G.; Hausmann, M.; Trunk, U.; et al. Combinatorial synthesis of peptide arrays onto a microchip. Science 2007, 318, 1888–1888. [Google Scholar] [PubMed]
- Borsenberger, P.M.; Weiss, D.S. Organic Photoreceptors for Imaging Systems; Optical Engineering; M. Dekker: New York, NY, USA, 1993; Volume xvi, p. 447. [Google Scholar]
- Pai, D.M.; Springett, B.E. Physics of Electrophotography. Rev. Modern Phys. 1993, 65, 163–211. [Google Scholar] [CrossRef]
- Löffler, F.; Cheng, Y.-C.; Förtsch, T.; Dörsam, E.; Bischoff, R.; Breitling, F.; Nesterov-Müller, A. Biofunctional xerography. In Biotechnology of Biopolymers; Elnashar, M., Ed.; InTech: Rijeka, Croatia, 2011. [Google Scholar]
- Feinberg, R.S.; Merrifie, R.B. Zinc chloride-catalyzed chloromethylation of resins for solid-phase peptide-synthesis. Tetrahedron 1974, 30, 3209–3212. [Google Scholar] [CrossRef]
- Stadler, V.; Felgenhauer, T.; Beyer, M.; Fernandez, S.; Leibe, K.; Guttler, S.; Groning, M.; Konig, K.; Torralba, G.; Hausmann, M.; et al. Combinatorial synthesis of peptide arrays with a laser printer. Angew. Chem. Int. Ed. Engl. 2008, 47, 7132–7135. [Google Scholar] [CrossRef] [PubMed]
- Pepperprint. Available online: http://www.pepperprint.com/ (accessed on 1 September 2014).
- Loeffler, F.F.; Cheng, Y.C.; Muenster, B.; Striffler, J.; Liu, F.C.; Ralf Bischoff, F.; Doersam, E.; Breitling, F.; Nesterov-Mueller, A. Printing Peptide arrays with a complementary metal oxide semiconductor chip. Adv. Biochem. Eng. Biotechnol. 2013, 137, 1–23. [Google Scholar] [PubMed]
- Löffler, F.; Wagner, J.; König, K.; Märkle, F.; Fernandez, S.; Schirwitz, C.; Torralba, G.; Hausmann, M.; Lindenstruth, V.; Bischoff, F.R.; et al. High-precision combinatorial deposition of micro particle patterns on a microelectronic chip. Aerosol. Sci. Technol. 2011, 45, 65–74. [Google Scholar] [CrossRef]
- Loeffler, F.; Schirwitz, C.; Wagner, J.; Koenig, K.; Maerkle, F.; Torralba, G.; Hausmann, M.; Bischoff, F.R.; Nesterov-Mueller, A.; Breitling, F. Biomolecule Arrays using functional combinatorial particle patterning on microchips. Adv. Funct. Mater. 2012, 22, 2503–2508. [Google Scholar] [CrossRef]
- Schmidt, R.; Jacak, J.; Schirwitz, C.; Stadler, V.; Michel, G.; Marme, N.; Schutz, G.J.; Hoheisel, J.D.; Knemeyer, J.P. Single-molecule detection on a protein-array assay platform for the exposure of a tuberculosis antigen. J. Proteome Res. 2011, 10, 1316–1322. [Google Scholar] [CrossRef] [PubMed]
- Stafford, P.; Halperin, R.; Legutki, J.B.; Magee, D.M.; Galgiani, J.; Johnston, S.A. Physical characterization of the “Immunosignaturing Effect”. Mol. Cell. Proteomics 2012, 11. [Google Scholar] [CrossRef] [PubMed]
- Grillberger, R.; Casina, V.C.; Turecek, P.L.; Zheng, X.L.; Rottensteiner, H.; Scheiflinger, F. Anti-ADAMTS13 IgG autoantibodies present in healthy individuals share linear epitopes with those in patients with thrombotic thrombocytopenic purpura. Haematologica 2014, 99, E58–E60. [Google Scholar] [CrossRef] [PubMed]
- Musset, L.; Miyara, M.; Benveniste, O.; Charuel, J.L.; Shikhman, A.; Boyer, O.; Fowler, R.; Mammen, A.; Phillips, J.; Mahler, M. Analysis of Autoantibodies to 3-Hydroxy-3-methylglutaryl-coenzyme a reductase using different technologies. J. Immunol. Res. 2014. [Google Scholar] [CrossRef]
- Borgwardt, D.S.; Martin, A.D.; Van Hemert, J.R.; Yang, J.Y.; Fischer, C.L.; Recker, E.N.; Nair, P.R.; Vidva, R.; Chandrashekaraiah, S.; Progulske-Fox, A.; et al. Histatin 5 binds to Porphyromonas gingivalis hemagglutinin B (HagB) and alters HagB-induced chemokine responses. Sci. Rep. 2014, 4, 3904. [Google Scholar] [CrossRef] [PubMed]
- Nesterov, A.; Konig, K.; Felgenhauer, T.; Lindenstruth, V.; Trunk, U.; Fernandez, S.; Hausmann, M.; Bischoff, F.R.; Breitling, F.; Stadler, V. Precise selective deposition of microparticles on electrodes of microelectronic chips. Rev. Sci. Instrum. 2008, 79, 035106. [Google Scholar] [CrossRef] [PubMed]
- Maerkle, F.; Loeffler, F.F.; Schillo, S.; Foertsch, T.; Muenster, B.; Striffler, J.; Schirwitz, C.; Bischoff, F.R.; Breitling, F.; Nesterov-Mueller, A. High-density peptide arrays with combinatorial laser fusing. Adv. Mater. 2014, 26, 3730–3734. [Google Scholar] [CrossRef] [PubMed]
- Maerkle, F. Laserbasierte Verfahren zur Herstellung hochdichter Peptidarrays Schriften des Instituts für Mikrostrukturtechnik. Ph.D. Thesis, Karlsruher Institut für Technologie, Karlsruhe, Germany, 2014; KIT Scientific Publishing, Karlsruhe, Germany, 2014.
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nesterov-Mueller, A.; Maerkle, F.; Hahn, L.; Foertsch, T.; Schillo, S.; Bykovskaya, V.; Sedlmayr, M.; Weber, L.K.; Ridder, B.; Soehindrijo, M.; et al. Particle-Based Microarrays of Oligonucleotides and Oligopeptides. Microarrays 2014, 3, 245-262. https://doi.org/10.3390/microarrays3040245
Nesterov-Mueller A, Maerkle F, Hahn L, Foertsch T, Schillo S, Bykovskaya V, Sedlmayr M, Weber LK, Ridder B, Soehindrijo M, et al. Particle-Based Microarrays of Oligonucleotides and Oligopeptides. Microarrays. 2014; 3(4):245-262. https://doi.org/10.3390/microarrays3040245
Chicago/Turabian StyleNesterov-Mueller, Alexander, Frieder Maerkle, Lothar Hahn, Tobias Foertsch, Sebastian Schillo, Valentina Bykovskaya, Martyna Sedlmayr, Laura K. Weber, Barbara Ridder, Miriam Soehindrijo, and et al. 2014. "Particle-Based Microarrays of Oligonucleotides and Oligopeptides" Microarrays 3, no. 4: 245-262. https://doi.org/10.3390/microarrays3040245