
Antioxidant Synergy of Mitochondrial Phospholipase PNPLA8/iPLA2γ with Fatty Acid–Conducting SLC25 Gene Family Transporters



Abstract
1. Introduction
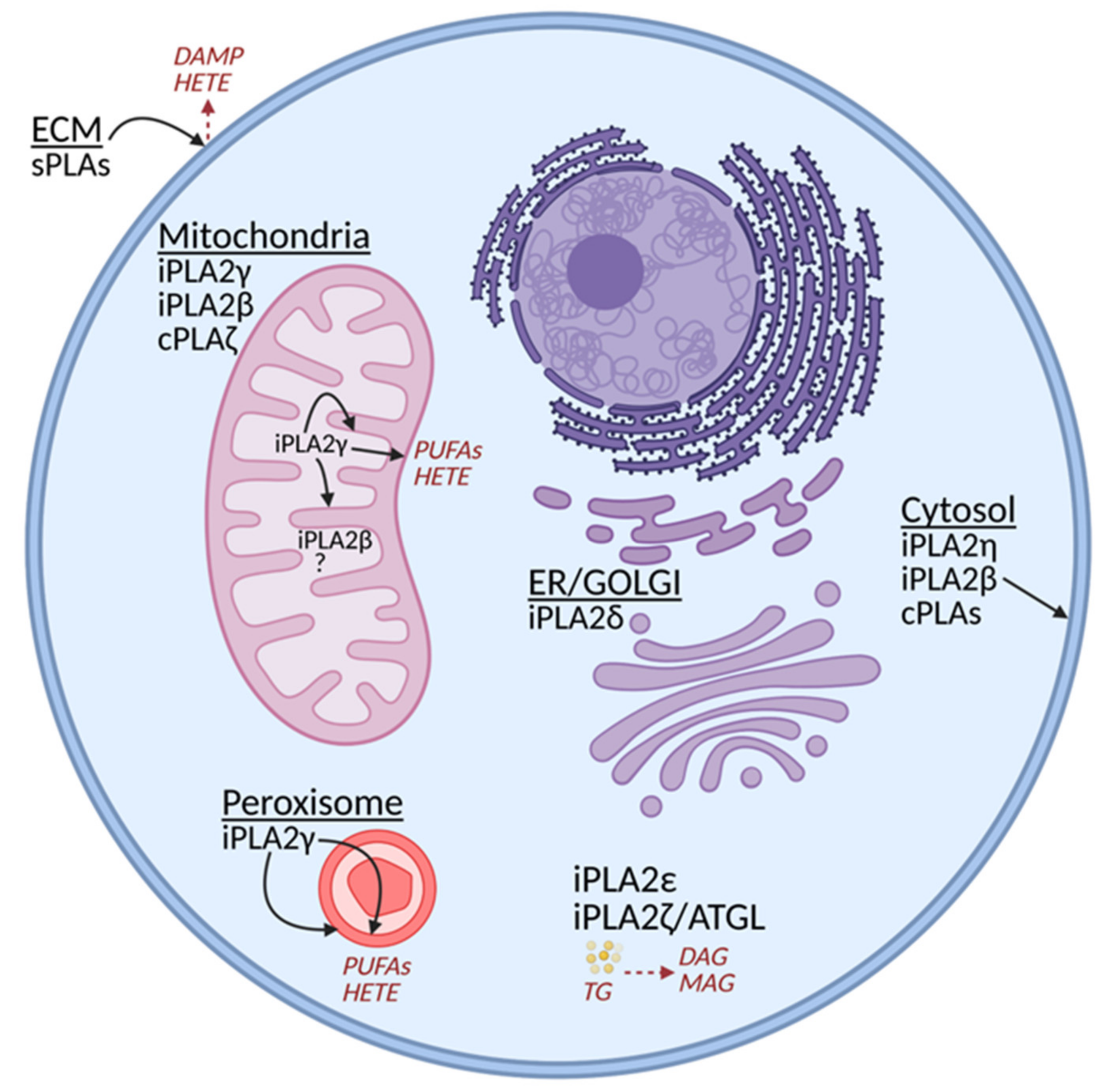
2. Phospholipase A2 Group VI, Patatin-Like Phospholipase Domain-Containing Proteins (PNPLAs)
2.1. Classification of Phospholipases A2
2.2. Phospholipase PNPLA9/ iPLA2β
2.3. Phospholipase PNPLA8/iPLA2γ
2.4. Redox Activation of Phospholipase iPLA2γ
3. Antioxidant Synergy of iPLA2γ and Mitochondrial Uncoupling Proteins
3.1. Nascent FAs—Spatiotemporal FA Release Initiates Mild Uncoupling by UCPs
3.2. Mechanism of Uncoupling Protein-Mediated Suppression of Mitochondrial Superoxide Formation
3.3. Antioxidant Synergy of iPLA2γ and Uncoupling Proteins
3.4. Mutual Influence of Mitochondrial and Cytosolic Redox State
3.5. Mechanism of Suppression of Mitochondrial Superoxide Formation by the Action of ANT or Other SLC25 Family Proteins
3.6. Antioxidant Synergy of iPLA2γ and ANT or iPLA2γ and Other SLC25 Family Proteins
4. Redox Activation of iPLA2γ
4.1. Direct Activation of iPLA2γ by Oxidants
4.2. iPLA2γ vs. Lipid Hydroperoxides and Other Products of Lipid Peroxidation
5. Dependence on Lipid Bilayer Structure and Cardiolipin
Cardiolipin and iPLA2γ
6. Fatty Acid–Mediated Signaling by iPLA2γ
6.1. Mitochondrial FAs as Messengers in Information Signaling
6.2. Mitochondrial Lysophospholipids as Messengers in Information Signaling
6.3. Hydroperoxy FAs, Hydroxy FAs, and other Lipid Peroxidation Products as Messengers in Information Signaling
7. Physiological Cytoprotective and Regulatory Roles of iPLA2γ
7.1. Mitochondrial iPLA2γ in Heart Physiology and Pathology
7.2. Mitochondrial iPLA2γ in Brain Physiology and Pathology
7.3. Mitochondrial iPLA2γ in BAT Physiology and Pathology
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ježek, P.; Hlavatá, L. Mitochondria in homeostasis of reactive oxygen species in cell, tissues, and organism. Int. J. Biochem. Cell Biol. 2005, 37, 2478–2503. [Google Scholar] [CrossRef] [PubMed]
- Ježek, P.; Plecitá-Hlavatá, L. Mitochondrial reticulum network dynamics in relation to oxidative stress, redox regulation, and hypoxia. Int. J. Biochem. Cell Biol. 2009, 41, 1790–1804. [Google Scholar] [CrossRef] [PubMed]
- Ježek, P.; Olejár, T.; Smolková, K.; Ježek, J.; Dlasková, A.; Plecitá-Hlavatá, L.; Zelenka, J.; Špaček, T.; Engstová, H.; Pajuelo Reguera, D.; et al. Antioxidant and regulatory role of mitochondrial uncoupling protein UCP2 in pancreatic beta-cells. Physiol. Res. 2014, 63 (Suppl. 1), S73–S91. [Google Scholar] [CrossRef]
- Plecitá-Hlavatá, L.; Ježek, P. Integration of superoxide formation and cristae morphology for mitochondrial redox signaling. Int. J. Biochem. Cell Biol. 2016, 80, 31–50. [Google Scholar] [CrossRef]
- Ježek, P.; Holendová, B.; Garlid, K.D.; Jabůrek, M. Mitochondrial uncoupling proteins: Subtle regulators of cellular redox signaling. Antioxidants Redox Signal. 2018, 29, 667–714. [Google Scholar] [CrossRef]
- Ježek, P.; Holendová, B.; Plecitá-Hlavatá, L. Redox signaling from mitochondria: Signal propagation and its targets. Biomolecules 2020, 10, 93. [Google Scholar] [CrossRef]
- Ježek, P.; Holendová, B.; Jabůrek, M.; Tauber, J.; Dlasková, A.; Plecitá-Hlavatá, L. The pancreatic β-cell: The perfect redox system. Antioxidants 2021, 10, 197. [Google Scholar] [CrossRef] [PubMed]
- Ursini, F.; Maiorino, M.; Forman, H.J. Redox homeostasis: The Golden Mean of healthy living. Redox Biol. 2016, 8, 205–215. [Google Scholar] [CrossRef]
- Forman, H.J. Redox signaling: An evolution from free radicals to aging. Free Radic. Biol. Med. 2016, 97, 398–407. [Google Scholar] [CrossRef] [PubMed]
- Skulachev, V.P. Fatty acid circuit as a physiological mechanism of uncoupling of oxidative phosphorylation. FEBS Lett. 1991, 294, 158–162. [Google Scholar] [CrossRef]
- Skulachev, V.P. Uncoupling: New approaches to an old problem of bioenergetics. Biochim. Biophys. Acta Bioenerg. 1998, 1363, 100–124. [Google Scholar] [CrossRef]
- Bertholet, A.M.; Chouchani, E.T.; Kazak, L.; Angelin, A.; Fedorenko, A.; Long, J.Z.; Vidoni, S.; Garrity, R.; Cho, J.; Terada, N.; et al. H+ transport is an integral function of the mitochondrial ADP/ATP carrier. Nature 2019, 571, 515–520. [Google Scholar] [CrossRef]
- Fedorenko, A.; Lishko, P.V.; Kirichok, Y. Mechanism of fatty-acid-dependent UCP1 uncoupling in brown fat mitochondria. Cell 2012, 151, 400–413. [Google Scholar] [CrossRef]
- Ramanadham, S.; Tomader, A.; Ashley, J.W.; Bone, R.N.; Hancock, W.D.; Lei, X. Calcium-independent phospholipases A2 and their roles in biological processes and diseases. J. Lipid Res. 2015, 56, 1643–1668. [Google Scholar] [CrossRef]
- Murakami, M.; Sato, H.; Taketomi, Y. Updating phospholipase A2 biology. Biomolecules 2020, 10, 1457. [Google Scholar] [CrossRef] [PubMed]
- Hara, S.; Yoda, E.; Sasaki, Y.; Nakatani, Y.; Kuwata, H. Calcium-independent phospholipase A2γ (iPLA2γ) and its roles in cellular functions and diseases. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2019, 1864, 861–868. [Google Scholar] [CrossRef]
- Ježek, J.; Jabůrek, M.; Zelenka, J.; Ježek, P. Mitochondrial phospholipase A2 activated by reactive oxygen species in heart mitochondria induces mild uncoupling. Physiol. Res. 2010, 59, 737–747. [Google Scholar] [CrossRef] [PubMed]
- Jabůrek, M.; Ježek, J.; Zelenka, J.; Ježek, P. Antioxidant activity by a synergy of redox-sensitive mitochondrial phospholipase A2 and uncoupling protein-2 in lung and spleen. Int. J. Biochem. Cell Biol. 2013, 45, 816–825. [Google Scholar] [CrossRef]
- Ježek, J.; Dlasková, A.; Zelenka, J.; Jabůrek, M.; Ježek, P. H2O2-activated mitochondrial phospholipase iPLA2γ prevents lipotoxic oxidative stress in synergy with UCP2, amplifies signaling via G-protein-coupled receptor GPR40, and regulates insulin secretion in pancreatic β-cells. Antioxid. Redox Signal. 2015, 23, 958–972. [Google Scholar] [CrossRef] [PubMed]
- Jabůrek, M.; Ježek, J.; Ježek, P. Cytoprotective activity of mitochondrial uncoupling protein-2 in lung and spleen. FEBS Open Bio 2018, 8, 692–701. [Google Scholar] [CrossRef]
- Murakami, M.; Taketomi, Y.; Miki, Y.; Sato, H.; Hirabayashi, T.; Yamamoto, K. Recent progress in phospholipase A2 research: From cells to animals to humans. Prog. Lipid Res. 2011, 50, 152–192. [Google Scholar] [CrossRef]
- Van Tienhoven, M.; Atkins, J.; Li, Y.; Glynn, P. Human neuropathy target esterase catalyzes hydrolysis of membrane lipids. J. Biol. Chem. 2002, 277, 20942–20948. [Google Scholar] [CrossRef] [PubMed]
- Baulande, S.; Lasnier, F.; Lucas, M.; Pairault, J. Adiponutrin, a Transmembrane Protein Corresponding to a Novel Dietary- and Obesity-linked mRNA Specifically Expressed in the Adipose Lineage. J. Biol. Chem. 2001, 276, 33336–33344. [Google Scholar] [CrossRef]
- Zimmermann, R.; Strauss, J.G.; Haemmerle, G.; Schoiswohl, G.; Birner-Gruenberger, R.; Riederer, M.; Lass, A.; Neuberger, G.; Eisenhaber, F.; Hermetter, A.; et al. Fat mobilization in adipose tissue is promoted by adipose triglyceride lipase. Science 2004, 306, 1383–1386. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, C.M.; Mancuso, D.J.; Yan, W.; Sims, H.F.; Gibson, B.; Gross, R.W. Identification, cloning, expression, and purification of three novel human calcium-independent phospholipase A2 family members possessing triacylglycerol lipase and acylglycerol transacylase activities. J. Biol. Chem. 2004, 279, 48968–48975. [Google Scholar] [CrossRef]
- Mancuso, D.J.; Jenkins, C.M.; Sims, H.F.; Cohen, J.M.; Yang, J.; Gross, R.W. Complex transcriptional and translational regulation of iPLA 2γ resulting in multiple gene products containing dual competing sites for mitochondrial or peroxisomal localization. Eur. J. Biochem. 2004, 271, 4709–4724. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Bao, S.; Lei, X.; Jin, C.; Zhang, S.; Turk, J.; Ramanadham, S. Evidence for proteolytic processing and stimulated organelle redistribution of iPLA2β. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2010, 1801, 547–558. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Zhang, X.; Zhao, C.; Choi, J.; Shi, J.; Song, K.; Turk, J.; Ma, Z.A. Protection of pancreatic β-cells by group VIA phospholipase A 2-mediated repair of mitochondrial membrane peroxidation. Endocrinology 2010, 151, 3038–3048. [Google Scholar] [CrossRef]
- Moon, S.H.; Liu, X.; Cedars, A.M.; Yang, K.; Kiebish, M.A.; Joseph, S.M.; Kelley, J.; Jenkins, C.M.; Gross, R.W. Heart failure-induced activation of phospholipase iPLA2γ generates hydroxyeicosatetraenoic acids opening the mitochondrial permeability transition pore. J. Biol. Chem. 2018, 293, 115–129. [Google Scholar] [CrossRef]
- Lio, Y.C.; Dennis, E.A. Interfacial activation, lysophospholipase and transacylase activity of Group VI Ca2+-independent phospholipase A2. Biochim. Biophys. Acta Lipids Lipid Metab. 1998, 1392, 320–332. [Google Scholar] [CrossRef]
- Jenkins, C.M.; Yan, W.; Mancuso, D.J.; Gross, R.W. Highly selective hydrolysis of fatty acyl-CoAs by calcium-independent phospholipase A2β: Enzyme autoacylation and acyl-CoA-mediated reversal of calmodulin inhibition of phospholipase A2 activity. J. Biol. Chem. 2006, 281, 15615–15624. [Google Scholar] [CrossRef]
- Ma, Z.; Ramanadham, S.; Kempe, K.; Chi, X.S.; Ladenson, J.; Turk, J. Pancreatic islets express a Ca2+-independent phospholipase A2 enzyme that contains a repeated structural motif homologous to the integral membrane protein binding domain of ankyrin. J. Biol. Chem. 1997, 272, 11118–11127. [Google Scholar] [CrossRef]
- Malley, K.R.; Koroleva, O.; Miller, I.; Sanishvili, R.; Jenkins, C.M.; Gross, R.W.; Korolev, S. The structure of iPLA2β reveals dimeric active sites and suggests mechanisms of regulation and localization. Nat. Commun. 2018, 9, 765. [Google Scholar] [CrossRef]
- Bucher, D.; Hsu, Y.H.; Mouchlis, V.D.; Dennis, E.A.; McCammon, J.A. Insertion of the Ca2+-Independent Phospholipase A2 into a Phospholipid Bilayer via Coarse-Grained and Atomistic Molecular Dynamics Simulations. PLoS Comput. Biol. 2013, 9, e1003156. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.; Jenkins, C.M.; Han, X.; Mancuso, D.J.; Sims, H.F.; Yang, K.; Gross, R.W. The highly selective production of 2-arachidonoyl lysophosphatidylcholine catalyzed by purified calcium-independent phospholipase A2γ: Identification of a novel enzymatic mediator for the generation of a key branch point intermediate in eicosanoid signali. J. Biol. Chem. 2005, 280, 26669–26679. [Google Scholar] [CrossRef] [PubMed]
- Mancuso, D.J.; Han, X.; Jenkins, C.M.; Lehman, J.J.; Sambandam, N.; Sims, H.F.; Yang, J.; Yan, W.; Yang, K.; Green, K.; et al. Dramatic accumulation of triglycerides and precipitation of cardiac hemodynamic dysfunction during brief caloric restriction in transgenic myocardium expressing human calcium-independent phospholipase A2γ. J. Biol. Chem. 2007, 282, 9216–9227. [Google Scholar] [CrossRef] [PubMed]
- Mancuso, D.J.; Jenkins, C.M.; Gross, R.W. The genomic organization, complete mRNA sequence, cloning, and expression of a novel human intracellular membrane-associated calcium-independent phospholipase A(2). J. Biol. Chem. 2000, 275, 9937–9945. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Takeya, R.; Sumimoto, H. A novel intracellular membrane-bound calcium-independent phospholipase A2. Biochem. Biophys. Res. Commun. 2000, 272, 320–326. [Google Scholar] [CrossRef]
- Liu, X.; Sims, H.F.; Jenkins, C.M.; Guan, S.; Dilthey, B.G.; Gross, R.W. 12-LOX catalyzes the oxidation of 2-arachidonoyl-lysolipids in platelets generating eicosanoid-lysolipids that are attenuated by iPLA2γ knockout. J. Biol. Chem. 2020, 295, 5307–5320. [Google Scholar] [CrossRef]
- Jabůrek, M.; Holendová, B.; Průchová, P.; Ježek, P. Cardiolipin hydroperoxides are both substrates and redox activators of phospholipase iPLA2γ. Free Radic. Biol. Med. 2018, 120, S65. [Google Scholar] [CrossRef]
- Kühlbrandt, W. Structure and function of mitochondrial membrane protein complexes. BMC Biol. 2015, 13, 89. [Google Scholar] [CrossRef]
- Dlasková, A.; Špaček, T.; Engstová, H.; Špačková, J.; Schröfel, A.; Holendová, B.; Smolková, K.; Plecitá-Hlavatá, L.; Ježek, P. Mitochondrial cristae narrowing upon higher 2-oxoglutarate load. Biochim. Biophys. Acta Bioenerg. 2019, 1860, 659–678. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Minakami, R.; Kanaya, H.; Sumimoto, H. Catalytic residues of group VIB calcium-independent phospholipase A 2 (iPLA2γ). Biochem. Biophys. Res. Commun. 2004, 320, 1284–1290. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.-Y.Y.; Moon, S.H.; Jenkins, C.M.; Li, M.; Sims, H.F.; Guan, S.; Gross, R.W.; Ho Moon, S.; Jenkins, C.M.; Li, M.; et al. The phospholipase iPLA2 is a major mediator releasing oxidized aliphatic chains from cardiolipin, integrating mitochondrial bioenergetics and signaling. J. Biol. Chem. 2017, 292, 10672–10684. [Google Scholar] [CrossRef] [PubMed]
- Yoda, E.; Hachisu, K.; Taketomi, Y.; Yoshida, K.; Nakamura, M.; Ikeda, K.; Taguchi, R.; Nakatani, Y.; Kuwata, H.; Murakami, M.; et al. Mitochondrial dysfunction and reduced prostaglandin synthesis in skeletal muscle of Group VIB Ca2+-independent phospholipase A2gamma-deficient mice. J. Lipid Res. 2010, 51, 3003–3015. [Google Scholar] [CrossRef] [PubMed]
- Moon, S.H.; Dilthey, B.G.; Liu, X.; Guan, S.; Sims, H.F.; Gross, R.W. High-Fat diet activates liver iPLA2γ generating eicosanoids that mediate metabolic stress. J. Lipid Res. 2021, 62, 100052. [Google Scholar] [CrossRef] [PubMed]
- Jabůrek, M.; Garlid, K.D. Reconstitution of recombinant uncoupling proteins. UCP1, -2, and -3 have similar affinities for ATP and are unaffected by coenzyme Q10. J. Biol. Chem. 2003, 278, 25825–25831. [Google Scholar] [CrossRef] [PubMed]
- Jabůrek, M.; Vařecha, M.; Gimeno, R.E.; Dembski, M.; Ježek, P.; Zhang, M.; Burn, P.; Tartaglia, L.A.; Garlid, K.D. Transport function and regulation of mitochondrial uncoupling proteins 2 and 3. J. Biol. Chem. 1999, 274, 26003–26007. [Google Scholar] [CrossRef]
- Jabůrek, M.; Miyamoto, S.; Di Mascio, P.; Garlid, K.D.; Ježek, P. Hydroperoxy fatty acid cycling mediated by mitochondrial uncoupling protein UCP2. J. Biol. Chem. 2004, 279, 53097–53102. [Google Scholar] [CrossRef]
- Bertholet, A.M.; Kazak, L.; Chouchani, E.T.; Bogaczyńska, M.G.; Paranjpe, I.; Wainwright, G.L.; Bétourné, A.; Kajimura, S.; Spiegelman, B.M.; Kirichok, Y. Mitochondrial Patch Clamp of Beige Adipocytes Reveals UCP1-Positive and UCP1-Negative Cells Both Exhibiting Futile Creatine Cycling. Cell Metab. 2017, 25, 811–822.e4. [Google Scholar] [CrossRef]
- Wojtczak, L.; Wiȩckowski, M.R. The mechanisms of fatty acid-induced proton permeability of the inner mitochondrial membrane. J. Bioenerg. Biomembr. 1999, 31, 447–455. [Google Scholar] [CrossRef]
- Wojtczak, L.; Wiȩckowski, M.R.; Schönfeld, P. Protonophoric activity of fatty acid analogs and derivatives in the inner mitochondrial membrane: A further argument for the fatty acid cycling model. Arch. Biochem. Biophys. 1998, 357, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Brustovetsky, N.; Klingenberg, M. The reconstituted ADP/ATP carrier can mediate H+ transport by free fatty acids, which is further stimulated by mersalyl. J. Biol. Chem. 1994, 269, 27329–27336. [Google Scholar] [CrossRef]
- Capaldi, R.A. Arrangement of proteins in the mitochondrial inner membrane. BBA Rev. Biomembr. 1982, 694, 291–306. [Google Scholar] [CrossRef]
- Elimam, H.; Papillon, J.; Kaufman, D.R.; Guillemette, J.; Aoudjit, L.; Gross, R.W.; Takano, T.; Cybulsky, A.V. Genetic ablation of calcium-independent phospholipase A2γ induces glomerular injury in mice. J. Biol. Chem. 2016, 291, 14468–14482. [Google Scholar] [CrossRef] [PubMed]
- Elimam, H.; Papillon, J.; Guillemette, J.; Navarro-Betancourt, J.R.; Cybulsky, A.V. Genetic Ablation of Calcium-independent Phospholipase A2γ Exacerbates Glomerular Injury in Adriamycin Nephrosis in Mice. Sci. Rep. 2019, 9, 16229. [Google Scholar] [CrossRef]
- Peterson, B.; Knotts, T.; Cummings, B.S. Involvement of Ca2+-independent phospholipase A2 isoforms in oxidant-induced neural cell death. Neurotoxicology 2007, 28, 150–160. [Google Scholar] [CrossRef]
- Rauckhorst, A.J.; Pfeiffer, D.R.; Broekemeier, K.M. The iPLA2γ is identified as the membrane potential sensitive phospholipase in liver mitochondria. FEBS Lett. 2015, 589, 2367–2371. [Google Scholar] [CrossRef] [PubMed]
- Garlid, K.D.; Jabůrek, M.; Ježek, P. Mechanism of uncoupling protein action. Biochem. Soc. Trans. 2001, 29, 803–806. [Google Scholar] [CrossRef]
- Skulachev, V.P. Membrane-linked systems preventing superoxide formation. Biosci. Rep. 1997, 17, 347–366. [Google Scholar] [CrossRef]
- Jastroch, M.; Divakaruni, A.S.; Mookerjee, S.; Treberg, J.R.; Brand, M.D. Mitochondrial proton and electron leaks. Essays Biochem. 2010, 47, 53–67. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, D.G. The Effective Proton Conductance of the Inner Membrane of Mitochondria from Brown Adipose Tissue: Dependency on Proton Electrochemical Potential Gradient. Eur. J. Biochem. 1977, 77, 349–356. [Google Scholar] [CrossRef]
- Ježek, P.; Žáčková, M.; Růžička, M.; Škobisová, E.; Jabůrek, M. Mitochondrial Uncoupling Proteins—Facts and Fantasies. Physiol. Res. 2004, 53, S199–S211. [Google Scholar]
- Garlid, K.D.; Beavis, A.D.; Ratkje, S.K. On the nature of ion leaks in energy-transducing membranes. BBA Bioenerg. 1989, 976, 109–120. [Google Scholar] [CrossRef]
- Garlid, K.D.; Orosz, D.E.; Modrianský, M.; Vassanelli, S.; Ježek, P. On the mechanism of fatty acid-induced proton transport by mitochondrial uncoupling protein. J. Biol. Chem. 1996, 271, 2615–2620. [Google Scholar] [CrossRef] [PubMed]
- Jabůrek, M.; Vařecha, M.; Ježek, P.; Garlid, K.D. Alkylsulfonates as probes of uncoupling protein transport mechanism. Ion pair transport demonstrates that direct H+ translocation by UCP1 is not necessary for uncoupling. J. Biol. Chem. 2001, 276, 31897–31905. [Google Scholar] [CrossRef] [PubMed]
- Brand, M.D. Mitochondrial generation of superoxide and hydrogen peroxide as the source of mitochondrial redox signaling. Free Radic. Biol. Med. 2016, 100, 14–31. [Google Scholar] [CrossRef]
- Korshunov, S.S.; Skulachev, V.P.; Starkov, A.A. High protonic potential actuates a mechanism of production of reactive oxygen species in mitochondria. FEBS Lett. 1997, 416, 15–18. [Google Scholar] [CrossRef]
- Vyssokikh, M.Y.; Holtze, S.; Averina, O.A.; Lyamzaev, K.G.; Panteleeva, A.A.; Marey, M.V.; Zinovkin, R.A.; Severin, F.F.; Skulachev, M.V.; Fasel, N.; et al. Mild depolarization of the inner mitochondrial membrane is a crucial component of an anti-aging program. Proc. Natl. Acad. Sci. USA 2020, 117, 6491–6501. [Google Scholar] [CrossRef]
- Komlódi, T.; Geibl, F.F.; Sassani, M.; Ambrus, A.; Tretter, L. Membrane potential and delta pH dependency of reverse electron transport-associated hydrogen peroxide production in brain and heart mitochondria. J. Bioenerg. Biomembr. 2018, 50, 355–365. [Google Scholar] [CrossRef] [PubMed]
- Rupprecht, A.; Sokolenko, E.A.; Beck, V.; Ninnemann, O.; Jabůrek, M.; Trimbuch, T.; Klishin, S.S.; Ježek, P.; Skulachev, V.P.; Pohl, E.E. Role of the transmembrane potential in the membrane proton leak. Biophys. J. 2010, 98, 1503–1511. [Google Scholar] [CrossRef] [PubMed]
- Rana, M.; De Coo, I.; Diaz, F.; Smeets, H.; Moraes, C.T. An out-of-frame cytochrome b gene deletion from a patient with parkinsonism is associated with impaired complex III assembly and an increase in free radical production. Ann. Neurol. 2000, 48, 774–781. [Google Scholar] [CrossRef]
- Borek, A.; Kuleta, P.; Ekiert, R.; Pietras, R.; Sarewicz, M.; Osyczka, A. Mitochondrial disease-related mutation G167P in cytochrome b of Rhodobacter capsulatus cytochrome bc1 (S151P in human) affects the equilibrium distribution of [2Fe-2S] cluster and generation of superoxide. J. Biol. Chem. 2015, 290, 23781–23792. [Google Scholar] [CrossRef]
- Yin, Z.; Burger, N.; Kula-Alwar, D.; Aksentijević, D.; Bridges, H.R.; Prag, H.A.; Grba, D.N.; Viscomi, C.; James, A.M.; Mottahedin, A.; et al. Structural basis for a complex I mutation that blocks pathological ROS production. Nat. Commun. 2021, 12, 707. [Google Scholar] [CrossRef] [PubMed]
- Kukat, A.; Dogan, S.A.; Edgar, D.; Mourier, A.; Jacoby, C.; Maiti, P.; Mauer, J.; Becker, C.; Senft, K.; Wibom, R.; et al. Loss of UCP2 Attenuates Mitochondrial Dysfunction without Altering ROS Production and Uncoupling Activity. PLoS Genet. 2014, 10, e1004385. [Google Scholar] [CrossRef]
- Andreyev, A.Y.; Bondareva, T.O.; Dedukhova, V.I.; Mokhova, E.N.; Skulachev, V.P.; Tsofina, L.M.; Volkov, N.I.; Vygodina, T.V. The ATP/ADP-antiporter is involved in the uncoupling effect of fatty acids on mitochondria. Eur. J. Biochem. 1989, 182, 585–592. [Google Scholar] [CrossRef] [PubMed]
- Engstová, H.; Žáčková, M.; Růžiča, M.; Meinhardt, A.; Hanuš, J.; Krämer, R.; Ježek, P. Natural and Azido Fatty Acids Inhibit Phosphate Transport and Activate Fatty Acid Anion Uniport Mediated by the Mitochondrial Phosphate Carrier. J. Biol. Chem. 2001, 276, 4683–4691. [Google Scholar] [CrossRef]
- Samartsev, V.N.; Smirnov, A.V.; Zeldi, I.P.; Markova, O.V.; Mokhova, E.N.; Skulachev, V.P. Involvement of aspartate/glutamate antiporter in fatty acid-induced uncoupling of liver mitochondria. Biochim. Biophys. Acta Bioenerg. 1997, 1319, 251–257. [Google Scholar] [CrossRef]
- Samartsev, V.N.; Mokhova, E.N.; Skulachev, V.P. The pH-dependent reciprocal changes in contributions of ADP/ATP antiporter and aspartate/glutamate antiporter to the fatty acid-induced uncoupling. FEBS Lett. 1997, 412, 179–182. [Google Scholar] [CrossRef]
- Khailova, L.S.; Prikhodko, E.A.; Dedukhova, V.I.; Mokhova, E.N.; Popov, V.N.; Skulachev, V.P. Participation of ATP/ADP antiporter in oleate- and oleate hydroperoxide-induced uncoupling suppressed by GDP and carboxyatractylate. Biochim. Biophys. Acta Bioenerg. 2006, 1757, 1324–1329. [Google Scholar] [CrossRef]
- Korshunov, S.S.; Korkina, O.V.; Ruuge, E.K.; Skulachev, V.P.; Starkov, A.A. Fatty acids as natural uncouplers preventing generation of O(·-)2 and H2O2 by mitochondria in the resting state. FEBS Lett. 1998, 435, 215–218. [Google Scholar] [CrossRef]
- Shabalina, I.G.; Kramarova, T.V.; Nedergaard, J.; Cannon, B. Carboxyatractyloside effects on brown-fat mitochondria imply that the adenine nucleotide translocator isoforms ANT1 and ANT2 may be responsible for basal and fatty-acid-induced uncoupling respectively. Biochem. J. 2006, 399, 405–414. [Google Scholar] [CrossRef]
- Kreiter, J.; Rupprecht, A.; Škulj, S.; Brkljača, Z.; Žuna, K.; Knyazev, D.G.; Bardakji, S.; Vazdar, M.; Pohl, E.E. Ant1 activation and inhibition patterns support the fatty acid cycling mechanism for proton transport. Int. J. Mol. Sci. 2021, 22, 2490. [Google Scholar] [CrossRef] [PubMed]
- Nordmann, C.; Strokin, M.; Schönfeld, P.; Reiser, G. Putative roles of Ca2+-independent phospholipase A2 in respiratory chain-associated ROS production in brain mitochondria: Infuence of docosahexaenoic acid and bromoenol lactone. J. Neurochem. 2014, 131, 163–176. [Google Scholar] [CrossRef]
- Průchová, P.; Leguina-Ruzzi, A.; Galkin, A.; Ježek, P.; Jabůrek, M. Antioxidant activity of calcium-independent phospholipase A2γ in brain mitochondria. Free Radic. Biol. Med. 2018, 128, S86. [Google Scholar] [CrossRef]
- Winterbourn, C.C. Are free radicals involved in thiol-based redox signaling? Free Radic. Biol. Med. 2015, 80, 164–170. [Google Scholar] [CrossRef] [PubMed]
- Requejo, R.; Hurd, T.R.; Costa, N.J.; Murphy, M.P. Cysteine residues exposed on protein surfaces are the dominant intramitochondrial thiol and may protect against oxidative damage. FEBS J. 2010, 277, 1465–1480. [Google Scholar] [CrossRef]
- Nietzel, T.; Mostertz, J.; Hochgräfe, F.; Schwarzländer, M. Redox regulation of mitochondrial proteins and proteomes by cysteine thiol switches. Mitochondrion 2017, 33, 72–83. [Google Scholar] [CrossRef] [PubMed]
- Poole, L.B. The basics of thiols and cysteines in redox biology and chemistry. Free Radic. Biol. Med. 2015, 80, 148–157. [Google Scholar] [CrossRef]
- Paulsen, C.E.; Carroll, K.S. Cysteine-mediated redox signaling: Chemistry, biology, and tools for discovery. Chem. Rev. 2013, 113, 4633–4679. [Google Scholar] [CrossRef]
- Poole, L.B.; Furdui, C.M.; King, S.B. Introduction to approaches and tools for the evaluation of protein cysteine oxidation. Essays Biochem. 2020, 64, 1–17. [Google Scholar] [CrossRef]
- Rhee, S.G.; Woo, H.A.; Kang, D. The Role of Peroxiredoxins in the Transduction of H2O2 Signals. Antioxidants Redox Signal. 2018, 28, 537–557. [Google Scholar] [CrossRef] [PubMed]
- Codreanu, S.G.; Liebler, D.C. Novel approaches to identify protein adducts produced by lipid peroxidation. Free Radic. Res. 2015, 49, 881–887. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Higdon, A.; Diers, A.R.; Oh, J.Y.; Landar, A.; Darley-Usmar, V.M. Cell signalling by reactive lipid species: New concepts and molecular mechanisms. Biochem. J. 2012, 442, 453–464. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Jedrychowski, M.P.; Schweppe, D.K.; Huttlin, E.L.; Yu, Q.; Heppner, D.E.; Li, J.; Long, J.; Mills, E.L.; Szpyt, J.; et al. A Quantitative Tissue-Specific Landscape of Protein Redox Regulation during Aging. Cell 2020, 180, 968–983.e24. [Google Scholar] [CrossRef] [PubMed]
- Elimam, H.; Papillon, J.; Takano, T.; Cybulsky, A.V. Complement-mediated activation of calcium-independent phospholipase A 2γ: Role of protein kinases and phosphorylation. J. Biol. Chem. 2013, 288, 3871–3885. [Google Scholar] [CrossRef]
- Sadžak, A.; Mravljak, J.; Maltar-Strmečki, N.; Arsov, Z.; Baranović, G.; Erceg, I.; Kriechbaum, M.; Strasser, V.; Přibyl, J.; Šegota, S. The structural integrity of the model lipid membrane during induced lipid peroxidation: The role of flavonols in the inhibition of lipid peroxidation. Antioxidants 2020, 9, 430. [Google Scholar] [CrossRef]
- Cozza, G.; Rossetto, M.; Bosello-Travain, V.; Maiorino, M.; Roveri, A.; Toppo, S.; Zaccarin, M.; Zennaro, L.; Ursini, F. Glutathione peroxidase 4-catalyzed reduction of lipid hydroperoxides in membranes: The polar head of membrane phospholipids binds the enzyme and addresses the fatty acid hydroperoxide group toward the redox center. Free Radic. Biol. Med. 2017, 112, 1–11. [Google Scholar] [CrossRef]
- Tsubone, T.M.; Junqueira, H.C.; Baptista, M.S.; Itri, R. Contrasting roles of oxidized lipids in modulating membrane microdomains. Biochim. Biophys. Acta Biomembr. 2019, 1861, 660–669. [Google Scholar] [CrossRef]
- Sevanian, A.; Wratten, M.; McLeod, L.L.; Kim, E. Lipid peroxidation and phospholipase A2 activity in liposomes composed of unsaturated phospholipids: A structural basis for enzyme activation. Biochim. Biophys. Acta 1988, 961, 316–327. [Google Scholar] [CrossRef]
- McLean, L.R.; Hagaman, K.A.; Davidson, W.S. Role of lipid structure in the activation of phospholipase A2 by peroxidized phospholipids. Lipids 1993, 28, 505–509. [Google Scholar] [CrossRef] [PubMed]
- Niki, E.; Yoshida, Y.; Saito, Y.; Noguchi, N. Lipid peroxidation: Mechanisms, inhibition, and biological effects. Biochem. Biophys. Res. Commun. 2005, 338, 668–676. [Google Scholar] [CrossRef] [PubMed]
- Pennington, E.R.; Funai, K.; Brown, D.A.; Shaikh, S.R. The role of cardiolipin concentration and acyl chain composition on mitochondrial inner membrane molecular organization and function. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2019, 1864, 1039–1052. [Google Scholar] [CrossRef] [PubMed]
- Tatsuta, T.; Langer, T. Intramitochondrial phospholipid trafficking. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2017, 1862, 81–89. [Google Scholar] [CrossRef]
- Kiebish, M.A.; Yang, K.; Liu, X.; Mancuso, D.J.; Guan, S.; Zhao, Z.; Sims, H.F.; Cerqua, R.; Cade, W.T.; Han, X.; et al. Dysfunctional cardiac mitochondrial bioenergetic, lipidomic, and signaling in a murine model of Barth syndrome. J. Lipid Res. 2013, 54, 1312–1325. [Google Scholar] [CrossRef] [PubMed]
- Casares, D.; Escribá, P.V.; Rosselló, C.A. Membrane lipid composition: Effect on membrane and organelle structure, function and compartmentalization and therapeutic avenues. Int. J. Mol. Sci. 2019, 20, 2167. [Google Scholar] [CrossRef]
- Valianpour, F.; Wanders, R.J.A.; Barth, P.G.; Overmars, H.; Van Gennip, A.H. Quantitative and compositional study of cardiolipin in platelets by electrospray ionization mass spectrometry: Application for the identification of Barth syndrome patients. Clin. Chem. 2002, 48, 1390–1397. [Google Scholar] [CrossRef] [PubMed]
- Kiebish, M.A.; Han, X.; Cheng, H.; Chuang, J.H.; Seyfried, T.N. Cardiolipin and electron transport chain abnormalities in mouse brain tumor mitochondria: Lipidomic evidence supporting the Warburg theory of cancer. J. Lipid Res. 2008, 49, 2545–2556. [Google Scholar] [CrossRef] [PubMed]
- Tyurina, Y.Y.; Shrivastava, I.; Tyurin, V.A.; Mao, G.; Dar, H.H.; Watkins, S.; Epperly, M.; Bahar, I.; Shvedova, A.A.; Pitt, B.; et al. Only a Life Lived for Others Is Worth Living: Redox Signaling by Oxygenated Phospholipids in Cell Fate Decisions. Antioxidants Redox Signal. 2018, 29, 1333–1358. [Google Scholar] [CrossRef]
- Ikon, N.; Ryan, R.O. Cardiolipin and mitochondrial cristae organization. Biochim. Biophys. Acta Biomembr. 2017, 1859, 1156–1163. [Google Scholar] [CrossRef]
- Ban, T.; Heymann, J.A.W.; Song, Z.; Hinshaw, J.E.; Chan, D.C. OPA1 disease alleles causing dominant optic atrophy have defects in cardiolipin-stimulated GTP hydrolysis and membrane tubulation. Hum. Mol. Genet. 2010, 19, 2113–2122. [Google Scholar] [CrossRef] [PubMed]
- Jussupow, A.; Di Luca, A.; Kaila, V.R.I. How cardiolipin modulates the dynamics of respiratory complex I. Sci. Adv. 2019, 5, eaav1850. [Google Scholar] [CrossRef] [PubMed]
- Malkamäki, A.; Sharma, V. Atomistic insights into cardiolipin binding sites of cytochrome c oxidase. Biochim. Biophys. Acta Bioenerg. 2019, 1860, 224–232. [Google Scholar] [CrossRef] [PubMed]
- Duncan, A.L.; Ruprecht, J.J.; Kunji, E.R.S.; Robinson, A.J. Cardiolipin dynamics and binding to conserved residues in the mitochondrial ADP/ATP carrier. Biochim. Biophys. Acta Biomembr. 2018, 1860, 1035–1045. [Google Scholar] [CrossRef]
- Gasanov, S.E.; Kim, A.A.; Yaguzhinsky, L.S.; Dagda, R.K. Non-bilayer structures in mitochondrial membranes regulate ATP synthase activity. Biochim. Biophys. Acta Biomembr. 2018, 1860, 586–599. [Google Scholar] [CrossRef]
- Khosravi, S.; Harner, M.E. The MICOS complex, a structural element of mitochondria with versatile functions. Biol. Chem. 2020, 401, 765–778. [Google Scholar] [CrossRef]
- Kozjak-Pavlovic, V. The MICOS complex of human mitochondria. Cell Tissue Res. 2017, 367, 83–93. [Google Scholar] [CrossRef]
- Hsu, Y.H.; Dumlao, D.S.; Cao, J.; Dennis, E.A. Assessing Phospholipase A2 Activity toward Cardiolipin by Mass Spectrometry. PLoS ONE 2013, 8, e59267. [Google Scholar] [CrossRef]
- Gonzalez-Baro, M.R.; Coleman, R.A. Mitochondrial acyltransferases and glycerophospholipid metabolism. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2017, 1862, 49–55. [Google Scholar] [CrossRef]
- Cipolat, S.; Rudka, T.; Hartmann, D.; Costa, V.; Serneels, L.; Craessaerts, K.; Metzger, K.; Frezza, C.; Annaert, W.; D’Adamio, L.; et al. Mitochondrial Rhomboid PARL Regulates Cytochrome c Release during Apoptosis via OPA1-Dependent Cristae Remodeling. Cell 2006, 126, 163–175. [Google Scholar] [CrossRef]
- Lopaschuk, G.D.; Ussher, J.R.; Folmes, C.D.L.; Jaswal, J.S.; Stanley, W.C. Myocardial fatty acid metabolism in health and disease. Physiol. Rev. 2010, 90, 207–258. [Google Scholar] [CrossRef]
- Ford, D.A.; Hazen, S.L.; Saffitz, J.E.; Gross, R.W. The rapid and reversible activation of a calcium-independent plasmalogen-selective phospholipase A2 during myocardial ischemia. J. Clin. Investig. 1991, 88, 331–335. [Google Scholar] [CrossRef]
- Williams, S.D.; Gottlieb, R.A. Inhibition of mitochondrial calcium-independent phospholipase A2 (iPLA2) attenuates mitochondrial phospholipid loss and is cardioprotective. Biochem. J. 2002, 362, 23–32. [Google Scholar] [CrossRef]
- Moon, S.H.; Mancuso, D.J.; Sims, H.F.; Liu, X.; Nguyen, A.L.; Yang, K.; Guan, S.; Dilthey, B.G.; Jenkins, C.M.; Weinheimer, C.J.; et al. Cardiac myocyte-specific knock-out of calcium-independent phospholipase A2γ (iPLA2γ) decreases oxidized fatty acids during ischemia/reperfusion and reduces infarct size. J. Biol. Chem. 2016, 291, 19687–19700. [Google Scholar] [CrossRef]
- Garlid, A.O.; Jabůrek, M.; Jacobs, J.P.; Garlid, K.D. Mitochondrial reactive oxygen species: Which ROS signals cardioprotection? AJP Hear. Circ. Physiol. 2013, 305, H960–H968. [Google Scholar] [CrossRef] [PubMed]
- Wolf, M.J.; Izumi, Y.; Zorumski, C.F.; Gross, R.W. Long-term potentiation requires activation of calcium-independent phospholipase A2. FEBS Lett. 1995, 377, 358–362. [Google Scholar] [CrossRef] [PubMed]
- Adibhatla, R.M.; Hatcher, J.F. Phospholipase A2, reactive oxygen species, and lipid peroxidation in cerebral ischemia. Free Radic. Biol. Med. 2006, 40, 376–387. [Google Scholar] [CrossRef]
- Adibhatla, R.M.; Hatcher, J.F. Phospholipase A2, reactive oxygen species, and lipid peroxidation in CNS pathologies. J. Biochem. Mol. Biol. 2008, 41, 560–567. [Google Scholar] [CrossRef] [PubMed]
- Mancuso, D.J.; Kotzbauer, P.; Wozniak, D.F.; Sims, H.F.; Jenkins, C.M.; Guan, S.; Han, X.; Yang, K.; Sun, G.; Malik, I.; et al. Genetic ablation of calcium-independent phospholipase A2γ leads to alterations in hippocampal cardiolipin content and molecular species distribution, mitochondrial degeneration, autophagy, and cognitive dysfunction. J. Biol. Chem. 2009, 284, 35632–35644. [Google Scholar] [CrossRef] [PubMed]
- Chao, H.; Liu, Y.; Fu, X.; Xu, X.; Bao, Z.; Lin, C.; Li, Z.; Liu, Y.; Wang, X.; You, Y.; et al. Lowered iPLA2γ activity causes increased mitochondrial lipid peroxidation and mitochondrial dysfunction in a rotenone-induced model of Parkinson’s disease. Exp. Neurol. 2018, 300, 74–86. [Google Scholar] [CrossRef] [PubMed]
- Chao, H.; Anthonymuthu, T.S.; Kenny, E.M.; Amoscato, A.A.; Cole, L.K.; Hatch, G.M.; Ji, J.; Kagan, V.E.; Bayır, H. Disentangling oxidation/hydrolysis reactions of brain mitochondrial cardiolipins in pathogenesis of traumatic injury. JCI Insight 2018, 3, e97677. [Google Scholar] [CrossRef]
- Ježek, P.; Jabůrek, M.; Porter, R.K. Uncoupling mechanism and redox regulation of mitochondrial uncoupling protein 1 (UCP1). Elsevier B.V. 2019, 1860, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Mancuso, D.J.; Sims, H.F.; Han, X.; Jenkins, C.M.; Shao, P.G.; Yang, K.; Sung, H.M.; Pietka, T.; Abumrad, N.A.; Schlesinger, P.H.; et al. Genetic ablation of calcium-independent phospholipase A2γ leads to alterations in mitochondrial lipid metabolism and function resulting in a deficient mitochondrial bioenergetic phenotype. J. Biol. Chem. 2007, 282, 34611–34622. [Google Scholar] [CrossRef] [PubMed]
- Schweizer, S.; Liebisch, G.; Oeckl, J.; Hoering, M.; Seeliger, C.; Schiebel, C.; Klingenspor, M.; Ecker, J. The lipidome of primary murine white, brite, and brown adipocytes—Impact of betaadrenergic stimulation. PLoS Biol. 2019, 17, e3000412. [Google Scholar] [CrossRef] [PubMed]
- Chouchani, E.T.; Kazak, L.; Jedrychowski, M.P.; Lu, G.Z.; Erickson, B.K.; Szpyt, J.; Pierce, K.A.; Laznik-Bogoslavski, D.; Vetrivelan, R.; Clish, C.B.; et al. Mitochondrial ROS regulate thermogenic energy expenditure and sulfenylation of UCP1. Nature 2016, 532, 112–116. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phospholipase A2 | Localization | Acts on Membrane | Targets | Cleavage Products | Main Functions | ||
|---|---|---|---|---|---|---|---|
| Group | Gene Name | Protein | |||||
| VIE | PNPLA2 | iPLA2ζ/ATGL | Adipocyte: Cytosolic | Plasma membrane OMM outer leaflet | Neutral lipid lipase: triacylglycerols | FAs + diacylglycerols | Nutrition regulation, Hydrolysis of triglycerides |
| VID | PNPLA3 | iPLA2ε | Hepatocyte: Cytosolic | Plasma membrane OMM outer leaflet | Neutral lipid lipase: triacylglycerols | FAs + diacylglycerols | Phosphatidic acid generation, Acyl-chain remodeling of triglycerides |
| VIF | PNPLA4 | iPLA2η | Ubiquitous: Cytosolic | Plasma membrane OMM outer leaflet | sn-1, sn-2 PL, LysoPL, triacylglycerols | FAs + LysoPL, FAs + diacylglycerols | Triacylglycerol lipase activity, Cytoprotection |
| VIC | PNPLA6, PNPLA7 | iPLA2δ, iPLA2θ | Neurons: Cytosolic | Plasma membrane OMM outer leaflet | ER, Golgi sn-1, sn-2 PL, LysoPL | FAs + LysoPL | Neuroprotection, Intracellular membrane trafficking |
| VIB | PNPLA8 | iPLA2γ | Ubiquitous: Mitochondrial Peroxisomal | IMM (outer or inner leaflet) OMM inner leaflet Peroxisome | sn-1, sn-2 PL, cardiolipin, hydroperoxy-PL, hydroperoxy-CL | Saturated or unsaturated FAs, PUFAs (or hydroperoxy) + PUFAlysPL or LysoPL | Antioxidant, Lipid second messengers, Eicosanoid signaling, Cardiolipin remodeling |
| VIA | PNPLA9 | iPLA2β | Ubiquitous: Mitochondrial Cytosolic | OMM (outer or inner leaflet) IMM (outer or inner leaflet) | sn-1, sn-2 PL, cardiolipin, hydroperoxy-PL, hydroperoxy-CL | Saturated or unsaturated FAs, PUFAs (or hydroperoxy) + PUFAlysPL or LysoPL | Cellular membrane homeostasis, Mitochondrial integrity, Signal transduction |
| IV | PLA2G4 | cPLA2s | Cytosolic | Plasma membrane inner leaflet; OMM outer leaflet | sn-2 PL | FAs, PUFAs | Membrane lipid remodeling, Biosynthesis of lipid mediators, inflammatory response |
| IVF | PLA2G4F | cPLA2ζ | Cytosolic, Mitochondrial | OMM (outer or inner leaflet) IMM (outer or inner leaflet) | sn-2 PL | FAs, PUFAs | Biosynthesis of lipid mediators, Arachidonate release, Cardioprotective eicosanoids |
| I, II, III V, X, XII | PLA2G1, PLA2G2, PLA2G3, PLA2G5, PLA2G10, PLA2G12 | sPLA2 | Extracellular matrix | Plasma membrane outer leaflet | sn-2 PL | FAs, PUFAs | Extracellular matrix remodeling, Lipid mediator secretion, Digestion, Immunity |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jabůrek, M.; Průchová, P.; Holendová, B.; Galkin, A.; Ježek, P. Antioxidant Synergy of Mitochondrial Phospholipase PNPLA8/iPLA2γ with Fatty Acid–Conducting SLC25 Gene Family Transporters. Antioxidants 2021, 10, 678. https://doi.org/10.3390/antiox10050678
Jabůrek M, Průchová P, Holendová B, Galkin A, Ježek P. Antioxidant Synergy of Mitochondrial Phospholipase PNPLA8/iPLA2γ with Fatty Acid–Conducting SLC25 Gene Family Transporters. Antioxidants. 2021; 10(5):678. https://doi.org/10.3390/antiox10050678
Chicago/Turabian StyleJabůrek, Martin, Pavla Průchová, Blanka Holendová, Alexander Galkin, and Petr Ježek. 2021. "Antioxidant Synergy of Mitochondrial Phospholipase PNPLA8/iPLA2γ with Fatty Acid–Conducting SLC25 Gene Family Transporters" Antioxidants 10, no. 5: 678. https://doi.org/10.3390/antiox10050678
APA StyleJabůrek, M., Průchová, P., Holendová, B., Galkin, A., & Ježek, P. (2021). Antioxidant Synergy of Mitochondrial Phospholipase PNPLA8/iPLA2γ with Fatty Acid–Conducting SLC25 Gene Family Transporters. Antioxidants, 10(5), 678. https://doi.org/10.3390/antiox10050678