
Mitochondrial Dysfunction and Endoplasmic Reticulum Stress in Age Related Macular Degeneration, Role in Pathophysiology, and Possible New Therapeutic Strategies

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Mitochondria and AMD
2.1. Retinal Pigment Epithelium and Mitochondria
2.2. Mitochondria and Retinal Senescence
2.3. Mitochondrial Dysfunction and AMD
2.4. Mitochondrial DNA Damage in AMD
3. Endoplasmic Reticulum and AMD
3.1. The Endoplasmic Reticulum Normal Functioning: Protein Folding
3.2. ER-Stress and Adaptive Mechanisms
3.2.1. ERAD
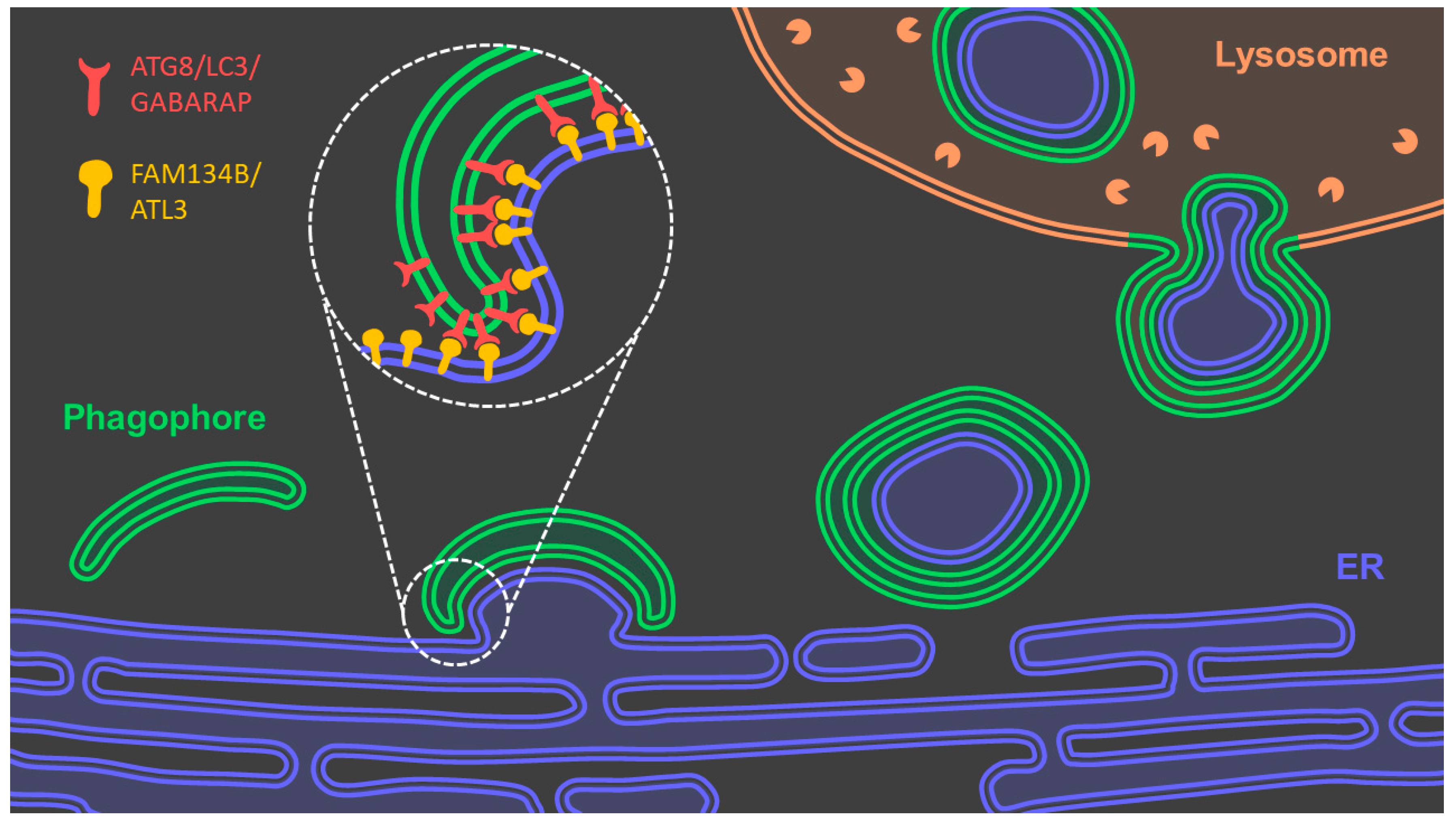
3.2.2. Reticulophagy
3.2.3. Unfolded Protein Response
3.3. ER-Stress and Aging
3.4. ER-Stress and AMD
3.5. ER-Stress and Mitochondrial Dysfunction in AMD
4. New Therapeutics for AMD
4.1. Bile Acids
4.2. Humanin
4.3. Resveratrol
4.4. Coenzyme Q10
4.5. Melatonin
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Mitchell, P.; Liew, G.; Gopinath, B.; Wong, T.Y. Age-related macular degeneration. Lancet 2018, 392, 1147–1159. [Google Scholar] [CrossRef]
- Chakravarthy, U.; Peto, T. Current Perspective on Age-Related Macular Degeneration. JAMA 2020, 324, 794. [Google Scholar] [CrossRef]
- Garcia-Layana, A.; Cabrera-López, F.; García-Arumí, J.; Arias-Barquet, L.; Ruiz-Moreno, J.M. Early and intermediate age-related macular degeneration: Update and clinical review. Clin. Interv. Aging 2017, 12, 1579–1587. [Google Scholar] [CrossRef] [Green Version]
- Li, J.Q.; Welchowski, T.; Schmid, M.; Mauschitz, M.M.; Holz, F.G.; Finger, R.P. Prevalence and incidence of age-related macular degeneration in Europe: A systematic review and meta-analysis. Br. J. Ophthalmol. 2019, 104, 1077–1084. [Google Scholar] [CrossRef]
- Sparrow, J.R.; Hicks, D.; Hamel, C.P. The retinal pigment epithelium in health and disease. Curr. Mol. Med. 2010, 10, 802–823. [Google Scholar] [CrossRef]
- Strauss, O. The Retinal Pigment Epithelium in Visual Function. Physiol. Rev. 2005, 85, 845–881. [Google Scholar] [CrossRef] [Green Version]
- Travis, G.H.; Golczak, M.; Moise, A.; Palczewski, K. Diseases Caused by Defects in the Visual Cycle: Retinoids as Potential Therapeutic Agents. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 469–512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez-Robredo, P.; González-Zamora, J.; Recalde, S.; Bilbao-Malavé, V.; Bezunartea, J.; Hernandez, M.; Garcia-Layana, A. Vitamin D Protects against Oxidative Stress and Inflammation in Human Retinal Cells. Antioxidants 2020, 9, 838. [Google Scholar] [CrossRef]
- Ebeling, M.C.; Polanco, J.R.; Qu, J.; Tu, C.; Montezuma, S.R.; Ferrington, D.A. Improving retinal mitochondrial function as a treatment for age-related macular degeneration. Redox Biol. 2020, 34, 101552. [Google Scholar] [CrossRef]
- Lutein, A.R. Zeaxanthin and Omega-3 Fatty Acids for Age-Related Macular Degeneration. JAMA 2013, 309, 2005–2015. [Google Scholar] [CrossRef]
- Miller, J.W.; Bagheri, S.; Demetrios, G.; Vavvas, D.G.; Arniranta, K. Advances in age related macular degeneration understanding and therapy. US Ophthalmic Rev. 2017, 10, 119–130. [Google Scholar] [CrossRef] [Green Version]
- Brown, E.; Lewin, A.; Ash, J.D. Mitochondria: Potential Targets for Protection in Age-Related Macular Degeneration. In Advances in Experimental Medicine and Biology; Springer Science and Business Media LLC: Berlin, Germany, 2018; Volume 1074, pp. 11–17. [Google Scholar]
- Ferrington, D.A.; Fisher, C.R.; Kowluru, R.A. Mitochondrial Defects Drive Degenerative Retinal Diseases. Trends Mol. Med. 2020, 26, 105–118. [Google Scholar] [CrossRef]
- Chapman, J.; Fielder, E.; Passos, J.F. Mitochondrial dysfunction and cell senescence: Deciphering a complex relationship. FEBS Lett. 2019, 593, 1566–1579. [Google Scholar] [CrossRef] [Green Version]
- Kaarniranta, K.; Uusitalo, H.; Blasiak, J.; Felszeghy, S.; Kannan, R.; Kauppinen, A.; Salminen, A.; Sinha, D.; Ferrington, D. Mechanisms of mitochondrial dysfunction and their impact on age-related macular degeneration. Prog. Retin. Eye Res. 2020, 79, 100858. [Google Scholar] [CrossRef]
- Lee, K.S.; Lin, S.; Copland, D.A.; Dick, A.D.; Liu, J. Cellular senescence in the aging retina and developments of senotherapies for age-related macular degeneration. J. Neuroinflamm. 2021, 18, 1–17. [Google Scholar] [CrossRef]
- Blasiak, J. Senescence in the pathogenesis of age-related macular degeneration. Cell. Mol. Life Sci. 2020, 77, 789–805. [Google Scholar] [CrossRef]
- DeAngelis, M.M.; Owen, L.; Morrison, M.A.; Morgan, D.J.; Li, M.; Shakoor, A.; Vitale, A.; Iyengar, S.; Stambolian, D.; Kim, I.; et al. Genetics of age-related macular degeneration (AMD). Hum. Mol. Genet. 2017, 26, 45–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marazita, M.C.; Dugour, A.; Ramella, M.D.M.; Figueroa, J.M.; Suburo, A.M. Oxidative stress-induced premature senescence dysregulates VEGF and CFH expression in retinal pigment epithelial cells: Implications for Age-related Macular Degeneration. Redox Biol. 2016, 7, 78–87. [Google Scholar] [CrossRef] [Green Version]
- Somasundaran, S.; Constable, I.J.; Mellough, C.B.; Carvalho, L.S. Retinal pigment epithelium and age-related macular degeneration: A review of major disease mechanisms. Clin. Exp. Ophthalmol. 2020, 48, 1043–1056. [Google Scholar] [CrossRef] [PubMed]
- Brown, E.; DeWeerd, A.J.; Ildefonso, C.J.; Lewin, A.; Ash, J.D. Mitochondrial oxidative stress in the retinal pigment epithelium (RPE) led to metabolic dysfunction in both the RPE and retinal photoreceptors. Redox Biol. 2019, 24, 101201. [Google Scholar] [CrossRef]
- Nita, M.; Grzybowski, A. Interplay between reactive oxygen species and autophagy in the course of age-related macular degeneration. EXCLI J. 2020, 19, 1353–1371. [Google Scholar] [PubMed]
- Kanow, M.A.; Giarmarco, M.M.; Jankowski, C.; Tsantilas, K.; Engel, A.L.; Du, J.; Linton, J.D.; Farnsworth, C.C.; Sloat, S.R.; Rountree, A.; et al. Biochemical adaptations of the retina and retinal pigment epithelium support a metabolic ecosystem in the vertebrate eye. eLife 2017, 6, e28899. [Google Scholar] [CrossRef] [PubMed]
- Karunadharma, P.P.; Nordgaard, C.L.; Olsen, T.W.; Ferrington, D. Mitochondrial DNA Damage as a Potential Mechanism for Age-Related Macular Degeneration. Investig. Opthalmol. Vis. Sci. 2010, 51, 5470–5479. [Google Scholar] [CrossRef] [Green Version]
- Mueller, E.E.; Schaier, E.; Brunner, S.M.; Eder, W.; Mayr, J.A.; Egger, S.F.; Nischler, C.; Oberkofler, H.; Reitsamer, H.A.; Patsch, W.; et al. Mitochondrial Haplogroups and Control Region Polymorphisms in Age-Related Macular Degeneration: A Case-Control Study. PLoS ONE 2012, 7, e30874. [Google Scholar] [CrossRef] [Green Version]
- Ferrington, D.A.; Kapphahn, R.J.; Leary, M.M.; Atilano, S.; Terluk, M.R.; Karunadharma, P.; Chen, G.K.-J.; Ratnapriya, R.; Swaroop, A.; Montezuma, S.R.; et al. Increased retinal mtDNA damage in the CFH variant associated with age-related macular degeneration. Exp. Eye Res. 2016, 145, 269–277. [Google Scholar] [CrossRef] [Green Version]
- Kanda, A.; Chen, W.; Othman, M.; Branham, K.; Brooks, M.; Khanna, R.; He, S.; Lyons, R.; Abecasis, G.R.; Swaroop, A. A variant of mitochondrial protein LOC387715/ARMS2, not HTRA1, is strongly associated with age-related macular degeneration. Proc. Natl. Acad. Sci. USA 2007, 104, 16227–16232. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, D.S.; Blower, M.D. The endoplasmic reticulum: Structure, function and response to cellular signaling. Cell. Mol. Life Sci. 2016, 73, 79–94. [Google Scholar] [CrossRef] [Green Version]
- Hetz, C.; Zhang, K.; Kaufman, R.J. Mechanisms, regulation and functions of the unfolded protein response. Nat. Rev. Mol. Cell Biol. 2020, 21, 421–438. [Google Scholar] [CrossRef]
- Ren, J.; Bi, Y.; Sowers, J.R.; Hetz, C.; Zhang, Y. Endoplasmic reticulum stress and unfolded protein response in cardiovascular diseases. Nat. Rev. Cardiol. 2021, 18, 499–521. [Google Scholar] [CrossRef]
- Hetz, C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89–102. [Google Scholar] [CrossRef]
- Adams, C.J.; Kopp, M.C.; Larburu, N.; Nowak, P.R.; Ali, M.M.U. Structure and Molecular Mechanism of ER Stress Signaling by the Unfolded Protein Response Signal Activator IRE1. Front. Mol. Biosci. 2019, 6, 11. [Google Scholar] [CrossRef] [Green Version]
- Patel, C.; Saad, H.; Shenkman, M.; Lederkremer, G.Z. Oxidoreductases in Glycoprotein Glycosylation, Folding, and ERAD. Cells 2020, 9, 2138. [Google Scholar] [CrossRef]
- Christis, C.; Lubsen, N.H.; Braakman, I. Protein folding includes oligomerization—Examples from the endoplasmic reticulum and cytosol. FEBS J. 2008, 275, 4700–4727. [Google Scholar] [CrossRef]
- Surgucheva, I.; Ninkina, N.; Buchman, V.; Grasing, K.; Surguchov, A. Protein Aggregation in Retinal Cells and Approaches to Cell Protection. Cell. Mol. Neurobiol. 2005, 25, 1051–1066. [Google Scholar] [CrossRef]
- Li, C.; Xia, B.; Wang, S.; Xu, J. Folded or Degraded in Endoplasmic Reticulum. In Advances in Experimental Medicine and Biology; Springer Science and Business Media LLC: Berlin, Germany, 2020; Volume 1248, pp. 265–294. [Google Scholar]
- Vembar, S.S.; Brodsky, J.L. One step at a time: Endoplasmic reticulum-associated degradation. Nat. Rev. Mol. Cell Biol. 2008, 9, 944–957. [Google Scholar] [CrossRef]
- Chino, H.; Mizushima, N. ER-Phagy: Quality Control and Turnover of Endoplasmic Reticulum. Trends Cell Biol. 2020, 30, 384–398. [Google Scholar] [CrossRef]
- Xie, W.; Ng, D.T. ERAD substrate recognition in budding yeast. Semin. Cell Dev. Biol. 2010, 21, 533–539. [Google Scholar] [CrossRef]
- Wu, X.; Rapoport, T.A. Mechanistic insights into ER-associated protein degradation. Curr. Opin. Cell Biol. 2018, 53, 22–28. [Google Scholar] [CrossRef]
- Christianson, J.; Ye, Y. Cleaning up in the endoplasmic reticulum: Ubiquitin in charge. Nat. Struct. Mol. Biol. 2014, 21, 325–335. [Google Scholar] [CrossRef]
- Carvalho, P.; Stanley, A.M.; Rapoport, T.A. Retrotranslocation of a Misfolded Luminal ER Protein by the Ubiquitin-Ligase Hrd1p. Cell 2010, 143, 579–591. [Google Scholar] [CrossRef] [Green Version]
- Han, G.; Casson, R.J.; Chidlow, G.; Wood, J.P.M. The Mitochondrial Complex I Inhibitor Rotenone Induces Endoplasmic Reticulum Stress and Activation of GSK-3β in Cultured Rat Retinal Cells. Investig. Opthalmol. Vis. Sci. 2014, 55, 5616–5628. [Google Scholar] [CrossRef] [Green Version]
- Huyer, G.; Piluek, W.F.; Fansler, Z.; Kreft, S.G.; Hochstrasser, M.; Brodsky, J.L.; Michaelis, S. Distinct Machinery Is Required in Saccharomyces cerevisiae for the Endoplasmic Reticulum-associated Degradation of a Multispanning Membrane Protein and a Soluble Luminal Protein. J. Biol. Chem. 2004, 279, 38369–38378. [Google Scholar] [CrossRef] [Green Version]
- Bodnar, N.; Rapoport, T.A. Molecular Mechanism of Substrate Processing by the Cdc48 ATPase Complex. Cell 2017, 169, 722–735. [Google Scholar] [CrossRef] [Green Version]
- Vats, S.; Galli, T. Introducing secretory reticulophagy/ER-phagy (SERP), a VAMP7-dependent pathway involved in neurite growth. Autophagy 2021, 17, 1037–1039. [Google Scholar] [CrossRef]
- Read, A.; Schröder, M. The Unfolded Protein Response: An Overview. Biology 2021, 10, 384. [Google Scholar] [CrossRef] [PubMed]
- Grootjans, J.; Kaser, A.; Kaufman, R.J.; Blumberg, R.S. The unfolded protein response in immunity and inflammation. Nat. Rev. Immunol. 2016, 16, 469–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gardner, B.M.; Walter, P. Unfolded Proteins Are Ire1-Activating Ligands That Directly Induce the Unfolded Protein Response. Science 2011, 333, 1891–1894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hetz, C.; Saxena, S. ER stress and the unfolded protein response in neurodegeneration. Nat. Rev. Neurol. 2017, 13, 477–491. [Google Scholar] [CrossRef]
- Maurel, M.; Chevet, E.; Tavernier, J.; Gerlo, S. Getting RIDD of RNA: IRE1 in cell fate regulation. Trends Biochem. Sci. 2014, 39, 245–254. [Google Scholar] [CrossRef]
- Yamamoto, K.; Sato, T.; Matsui, T.; Sato, M.; Okada, T.; Yoshida, H.; Harada, A.; Mori, K. Transcriptional Induction of Mammalian ER Quality Control Proteins Is Mediated by Single or Combined Action of ATF6α and XBP1. Dev. Cell 2007, 13, 365–376. [Google Scholar] [CrossRef] [Green Version]
- Shoulders, M.D.; Ryno, L.M.; Genereux, J.C.; Moresco, J.; Tu, P.G.; Wu, C.; Yates, J.R.; Su, A.I.; Kelly, J.W.; Wiseman, R.L. Stress-Independent Activation of XBP1s and/or ATF6 Reveals Three Functionally Diverse ER Proteostasis Environments. Cell Rep. 2013, 3, 1279–1292. [Google Scholar] [CrossRef] [Green Version]
- Minakshi, R.; Rahman, S.; Jan, A.T.; Archana, A.; Kim, J. Implications of aging and the endoplasmic reticulum unfolded protein response on the molecular modality of breast cancer. Exp. Mol. Med. 2017, 49, e389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nuss, J.E.; Choksi, K.B.; DeFord, J.H.; Papaconstantinou, J. Decreased enzyme activities of chaperones PDI and BiP in aged mouse livers. Biochem. Biophys. Res. Commun. 2008, 365, 355–361. [Google Scholar] [CrossRef] [Green Version]
- Estébanez, B.; de Paz, J.A.; Cuevas, M.J.; González-Gallego, J. Endoplasmic Reticulum Unfolded Protein Response, Aging and Exercise: An Update. Front. Physiol. 2018, 9, 1744. [Google Scholar] [CrossRef] [Green Version]
- Naidoo, N. ER and aging—Protein folding and the ER stress response. Ageing Res. Rev. 2009, 8, 150–159. [Google Scholar] [CrossRef]
- Salminen, A.; Kaarniranta, K.; Kauppinen, A. ER stress activates immunosuppressive network: Implications for aging and Alzheimer’s disease. J. Mol. Med. 2020, 98, 633–650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikeyama, S.; Wang, X.-T.; Li, J.; Podlutsky, A.; Martindale, J.L.; Kokkonen, G.; van Huizen, R.; Gorospe, M.; Holbrook, N.J. Expression of the Pro-apoptotic Genegadd153/chop Is Elevated in Liver with Aging and Sensitizes Cells to Oxidant Injury. J. Biol. Chem. 2003, 278, 16726–16731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirkland, J.L.; Tchkonia, T.; Pirtskhalava, T.; Han, J.; Karagiannides, I. Adipogenesis and aging: Does aging make fat go MAD? Exp. Gerontol. 2002, 37, 757–767. [Google Scholar] [CrossRef]
- Gavilan, M.P.; Vela, J.; Castaño, A.; Ramos, B.; del Río, J.C.; Vitorica, J.; Ruano, D. Cellular environment facilitates protein accumulation in aged rat hippocampus. Neurobiol. Aging 2006, 27, 973–982. [Google Scholar] [CrossRef]
- Willy, J.A.; Young-Baird, S.; Stevens, J.L.; Masuoka, H.C.; Wek, R.C. CHOP links endoplasmic reticulum stress to NF-κB activation in the pathogenesis of nonalcoholic steatohepatitis. Mol. Biol. Cell 2015, 26, 2190–2204. [Google Scholar] [CrossRef]
- Suzuki, T.; Gao, J.; Ishigaki, Y.; Kondo, K.; Sawada, S.; Izumi, T.; Uno, K.; Kaneko, K.; Tsukita, S.; Takahashi, K.; et al. ER Stress Protein CHOP Mediates Insulin Resistance by Modulating Adipose Tissue Macrophage Polarity. Cell Rep. 2017, 18, 2045–2057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Liu, L.; Naik, I.; Braunstein, Z.; Zhong, J.; Ren, B. Transcription Factor C/EBP Homologous Protein in Health and Diseases. Front. Immunol. 2017, 8, 1612. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.G.; Ramaiah, K.V. Reduced eIF2α phosphorylation and increased proapoptotic proteins in aging. Biochem. Biophys. Res. Commun. 2007, 355, 365–370. [Google Scholar] [CrossRef] [PubMed]
- Cabral-Miranda, F.; Hetz, C. ER Stress and Neurodegenerative Disease: A Cause or Effect Relationship? In Current Topics in Microbiology and Immunology; Springer Science and Business Media LLC: Berlin, Germany, 2018; Volume 414, pp. 131–157. [Google Scholar]
- Druelle, C.; Drullion, C.; Deslé, J.; Martin, N.; Saas, L.; Cormenier, J.; Malaquin, N.; Huot, L.; Slomianny, C.; Bouali, F.; et al. ATF6α regulates morphological changes associated with senescence in human fibroblasts. Oncotarget 2016, 7, 67699–67715. [Google Scholar] [CrossRef] [Green Version]
- Oubaha, M.; Miloudi, K.; Dejda, A.; Guber, V.; Mawambo, G.; Germain, M.-A.; Bourdel, G.; Popovic, N.; Rezende, F.A.; Kaufman, R.J.; et al. Senescence-associated secretory phenotype contributes to pathological angiogenesis in retinopathy. Sci. Transl. Med. 2016, 8, 362ra144. [Google Scholar] [CrossRef]
- Cormenier, J.; Martin, N.; Deslé, J.; Salazar-Cardozo, C.; Pourtier, A.; Abbadie, C.; Pluquet, O. The ATF6α arm of the Unfolded Protein Response mediates replicative senescence in human fibroblasts through a COX2/prostaglandin E 2 intracrine pathway. Mech. Ageing Dev. 2018, 170, 82–91. [Google Scholar] [CrossRef]
- Kumar Barodia, S.; Kanthasamy, A.; Oakes, S.; Colla, E. Linking the Endoplasmic Reticulum to Parkinson’s Disease and Alpha-Synucleinopathy. Front. Neurosci. 2019, 13, 560. [Google Scholar] [CrossRef] [Green Version]
- Ben-Zvi, A.; Cohen, E.; Duennwald, M.L.; Lederkremer, G.Z.; Shacham, T.; Sharma, N. Protein Misfolding and ER Stress in Huntington’s Disease. Front. Mol. Biosci. 2019, 6, 20. [Google Scholar] [CrossRef] [Green Version]
- Cao, S.S.; Kaufman, R.J. Endoplasmic Reticulum Stress and Oxidative Stress in Cell Fate Decision and Human Disease. Antioxid. Redox Signal. 2014, 21, 396–413. [Google Scholar] [CrossRef]
- Yu, D.-Y.; Cringle, S.J. Retinal degeneration and local oxygen metabolism. Exp. Eye Res. 2005, 80, 745–751. [Google Scholar] [CrossRef]
- Abokyi, S.; To, C.-H.; Lam, T.T.; Tse, D.Y. Central Role of Oxidative Stress in Age-Related Macular Degeneration: Evidence from a Review of the Molecular Mechanisms and Animal Models. Oxidative Med. Cell. Longev. 2020, 2020, 1–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kheitan, S.; Minuchehr, Z.; Soheili, Z.-S. Exploring the cross talk between ER stress and inflammation in age-related macular degeneration. PLoS ONE 2017, 12, e0181667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salminen, A.; Kauppinen, A.; Hyttinen, J.M.T.; Toropainen, E.; Kaarniranta, K. Endoplasmic Reticulum Stress in Age-Related Macular Degeneration: Trigger for Neovascularization. Mol. Med. 2010, 16, 535–542. [Google Scholar] [CrossRef] [PubMed]
- Bechtel, T.J.; Weerapana, E. From structure to redox: The diverse functional roles of disulfides and implications in disease. Proteome 2017, 17, 1600391. [Google Scholar] [CrossRef] [Green Version]
- Libby, R.T.; Gould, D.B. Endoplasmic reticulum stress as a primary pathogenic mechanism leading to age-related macular degeneration. Adv. Exp. Med. Biol. 2010, 664, 403–409. [Google Scholar]
- Merksamer, P.I.; Trusina, A.; Papa, F.R. Real-Time Redox Measurements during Endoplasmic Reticulum Stress Reveal Interlinked Protein Folding Functions. Cell 2008, 135, 933–947. [Google Scholar] [CrossRef] [Green Version]
- Chakravarthi, S.; Jessop, C.E.; Bulleid, N.J. The role of glutathione in disulphide bond formation and endoplasmic-reticulum-generated oxidative stress. EMBO Rep. 2006, 7, 271–275. [Google Scholar] [CrossRef] [Green Version]
- Ellgaard, L.; Sevier, C.S.; Bulleid, N.J. How Are Proteins Reduced in the Endoplasmic Reticulum? Trends Biochem. Sci. 2018, 43, 32–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chong, W.C.; Shastri, M.D.; Eri, R. Endoplasmic Reticulum Stress and Oxidative Stress: A Vicious Nexus Implicated in Bowel Disease Pathophysiology. Int. J. Mol. Sci. 2017, 18, 771. [Google Scholar] [CrossRef]
- Li, Y.; Wang, Y.-S.; Shen, X.-F.; Hui, Y.-N.; Han, J.; Zhao, W.; Zhu, J. Alterations of activity and intracellular distribution of the 20S proteasome in ageing retinal pigment epithelial cells. Exp. Gerontol. 2008, 43, 1114–1122. [Google Scholar] [CrossRef]
- Zhang, X.; Zhou, J.; Fernandes, A.; Sparrow, J.R.; Pereira, P.; Taylor, A.; Shang, F. The Proteasome: A Target of Oxidative Damage in Cultured Human Retina Pigment Epithelial Cells. Investig. Opthalmol. Vis. Sci. 2008, 49, 3622–3630. [Google Scholar] [CrossRef] [PubMed]
- Terman, A.; Sandberg, S. Proteasome inhibition enhances lipofuscin formation. Ann. N. Y. Acad. Sci. USA 2002, 973, 309–312. [Google Scholar] [CrossRef] [PubMed]
- Nociari, M.M.; Kiss, S.; Rodriguez-Boulan, E. Lipofuscin Accumulation into and Clearance from Retinal Pigment Epithelium Lysosomes: Physiopathology and Emerging Therapeutics. In Lysosomes–Associated Diseases and Methods to Study Their Function; IntechOpen: London, UK, 2017. [Google Scholar]
- Fernandes, A.F.; Guo, W.; Zhang, X.; Gallagher, M.; Ivan, M.; Taylor, A.; Pereira, P.; Shang, F. Proteasome-dependent regulation of signal transduction in retinal pigment epithelial cells. Exp. Eye Res. 2006, 83, 1472–1481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, J.; Qiu, Y.; Frontera, E.; Jia, L.; Khan, N.W.; Klionsky, D.J.; Ferguson, T.A.; Thompson, D.; Zacks, D.N. Inhibiting autophagy reduces retinal degeneration caused by protein misfolding. Autophagy 2018, 14, 1226–1238. [Google Scholar] [CrossRef] [Green Version]
- Qiu, Y.; Yao, J.; Jia, L.; Thompson, D.; Zacks, D.N. Shifting the balance of autophagy and proteasome activation reduces proteotoxic cell death: A novel therapeutic approach for restoring photoreceptor homeostasis. Cell Death Dis. 2019, 10, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Tang, B.; Cai, J.; Sun, L.; Li, Y.; Qu, J.; Snider, B.J.; Wu, S. Proteasome Inhibitors Activate Autophagy Involving Inhibition of PI3K-Akt-mTOR Pathway as an Anti-Oxidation Defense in Human RPE Cells. PLoS ONE 2014, 9, e103364. [Google Scholar] [CrossRef]
- McLaughlin, T.; Falkowski, M.; Park, J.W.; Keegan, S.; Elliott, M.; Wang, J.J.; Zhang, S.X. Loss of XBP1 accelerates age-related decline in retinal function and neurodegeneration. Mol. Neurodegener. 2018, 13, 16. [Google Scholar] [CrossRef] [Green Version]
- Zhong, Y.; Li, J.; Wang, J.J.; Chen, C.; Tran, J.-T.A.; Saadi, A.; Yu, Q.; Le, Y.-Z.; Mandal, N.A.; Anderson, R.E.; et al. X-Box Binding Protein 1 Is Essential for the Anti-Oxidant Defense and Cell Survival in the Retinal Pigment Epithelium. PLoS ONE 2012, 7, e38616. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.X.; Sanders, E.; Fliesler, S.J.; Wang, J.J. Endoplasmic reticulum stress and the unfolded protein responses in retinal degeneration. Exp. Eye Res. 2014, 125, 30–40. [Google Scholar] [CrossRef] [Green Version]
- Athanasiou, D.; Aguila, M.; Bellingham, J.; Kanuga, N.; Adamson, P.; Cheetham, M.E. The role of the ER stress-response protein PERK in rhodopsin retinitis pigmentosa. Hum. Mol. Genet. 2017, 26, 4896–4905. [Google Scholar] [CrossRef] [Green Version]
- Ansar, M.; Santos-Cortez, R.L.P.; Saqib, M.A.N.; Zulfiqar, F.; Lee, K.; Ashraf, N.M.; Ullah, E.; Wang, X.; Sajid, S.; Khan, F.S.; et al. Mutation of ATF6 causes autosomal recessive achromatopsia. Hum. Genet. 2015, 134, 941–950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, W.; Zou, J.; Yoshida, S.; Jiang, B.; Zhou, Y. The Role of Inflammation in Age-Related Macular Degeneration. Int. J. Biol. Sci. 2020, 16, 2989–3001. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Kaufman, R.J. From endoplasmic-reticulum stress to the inflammatory response. Nat. Cell Biol. 2008, 454, 455–462. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Cao, T.; Luo, C.; Cai, J.; Zhou, X.; Xiao, X.; Liu, S. Crosstalk between ER stress, NLRP3 inflammasome, and inflammation. Appl. Microbiol. Biotechnol. 2020, 104, 6129–6140. [Google Scholar] [CrossRef]
- Rana, T.M.; Shinde, V.M.; Starr, C.R.; Kruglov, A.; Boitet, E.; Kotla, P.; Zolotukhin, S.; Gross, A.K.; Gorbatyuk, M. An activated unfolded protein response promotes retinal degeneration and triggers an inflammatory response in the mouse retina. Cell Death Dis. 2014, 5, e1578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khanna, S.; Komati, R.; Eichenbaum, D.; Hariprasad, I.; Ciulla, T.; Hariprasad, S.M. Current and upcoming anti-VEGF therapies and dosing strategies for the treatment of neovascular AMD: A comparative review. BMJ Open Ophthalmol. 2019, 4, e000398. [Google Scholar] [CrossRef] [Green Version]
- Ois Binet, F.; Sapieha, P. Cell Metabolism Review ER Stress and Angiogenesis. Cell Metab. 2015, 22, 560–575. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, R.; Lipson, K.L.; Sargent, K.E.; Mercurio, A.M.; Hunt, J.S.; Ron, D.; Urano, F. Transcriptional Regulation of VEGF-A by the Unfolded Protein Response Pathway. PLoS ONE 2010, 5, e9575. [Google Scholar] [CrossRef] [PubMed]
- Drogat, B.; Auguste, P.; Nguyen, D.T.; Bouchecareilh, M.; Pineau, R.; Nalbantoglu, J.; Kaufman, R.J.; Chevet, E.; Bikfalvi, A.; Moenner, M. IRE1 Signaling Is Essential for Ischemia-Induced Vascular Endothelial Growth Factor-A Expression and Contributes to Angiogenesis and Tumor Growth In vivo. Cancer Res. 2007, 67, 6700–6707. [Google Scholar] [CrossRef] [Green Version]
- Zhu, D.; Xu, X.; Zheng, Z.; Gu, Q. Regulation of vascular endothelial growth factor and pigment epithelium-derived factor in rat retinal explants under retinal acidification. Eye 2009, 23, 2105–2111. [Google Scholar] [CrossRef] [Green Version]
- Koyama, Y.; Matsuzaki, S.; Gomi, F.; Yamada, K.; Katayama, T.; Sato, K.; Kumada, T.; Fukuda, A.; Matsuda, S.; Tano, Y.; et al. Induction of Amyloid β Accumulation by ER Calcium Disruption and Resultant Upregulation of Angiogenic Factors in ARPE19 Cells. Investig. Opthalmol. Vis. Sci. 2008, 49, 2376–2383. [Google Scholar] [CrossRef] [Green Version]
- Cano, M.; Wang, L.; Wan, J.; Barnett, B.P.; Ebrahimi, K.; Qian, J.; Handa, J.T. Oxidative stress induces mitochondrial dysfunction and a protective unfolded protein response in RPE cells. Free. Radic. Biol. Med. 2014, 69, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Dou, G.; Sreekumar, P.G.; Spee, C.; He, S.; Ryan, S.J.; Kannan, R.; Hinton, D.R. Deficiency of αB crystallin augments ER stress-induced apoptosis by enhancing mitochondrial dysfunction. Free. Radic. Biol. Med. 2012, 53, 1111–1122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernandez, M.; Recalde, S.; González-Zamora, J.; Bilbao-Malavé, V.; de Viteri, M.S.; Bezunartea, J.; Moreno-Orduña, M.; Belza, I.; Barrio-Barrio, J.; Fernandez-Robredo, P.; et al. Anti-Inflammatory and Anti-Oxidative Synergistic Effect of Vitamin D and Nutritional Complex on Retinal Pigment Epithelial and Endothelial Cell Lines against Age-Related Macular Degeneration. Nutrients 2021, 13, 1423. [Google Scholar] [CrossRef]
- De Viteri, M.S.; Hernandez, M.; Bilbao-Malavé, V.; Fernandez-Robredo, P.; González-Zamora, J.; Garcia-Garcia, L.; Ispizua, N.; Recalde, S.; Garcia-Layana, A. A Higher Proportion of Eicosapentaenoic Acid (EPA) When Combined with Docosahexaenoic Acid (DHA) in Omega-3 Dietary Supplements Provides Higher Antioxidant Effects in Human Retinal Cells. Antioxidants 2020, 9, 828. [Google Scholar] [CrossRef] [PubMed]
- Monte, M.J.; Marin, J.; Antelo, A.; Vazquez-Tato, J. Bile acids: Chemistry, physiology, and pathophysiology. World J. Gastroenterol. 2009, 15, 804–816. [Google Scholar] [CrossRef]
- Warden, C.; Brantley, M. Glycine-Conjugated Bile Acids Protect RPE Tight Junctions against Oxidative Stress and Inhibit Choroidal Endothelial Cell Angiogenesis In Vitro. Biomolecules 2021, 11, 626. [Google Scholar] [CrossRef] [PubMed]
- European Association for the Study of the Liver. EASL Clinical Practice Guidelines: Management of cholestatic liver diseases. J. Hepatol. 2009, 51, 237–267. [Google Scholar] [CrossRef] [PubMed]
- Vang, S.; Longley, K.; Steer, C.J.; Low, W.C. The Unexpected Uses of Urso- and Tauroursodeoxycholic Acid in the Treatment of Non-liver Diseases. Glob. Adv. Health Med. 2014, 3, 58–69. [Google Scholar] [CrossRef] [Green Version]
- Oveson, B.C.; Iwase, T.; Hackett, S.F.; Lee, S.Y.; Usui, S.; Sedlak, T.W.; Snyder, S.H.; Campochiaro, P.A.; Sung, J.U. Constituents of bile, bilirubin and TUDCA, protect against oxidative stress-induced retinal degeneration. J. Neurochem. 2010, 116, 144–153. [Google Scholar] [CrossRef] [Green Version]
- Boatright, J.H.; Moring, A.G.; McElroy, C.; Phillips, M.J.; Do, V.T.; Chang, B.; Hawes, N.L.; Boyd, A.P.; Sidney, S.S.; Stewart, R.E.; et al. Tool from ancient pharmacopoeia prevents vision loss. Mol. Vis. 2006, 12, 1706–1714. [Google Scholar] [PubMed]
- Phillips, M.J.; Walker, T.A.; Choi, H.-Y.; Faulkner, A.E.; Kim, M.K.; Sidney, S.S.; Boyd, A.P.; Nickerson, J.M.; Boatright, J.H.; Pardue, M.T. Tauroursodeoxycholic Acid Preservation of Photoreceptor Structure and Function in therd10Mouse through Postnatal Day 30. Investig. Opthalmol. Vis. Sci. 2008, 49, 2148–2155. [Google Scholar] [CrossRef] [Green Version]
- Boatright, J.H.; Nickerson, J.M.; Moring, A.G.; Pardue, M.T. Bile acids in treatment of ocular disease. J. Ocul. Biol. Dis. Inform. 2009, 2, 149–159. [Google Scholar] [CrossRef] [Green Version]
- Fernández-Sánchez, L.; Lax, P.; Pinilla, I.; Martín-Nieto, J.; Cuenca, N. Tauroursodeoxycholic Acid Prevents Retinal Degeneration in Transgenic P23H Rats. Investig. Opthalmol. Vis. Sci. 2011, 52, 4998–5008. [Google Scholar] [CrossRef] [PubMed]
- Drack, A.; Dumitrescu, A.; Bhattarai, S.; Gratie, D.; Stone, E.M.; Mullins, R.; Sheffield, V.C. TUDCA Slows Retinal Degeneration in Two Different Mouse Models of Retinitis Pigmentosa and Prevents Obesity in Bardet-Biedl Syndrome Type 1 Mice. Investig. Opthalmol. Vis. Sci. 2012, 53, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Baehr, W.; Fu, Y. Chemical Chaperone TUDCA Preserves Cone Photoreceptors in a Mouse Model of Leber Congenital Amaurosis. Investig. Opthalmol Vis. Sci. 2012, 53, 3349–3356. [Google Scholar] [CrossRef] [Green Version]
- Mantopoulos, D.; Murakami, Y.; Comander, J.; Thanos, A.; Roh, M.; Miller, J.W.; Vavvas, D.G. Tauroursodeoxycholic Acid (TUDCA) Protects Photoreceptors from Cell Death after Experimental Retinal Detachment. PLoS ONE 2011, 6, e24245. [Google Scholar] [CrossRef]
- Gómez-Vicente, V.; Lax, P.D.; Fernández-Sánchez, L.; Rondón, N.; Esquiva, G.; Germain, F.; De La Villa, P.; Cuenca, N. Neuroprotective Effect of Tauroursodeoxycholic Acid on N-Methyl-D-Aspartate-Induced Retinal Ganglion Cell Degeneration. PLoS ONE 2015, 10, e0137826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De La Barca, J.M.C.; Simard, G.; Amati-Bonneau, P.; Safiedeen, Z.; Prunier-Mirebeau, D.; Chupin, S.; Gadras, C.; Tessier, L.; Gueguen, N.; Chevrollier, A.; et al. The metabolomic signature of Leber’s hereditary optic neuropathy reveals endoplasmic reticulum stress. Brain 2016, 139, 2864–2876. [Google Scholar] [CrossRef]
- Gaspar, J.; Martins, A.; Cruz, R.; Rodrigues, C.; Ambrósio, A.F.; Santiago, A.R. Tauroursodeoxycholic acid protects retinal neural cells from cell death induced by prolonged exposure to elevated glucose. Neuroscience 2013, 253, 380–388. [Google Scholar] [CrossRef]
- Daruich, A.; Picard, E.; Boatright, J.H.; Behar-Cohen, F. Review: The bile acids urso- and tauroursodeoxycholic acid as neuroprotective therapies in retinal disease. Mol. Vis. 2019, 25, 610–624. [Google Scholar]
- Osborn, M.P.; Park, Y.; Parks, M.B.; Burgess, L.G.; Uppal, K.; Lee, K.; Jones, D.P.; Brantley, M.A. Metabolome-Wide Association Study of Neovascular Age-Related Macular Degeneration. PLoS ONE 2013, 8, e72737. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, S.L.; Uppal, K.; Williamson, S.M.; Liu, K.; Burgess, L.G.; Tran, V.; Umfress, A.C.; Jarrell, K.L.; Bailey, J.C.; Agarwal, A.; et al. The Carnitine Shuttle Pathway is Altered in Patients with Neovascular Age-Related Macular Degeneration. Investig. Opthalmol. Vis. Sci. 2018, 59, 4978–4985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alhasani, R.H.; Almarhoun, M.; Zhou, X.; Reilly, J.; Patterson, S.; Zeng, Z.; Shu, X. Tauroursodeoxycholic Acid Protects Retinal Pigment Epithelial Cells from Oxidative Injury and Endoplasmic Reticulum Stress In Vitro. Biomedicine 2020, 8, 367. [Google Scholar] [CrossRef]
- Murase, H.; Tsuruma, K.; Shimazawa, M.; Hara, H. TUDCA Promotes Phagocytosis by Retinal Pigment Epithelium via MerTK Activation. Investig. Opthalmol. Vis. Sci. 2015, 56, 2511–2518. [Google Scholar] [CrossRef] [Green Version]
- Warden, C.; Barnett, J.M.; Brantley, M.A. Taurocholic acid inhibits features of age-related macular degeneration in vitro. Exp. Eye Res. 2020, 193, 107974. [Google Scholar] [CrossRef]
- Woo, S.J.; Kim, J.H.; Yu, H.G. Ursodeoxycholic Acid and Tauroursodeoxycholic Acid Suppress Choroidal Neovascularization in a Laser-Treated Rat Model. J. Ocul. Pharmacol. Ther. 2010, 26, 223–229. [Google Scholar] [CrossRef] [PubMed]
- Maharjan, P.; Kim, D.; Jin, M.; Ko, H.J.; Song, Y.H.; Lee, Y.; Ahn, B.-N.; Kim, S.-K.; Lee, Y.; Shin, M.C.; et al. Preclinical Evaluation of UDCA-Containing Oral Formulation in Mice for the Treatment of Wet Age-Related Macular Degeneration. Pharmaceutics 2019, 11, 561. [Google Scholar] [CrossRef] [Green Version]
- Okabe, M.; Szakacs, G.; Reimers, M.A.; Suzuki, T.; Hall, M.D.; Abe, T.; Weinstein, J.N.; Gottesman, M.M. Profiling SLCO and SLC22 genes in the NCI-60 cancer cell lines to identify drug uptake transporters. Mol. Cancer Ther. 2008, 7, 3081–3091. [Google Scholar] [CrossRef] [Green Version]
- Suga, T.; Yamaguchi, H.; Sato, T.; Maekawa, M.; Goto, J.; Mano, N. Preference of Conjugated Bile Acids over Unconjugated Bile Acids as Substrates for OATP1B1 and OATP1B3. PLoS ONE 2017, 12, e0169719. [Google Scholar] [CrossRef]
- Zuccato, C.F.; Asad, A.S.; Candia, A.J.N.; Gottardo, M.F.; Ayala, M.A.M.; Theas, M.S.; Seilicovich, A.; Candolfi, M. Mitochondrial-derived peptide humanin as therapeutic target in cancer and degenerative diseases. Expert Opin. Ther. Targets 2018, 23, 117–126. [Google Scholar] [CrossRef]
- Sreekumar, P.G.; Kannan, R. Mechanisms of protection of retinal pigment epithelial cells from oxidant injury by humanin and other mitochondrial-derived peptides: Implications for age-related macular degeneration. Redox Biol. 2020, 37, 101663. [Google Scholar] [CrossRef] [PubMed]
- Nashine, S.; Cohen, P.; Chwa, M.; Lu, S.; Nesburn, A.B.; Kuppermann, B.D.; Kenney, M.C. Humanin G (HNG) protects age-related macular degeneration (AMD) transmitochondrial ARPE-19 cybrids from mitochondrial and cellular damage. Cell Death Dis. 2017, 8, e2951. [Google Scholar] [CrossRef]
- Sreekumar, P.G.; Ishikawa, K.; Spee, C.; Mehta, H.H.; Wan, J.; Yen, K.; Cohen, P.; Kannan, R.; Hinton, D.R. The Mitochondrial-Derived Peptide Humanin Protects RPE Cells from Oxidative Stress, Senescence, and Mitochondrial Dysfunction. Investig. Opthalmol. Vis. Sci. 2016, 57, 1238–1253. [Google Scholar] [CrossRef] [PubMed]
- Matsunaga, D.; Sreekumar, P.G.; Ishikawa, K.; Terasaki, H.; Barron, E.; Cohen, P.; Kannan, R.; Hinton, D.R. Humanin Protects RPE Cells from Endoplasmic Reticulum Stress-Induced Apoptosis by Upregulation of Mitochondrial Glutathione. PLoS ONE 2016, 11, e0165150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, Z.; Tas, E.; Muzumdar, R. Humanin and Age-Related Diseases: A New Link? Front. Endocrinol. 2014, 5, 210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nashine, S.; Kenney, M.C. Effects of Mitochondrial-Derived Peptides (MDPs) on Mitochondrial and Cellular Health in AMD. Cells 2020, 9, 1102. [Google Scholar] [CrossRef] [PubMed]
- Solanki, A.; Smalling, R.; Parola, A.H.; Nathan, I.; Kasher, R.; Pathak, Y.; Sutariya, V. Humanin Nanoparticles for Reducing Pathological Factors Characteristic of Age-Related Macular Degeneration. Curr. Drug Deliv. 2019, 16, 226–232. [Google Scholar] [CrossRef]
- Kaarniranta, K.; Sinha, D.; Blasiak, J.; Kauppinen, A.; Vereb, Z.; Salminen, A.; Boulton, M.E.; Petrovski, G. Autophagy and heterophagy dysregulation leads to retinal pigment epithelium dysfunction and development of age-related macular degeneration. Autophagy 2013, 9, 973–984. [Google Scholar] [CrossRef] [Green Version]
- Gong, Z.; Tasset, I.; Diaz, A.; Anguiano, J.; Tas, E.; Cui, L.; Kuliawat, R.; Liu, H.; Kühn, B.; Cuervo, A.M.; et al. Humanin is an endogenous activator of chaperone-mediated autophagy. J. Cell Biol. 2017, 217, 635–647. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Sreekumar, P.G.; Peddi, S.; Hinton, D.R.; Kannan, R.; MacKay, J.A. The humanin peptide mediates ELP nanoassembly and protects human retinal pigment epithelial cells from oxidative stress. Nanomed. Nanotechnol. Biol. Med. 2020, 24, 102111. [Google Scholar] [CrossRef]
- Surguchov, A.; Bernal, L.; Surguchev, A.A. Phytochemicals as Regulators of Genes Involved in Synucleinopathies. Biomolecules 2021, 11, 624. [Google Scholar] [CrossRef] [PubMed]
- Delmas, D.; Cornebise, C.; Courtaut, F.; Xiao, J.; Aires, V. New Highlights of Resveratrol: A Review of Properties against Ocular Diseases. Int. J. Mol. Sci. 2021, 22, 1295. [Google Scholar] [CrossRef] [PubMed]
- Baur, J.; Sinclair, D. Therapeutic potential of resveratrol: The in vivo evidence. Nat. Rev. Drug Discov. 2006, 5, 493–506. [Google Scholar] [CrossRef] [PubMed]
- Maugeri, A.; Barchitta, M.; Mazzone, M.G.; Giuliano, F.; Basile, G.; Agodi, A. Resveratrol Modulates SIRT1 and DNMT Functions and Restores LINE-1 Methylation Levels in ARPE-19 Cells under Oxidative Stress and Inflammation. Int. J. Mol. Sci. 2018, 19, 2118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- King, R.E.; Kent, K.D.; Bomser, J.A. Resveratrol reduces oxidation and proliferation of human retinal pigment epithelial cells via extracellular signal-regulated kinase inhibition. Chem. Interact. 2005, 151, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Wu, Z.-Z.; Cheng, Y.-L.; Lin, W.; Qu, C. Resveratrol protects against oxidative damage of retinal pigment epithelium cells by modulating SOD/MDA activity and activating Bcl-2 expression. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 378–388. [Google Scholar]
- Qin, S.; Lu, Y.; Rodrigues, G.A. Resveratrol protects RPE cells from sodium iodate by modulating PPARα and PPARδ. Exp. Eye Res. 2014, 118, 100–108. [Google Scholar] [CrossRef]
- Chan, C.-M.; Huang, C.-H.; Li, H.-J.; Hsiao, C.-Y.; Su, C.-C.; Lee, P.-L.; Hung, C.-F. Protective Effects of Resveratrol against UVA-Induced Damage in ARPE19 Cells. Int. J. Mol. Sci. 2015, 16, 5789–5802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheu, S.-J.; Liu, N.-C.; Chen, J.-L. Resveratrol Protects Human Retinal Pigment Epithelial Cells from Acrolein-Induced Damage. J. Ocul. Pharmacol. Ther. 2010, 26, 231–236. [Google Scholar] [CrossRef]
- Kenney, C.M.; Kuppermann, B.D.; Patil, J.A.; Seigel, G.M.; Sharma, A.; Gramajo, A.L. Effects of hydroquinone on retinal and vascular cells in vitro. Indian J. Ophthalmol. 2012, 60, 189–193. [Google Scholar] [CrossRef] [PubMed]
- Bhattarai, N.; Korhonen, E.; Toppila, M.; Koskela, A.; Kaarniranta, K.; Mysore, Y.; Kauppinen, A. Resvega Alleviates Hydroquinone-Induced Oxidative Stress in ARPE-19 Cells. Int. J. Mol. Sci. 2020, 21, 2066. [Google Scholar] [CrossRef] [Green Version]
- Neal, S.; Buehne, K.; Besley, N.A.; Yang, P.; Silinski, P.; Hong, J.; Ryde, I.T.; Meyer, J.; Jaffe, G.J. Resveratrol Protects Against Hydroquinone-Induced Oxidative Threat in Retinal Pigment Epithelial Cells. Investig. Opthalmol. Vis. Sci. 2020, 61, 32. [Google Scholar] [CrossRef] [Green Version]
- Koskela, A.; Reinisalo, M.; Petrovski, G.; Sinha, D.; Olmiere, C.; Karjalainen, R.; Kaarniranta, K. Nutraceutical with Resveratrol and Omega-3 Fatty Acids Induces Autophagy in ARPE-19 Cells. Nutrients 2016, 8, 284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Josifovska, N.; Albert, R.; Nagymihály, R.; Lytvynchuk, L.; Moe, M.C.; Kaarniranta, K.; Veréb, Z.J.; Petrovski, G. Resveratrol as Inducer of Autophagy, Pro-Survival, and Anti-Inflammatory Stimuli in Cultured Human RPE Cells. Int. J. Mol. Sci. 2020, 21, 813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alaimo, A.; Di Santo, M.C.; Rubio, A.P.D.; Chaufan, G.; Liñares, G.G.; Pérez, O.E. Toxic effects of A2E in human ARPE-19 cells were prevented by resveratrol: A potential nutritional bioactive for age-related macular degeneration treatment. Arch. Toxicol. 2020, 94, 553–572. [Google Scholar] [CrossRef]
- Kang, J.-H.; Choung, S.-Y. Protective effects of resveratrol and its analogs on age-related macular degeneration in vitro. Arch. Pharmacal. Res. 2016, 39, 1703–1715. [Google Scholar] [CrossRef] [PubMed]
- Nagineni, C.N.; Raju, R.; Nagineni, K.K.; Kommineni, V.K.; Cherukuri, A.; Kutty, R.K.; Hooks, J.J.; Detrick, B. Resveratrol Suppresses Expression of VEGF by Human Retinal Pigment Epithelial Cells: Potential Nutraceutical for Age-related Macular Degeneration. Aging Dis. 2014, 5, 88–100. [Google Scholar] [CrossRef]
- Lee, C.S.; Choi, E.Y.; Lee, S.C.; Koh, H.J.; Lee, J.H.; Chung, J.H. Resveratrol Inhibits Hypoxia-Induced Vascular Endothelial Growth Factor Expression and Pathological Neovascularization. Yonsei Med. J. 2015, 56, 1678–1685. [Google Scholar] [CrossRef] [Green Version]
- Dugas, B.; Charbonnier, S.; Baarine, M.; Ragot, K.; Delmas, D.; Ménétrier, F.; Lherminier, J.; Malvitte, L.; Khalfaoui, T.; Bron, A.; et al. Effects of oxysterols on cell viability, inflammatory cytokines, VEGF, and reactive oxygen species production on human retinal cells: Cytoprotective effects and prevention of VEGF secretion by resveratrol. Eur. J. Nutr. 2010, 49, 435–446. [Google Scholar] [CrossRef]
- Kanavi, M.R.; Darjatmoko, S.; Wang, S.; Azari, A.A.; Farnoodian, M.; Kenealey, J.D.; Van Ginkel, P.R.; Albert, D.M.; Sheibani, N.; Polans, A.S. The Sustained Delivery of Resveratrol or a Defined Grape Powder Inhibits New Blood Vessel Formation in a Mouse Model of Choroidal Neovascularization. Molecules 2014, 19, 17578–17603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.-M.; Li, X.-H.; Chen, M.; Luo, J. Intravitreal injection of resveratrol inhibits laser-induced murine choroidal neovascularization. Int. J. Ophthalmol. 2020, 13, 886–892. [Google Scholar] [CrossRef] [PubMed]
- Cornebise, C.; Courtaut, F.; Taillandier-Coindard, M.; Valls-Fonayet, J.; Richard, T.; Monchaud, D.; Aires, V.; Delmas, D. Red Wine Extract Inhibits VEGF Secretion and Its Signaling Pathway in Retinal ARPE-19 Cells to Potentially Disrupt AMD. Molecules 2020, 25, 5564. [Google Scholar] [CrossRef]
- Nagai, N.; Kubota, S.; Tsubota, K.; Ozawa, Y. Resveratrol prevents the development of choroidal neovascularization by modulating AMP-activated protein kinase in macrophages and other cell types. J. Nutr. Biochem. 2014, 25, 1218–1225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivanescu, A.A.; Fernández-Robredo, P.; Heras-Mulero, H.; Sádaba-Echarri, L.M.; García-García, L.; Fernández-García, V.; Moreno-Orduna, M.; Redondo-Exposito, A.; Recalde, S.; García-Layana, A. Modifying Choroidal Neovascularization Development with a Nutritional Supplement in Mice. Nutrients 2015, 7, 5423–5442. [Google Scholar] [CrossRef] [Green Version]
- Richer, S.; Stiles, W.; Ulanski, L.; Carroll, D.; Podella, C. Observation of Human Retinal Remodeling in Octogenarians with a Resveratrol Based Nutritional Supplement. Nutrients 2013, 5, 1989–2005. [Google Scholar] [CrossRef] [PubMed]
- Richer, S.; Patel, S.; Sockanathan, S.; Ulanski, L.J.; Miller, L.; Podella, C. Resveratrol Based Oral Nutritional Supplement Produces Long-Term Beneficial Effects on Structure and Visual Function in Human Patients. Nutrients 2014, 6, 4404–4420. [Google Scholar] [CrossRef]
- García-Layana, A.; Recalde, S.; Hernandez, M.; Abraldes, M.; Nascimento, J.; Hernández-Galilea, E.; Olmedilla-Alonso, B.; Escobar-Barranco, J.; Zapata, M.; Silva, R.; et al. A Randomized Study of Nutritional Supplementation in Patients with Unilateral Wet Age-Related Macular Degeneration. Nutrients 2021, 13, 1253. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, P.; Fnu, G.; Bhatia, D.; Shahid, A.; Sutariya, V. Nanodelivery of Resveratrol-Loaded PLGA Nanoparticles for Age-Related Macular Degeneration. AAPS Pharm. Sci. Tech. 2020, 21, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Subramani, M.; Ponnalagu, M.; Krishna, L.; Jeyabalan, N.; Chevour, P.; Sharma, A.; Jayadev, C.; Shetty, R.; Begum, N.; Archunan, G.; et al. Resveratrol reverses the adverse effects of bevacizumab on cultured ARPE-19 cells. Sci. Rep. 2017, 7, 12242. [Google Scholar] [CrossRef] [Green Version]
- Casado, D.; Quiles, J.L.; Casado, B.; González-García, P.; Battino, M.; López, L.C.; Varela-López, A. The Paradox of Coenzyme Q10 in Aging. Nutrients 2019, 11, 2221. [Google Scholar] [CrossRef] [Green Version]
- Garrido-Maraver, J.; Cordero, M.D.; Oropesa-Avila, M.; Vega, A.F.; Mata, M.D.; Pavon, A.D.; Alcocer-Gomez, E.; Calero, C.P.; Paz, M.V.; Alanis, M.; et al. Clinical applications of coenzyme Q10. Front. Biosci. 2014, 19, 619–633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Tohari, A.M.; Marcheggiani, F.; Zhou, X.; Reilly, J.; Tiano, L.; Shu, X. Therapeutic Potential of Co-enzyme Q10 in Retinal Diseases. Curr. Med. Chem. 2017, 24, 4329–4339. [Google Scholar] [CrossRef] [Green Version]
- Hernández-Camacho, J.D.; Bernier, M.; López-Lluch, G.; Navas, P. Coenzyme Q10 Supplementation in Aging and Disease. Front. Physiol. 2018, 9, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crane, F.L. Biochemical Functions of Coenzyme Q10. J. Am. Coll. Nutr. 2001, 20, 591–598. [Google Scholar] [CrossRef]
- Kagan, V.E.; Fabisiak, J.P.; Quinn, P.J. Coenzyme Q and vitamin E need each other as antioxidants. Protoplasma 2000, 214, 11–18. [Google Scholar] [CrossRef]
- Tomasetti, M.; Alleva, R.; Collins, A.R. In vivo supplementation with coenzyme Q10 enhances the recovery of human lymphocytes from oxidative DNA damage. FASEB J. 2001, 15, 1425–1427. [Google Scholar] [CrossRef]
- Manzar, H.; Abdulhussein, D.; Yap, T.E.; Cordeiro, M.F. Cellular Consequences of Coenzyme Q10 Deficiency in Neurodegeneration of the Retina and Brain. Int. J. Mol. Sci. 2020, 21, 9299. [Google Scholar] [CrossRef]
- Rotig, A.; Appelkvist, E.-L.; Geromel, V.; Chretien, D.; Kadhom, N.; Edery, P.; Lebideau, M.; Dallner, G.; Munnich, A.; Ernster, L.; et al. Quinone-responsive multiple respiratory-chain dysfunction due to widespread coenzyme Q10 deficiency. Lancet 2000, 356, 391–395. [Google Scholar] [CrossRef]
- Qu, J.; Kaufman, Y.; Washington, I. Coenzyme Q10 in the Human Retina. Investig. Opthalmol. Vis. Sci. 2009, 50, 1814–1818. [Google Scholar] [CrossRef] [Green Version]
- Navas, P.; Villalba, J.M.; de Cabo, R. The importance of plasma membrane coenzyme Q in aging and stress responses. Mitochondrion 2007, 7, 34–40. [Google Scholar] [CrossRef]
- De Barcelos, I.P.; Haas, R.H. CoQ10 and Aging. Biology 2019, 8, 28. [Google Scholar] [CrossRef] [Green Version]
- Blasi, M.A.; Bovina, C.; Carella, G.; Genova, M.L.; Jansen, A.M.; Lenaz, G.; Brancato, R. Does Coenzyme Q10 Play a Role in Opposing Oxidative Stress in Patients with Age-Related Macular Degeneration? Ophthalmology 2000, 215, 51–54. [Google Scholar] [CrossRef] [PubMed]
- Feher, J.; Papale, A.; Mannino, G.; Gualdi, L.; Gabrieli, C.B. Mitotropic Compounds for the Treatment of Age-Related Macular Degeneration. Ophthalmology 2003, 217, 351–357. [Google Scholar] [CrossRef]
- Feher, J.; Kovacs, B.; Kovács, I.; Schveoller, M.; Papale, A.; Gabrieli, C.B. Improvement of Visual Functions and Fundus Alterations in Early Age-Related Macular Degeneration Treated with a Combination of Acetyl-L-Carnitine, n-3 Fatty Acids, and Coenzyme Q10. Ophthalmologica 2005, 219, 154–166. [Google Scholar] [CrossRef] [PubMed]
- Garrido-Maraver, J.; Cordero, M.D.; Oropesa-Ávila, M.; Vega, A.F.; de la Mata, M.; Pavón, A.D.; De-Miguel, M.; Calero, C.P.; Paz, M.V.; Cotán, D.; et al. Coenzyme q10 therapy. Mol. Syndr. 2014, 5, 187–197. [Google Scholar] [CrossRef] [Green Version]
- Villalba, J.M.; Parrado-Fernández, C.; Santos-Gonzalez, M.; Alcain, F.J. Therapeutic use of coenzyme Q10 and coenzyme Q10-related compounds and formulations. Expert Opin. Investig. Drugs 2010, 19, 535–554. [Google Scholar] [CrossRef] [PubMed]
- Crooke, A.; Huete-Toral, F.; Colligris, B.; Pintor, J. The role and therapeutic potential of melatonin in age-related ocular diseases. J. Pineal Res. 2017, 63, e12430. [Google Scholar] [CrossRef] [Green Version]
- Acuña-Castroviejo, D.; Escames, G.; Venegas, C.; Casado, M.E.D.; Cabello, M.E.L.; Lopez, L.C.; Rosales-Corral, S.; Tan, D.-X.; Reiter, R.J. Extrapineal melatonin: Sources, regulation, and potential functions. Cell. Mol. Life Sci. 2014, 71, 2997–3025. [Google Scholar] [CrossRef]
- Wiechmann, A.F.; Summers, J.A. Circadian rhythms in the eye: The physiological significance of melatonin receptors in ocular tissues. Prog. Retin. Eye Res. 2008, 27, 137–160. [Google Scholar] [CrossRef]
- Fanjul-Moles, M.L.; Riquelme, G.O.L. Relationship between Oxidative Stress, Circadian Rhythms, and AMD. Oxidative Med. Cell. Longev. 2016, 2016, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Blasiak, J.; Reiter, R.J.; Kaarniranta, K. Melatonin in Retinal Physiology and Pathology: The Case of Age-Related Macular Degeneration. Oxidative Med. Cell. Longev. 2016, 2016, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Rastmanesh, R. Potential of melatonin to treat or prevent age-related macular degeneration through stimulation of telomerase activity. Med. Hypotheses 2011, 76, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Tan, D. Actions of Melatonin in the Reduction of Oxidative Stress. J. Biomed. Sci. 2000, 3900, 444–458. [Google Scholar]
- Stepicheva, N.A.; Weiss, J.; Shang, P.; Yazdankhah, M.; Ghosh, S.; Bhutto, I.A.; Hose, S.; Zigler, J.S.; Sinha, D. Melatonin as the Possible Link Between Age-Related Retinal Regeneration and the Disrupted Circadian Rhythm in Elderly. In Advances in Experimental Medicine and Biology; Springer: Berlin, Germany, 2019; Volume 1185, pp. 45–49. [Google Scholar]
- Bubenik, G.A.; Konturek, S.J. Melatonin and aging: Prospects for human treatment. J. Physiol. Pharmacol. 2011, 62, 13–19. [Google Scholar] [PubMed]
- Liang, F.-Q.; Green, L.; Wang, C.; Alssadi, R.; Godley, B.F. Melatonin protects human retinal pigment epithelial (RPE) cells against oxidative stress. Exp. Eye Res. 2004, 78, 1069–1075. [Google Scholar] [CrossRef] [PubMed]
- Rosen, R.B.; Hu, D.-N.; Chen, M.; McCormick, S.A.; Walsh, J.; Roberts, J.E. Effects of melatonin and its receptor antagonist on retinal pigment epithelial cells against hydrogen peroxide damage. Mol. Vis. 2012, 18, 1640–1648. [Google Scholar]
- Yan, G.; Yu, L.; Jiang, S.; Zhu, J. Melatonin antagonizes oxidative stress-induced mitochondrial dysfunction in retinal pigmented epithelium cells via melatonin receptor 1 (MT1). J. Toxicol. Sci. 2018, 43, 659–669. [Google Scholar] [CrossRef] [Green Version]
- Perepechaeva, M.L.; Stefanova, N.A.; Grishanova, A.Y. Expression of Genes for AhR and Nrf2 Signal Pathways in the Retina of OXYS Rats during the Development of Retinopathy and Melatonin-Induced Changes in This Process. Bull. Exp. Biol. Med. 2014, 157, 424–429. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-C.; Huang, T.-Y.; Chen, H.-Y.; Huang, T.-C.; Lin, L.-C.; Chang, Y.-J.; Hsia, S.-M. Protective Effect of Melatonin against Oxidative Stress-Induced Apoptosis and Enhanced Autophagy in Human Retinal Pigment Epithelium Cells. Oxidative Med. Cell. Longev. 2018, 2018, 1–12. [Google Scholar] [CrossRef]
- Diéguez, H.H.; Fleitas, M.F.G.; Aranda, M.L.; Calanni, J.S.; Sarmiento, M.I.K.; Chianelli, M.S.; Alaimo, A.; Sande, P.H.; Romeo, H.E.; Rosenstein, R.E.; et al. Melatonin protects the retina from experimental nonexudative age-related macular degeneration in mice. J. Pineal Res. 2020, 68, e12643. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Tian, Y.; Yao, A.; Zha, X.; Zhang, J.; Tao, Y. Intravitreal Delivery of Melatonin Is Protective Against the Photoreceptor Loss in Mice: A Potential Therapeutic Strategy for Degenerative Retinopathy. Front. Pharmacol. 2020, 10, 1633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaur, C.; Sivakumar, V.; Yong, Z.; Lü, J.; Foulds, W.; Ling, E. Blood–retinal barrier disruption and ultrastructural changes in the hypoxic retina in adult rats: The beneficial effect of melatonin administration. J. Pathol. 2007, 212, 429–439. [Google Scholar] [CrossRef]
- Xu, Y.; Cui, K.; Li, J.; Tang, X.; Lin, J.; Lu, X.; Huang, R.; Yang, B.; Shi, Y.; Ye, D.; et al. Melatonin attenuates choroidal neovascularization by regulating macrophage/microglia polarization via inhibition of RhoA/ROCK signaling pathway. J. Pineal Res. 2020, 69, e12660. [Google Scholar] [CrossRef] [PubMed]
- Bardak, H.; Uğuz, A.C.; Bardak, Y. Protective effects of melatonin and memantine in human retinal pigment epithelium (ARPE-19) cells against 2-ethylpyridine-induced oxidative stress: Implications for age-related macular degeneration. Cutan. Ocul. Toxicol. 2017, 37, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Yi, C.; Pan, X.; Xiaoyan, P.; Guo, M.; Pierpaoli, W. Effects of Melatonin in Age-Related Macular Degeneration. Ann. N. Y. Acad. Sci. USA 2005, 1057, 384–392. [Google Scholar] [CrossRef]
- Mehrzadi, S.; Hemati, K.; Reiter, R.J.; Hosseinzadeh, A. Mitochondrial dysfunction in age-related macular degeneration: Melatonin as a potential treatment. Expert Opin. Ther. Targets 2020, 24, 359–378. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bilbao-Malavé, V.; González-Zamora, J.; de la Puente, M.; Recalde, S.; Fernandez-Robredo, P.; Hernandez, M.; Layana, A.G.; Saenz de Viteri, M. Mitochondrial Dysfunction and Endoplasmic Reticulum Stress in Age Related Macular Degeneration, Role in Pathophysiology, and Possible New Therapeutic Strategies. Antioxidants 2021, 10, 1170. https://doi.org/10.3390/antiox10081170
Bilbao-Malavé V, González-Zamora J, de la Puente M, Recalde S, Fernandez-Robredo P, Hernandez M, Layana AG, Saenz de Viteri M. Mitochondrial Dysfunction and Endoplasmic Reticulum Stress in Age Related Macular Degeneration, Role in Pathophysiology, and Possible New Therapeutic Strategies. Antioxidants. 2021; 10(8):1170. https://doi.org/10.3390/antiox10081170
Chicago/Turabian StyleBilbao-Malavé, Valentina, Jorge González-Zamora, Miriam de la Puente, Sergio Recalde, Patricia Fernandez-Robredo, María Hernandez, Alfredo Garcia Layana, and Manuel Saenz de Viteri. 2021. "Mitochondrial Dysfunction and Endoplasmic Reticulum Stress in Age Related Macular Degeneration, Role in Pathophysiology, and Possible New Therapeutic Strategies" Antioxidants 10, no. 8: 1170. https://doi.org/10.3390/antiox10081170
APA StyleBilbao-Malavé, V., González-Zamora, J., de la Puente, M., Recalde, S., Fernandez-Robredo, P., Hernandez, M., Layana, A. G., & Saenz de Viteri, M. (2021). Mitochondrial Dysfunction and Endoplasmic Reticulum Stress in Age Related Macular Degeneration, Role in Pathophysiology, and Possible New Therapeutic Strategies. Antioxidants, 10(8), 1170. https://doi.org/10.3390/antiox10081170