
Theoretical Study of Radical Inactivation, LOX Inhibition, and Iron Chelation: The Role of Ferulic Acid in Skin Protection against UVA Induced Oxidative Stress


 ,
,  , and
, and
Abstract
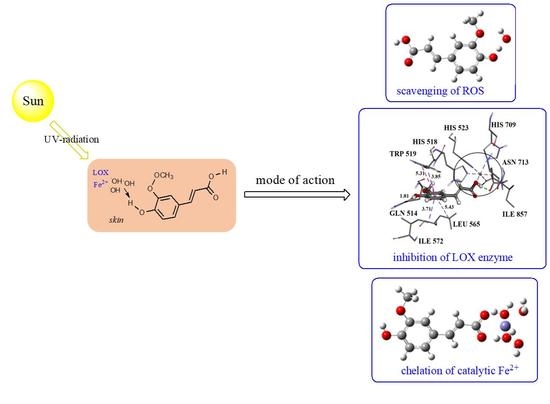
:
1. Introduction
2. Materials and Methods
2.1. DFT Calculations
2.2. Molecular Docking Simulation
3. Results and Discussion
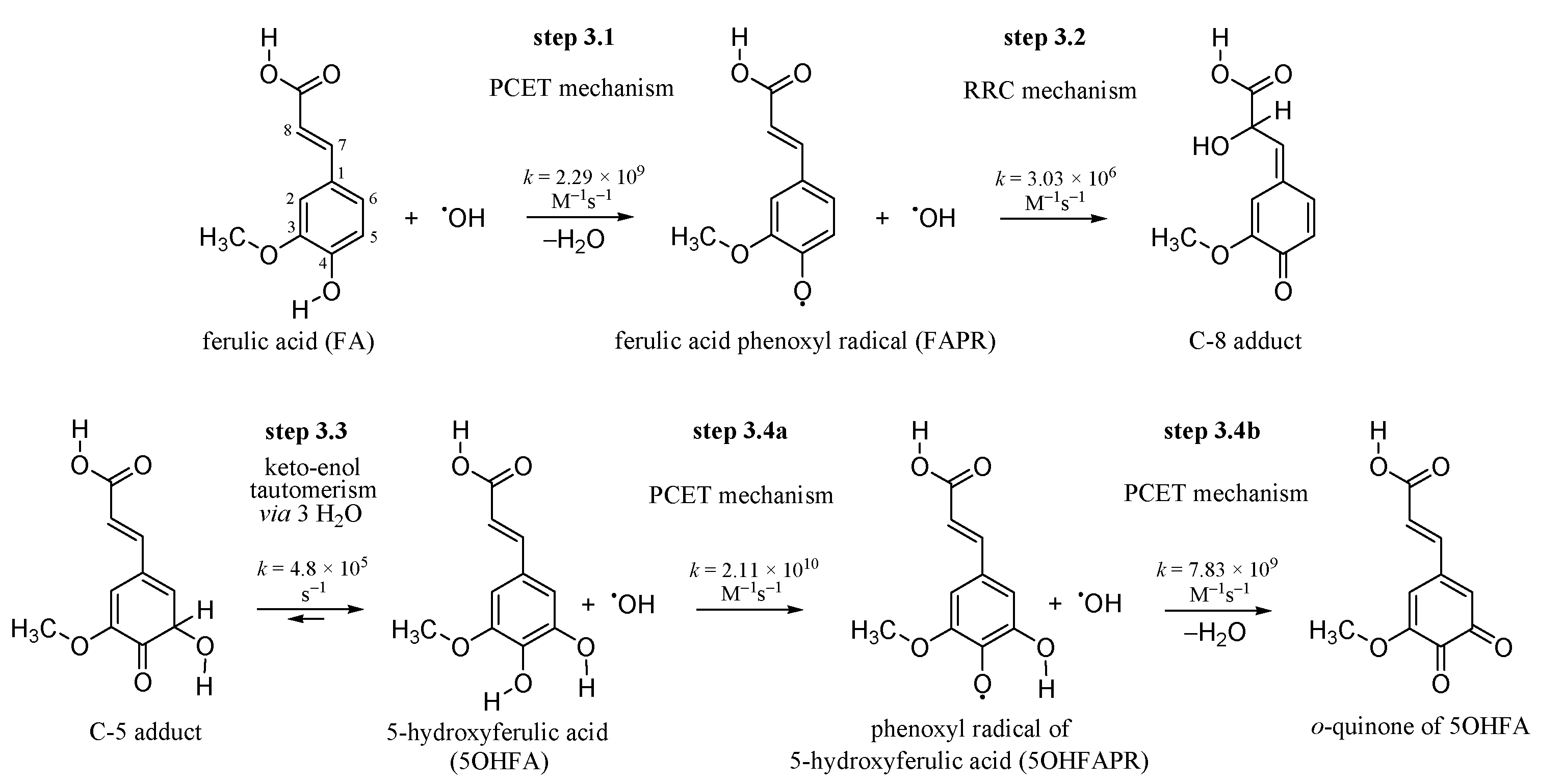
3.1. Inactivating the •OH Radical by FA via the PCET Mechanism
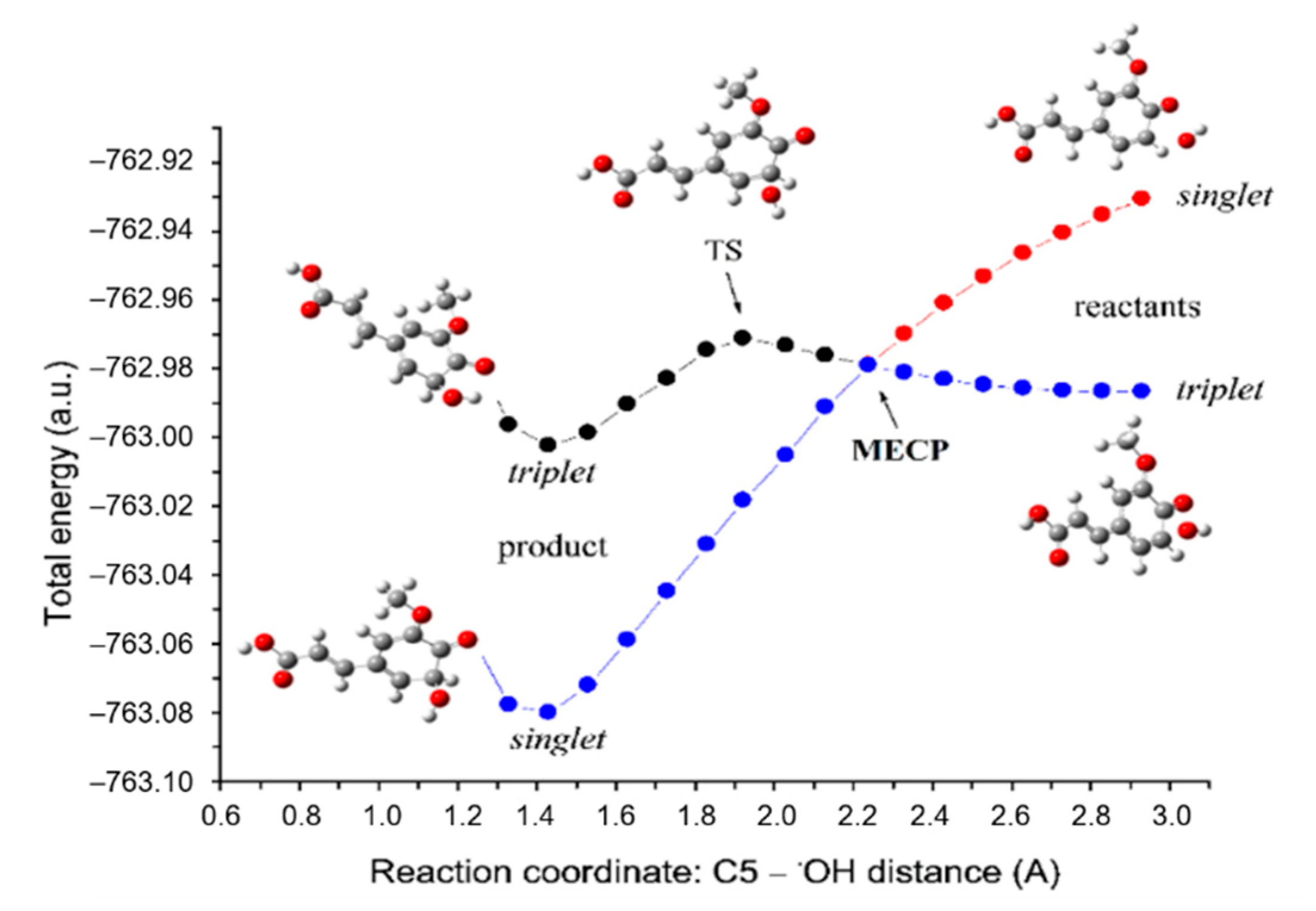
3.2. Inactivation of the •OH Radical by FAPR via the RRC Mechanism
3.3. Keto-enol Tautomerization: Proton Transfers along Hydrogen Bonds
3.4. 5OHFA Inactivates Two •OH Radicals via Double PCET Mechanism
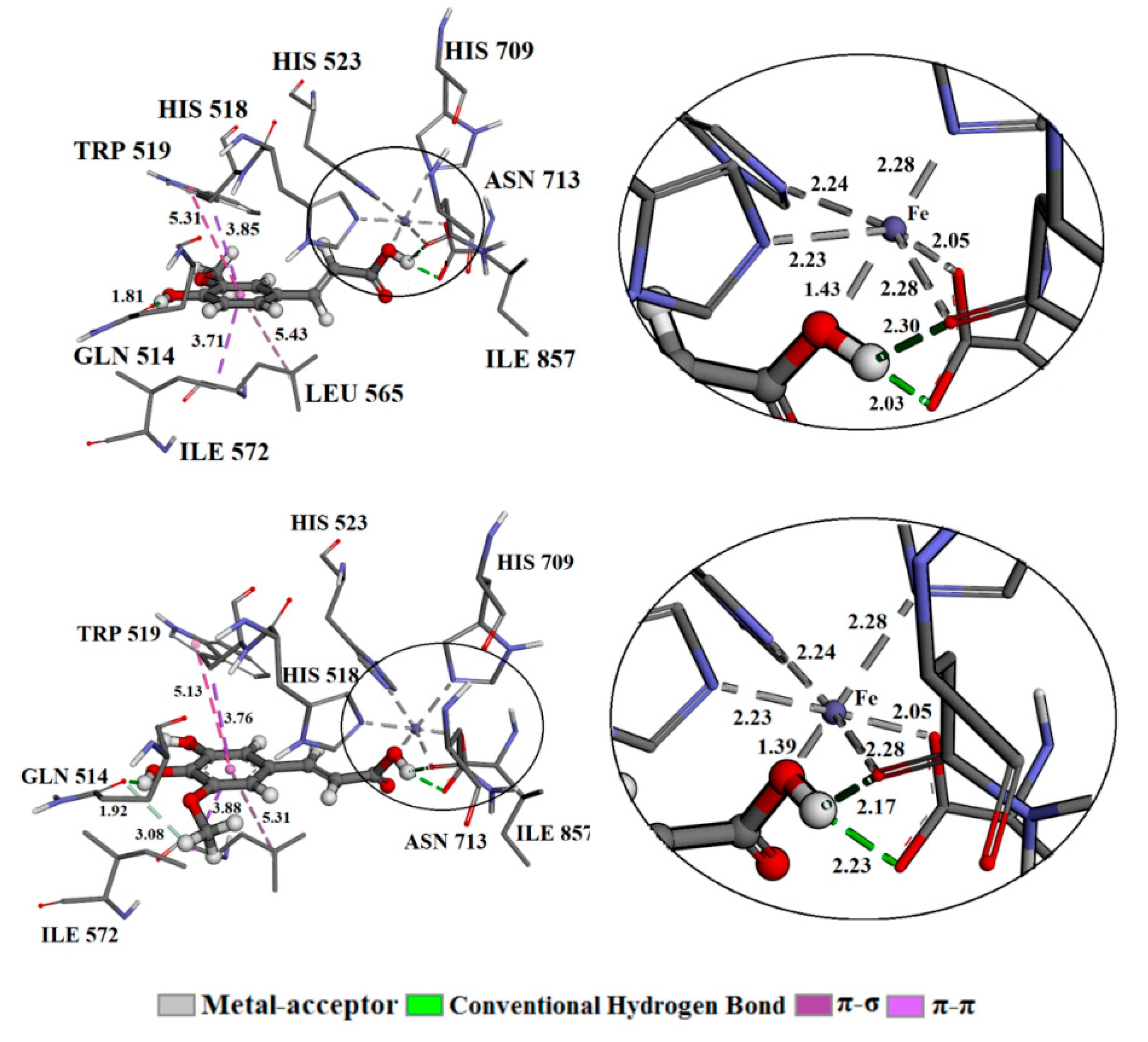
3.5. Molecular Docking
3.6. Chelation of Catalytic Fe2+ Ion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bais, A.F.; Lucas, R.M.; Bornman, J.F.; Williamson, C.E.; Sulzberger, B.; Austin, A.T.; Wilson, S.R.; Andrady, A.L.; Bernhard, G.; McKenzie, R.L.; et al. Environmental effects of ozone depletion, UV radiation and interactions with climate change: UNEP Environmental Effects Assessment Panel, update 2017. Photochem. Photobiol. Sci. 2018, 17, 127–179. [Google Scholar] [CrossRef]
- Skarupova, D.; Vostalova, J.; Rajnochova Svobodova, A. Ultraviolet A protective potential of plant extracts and phytochemicals. Biomed. Pap. Med. Fac. Palacky Univ. Olomouc 2020, 164, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Ou-Yang, H.; Stamatas, G.; Saliou, C.; Kollias, N. A chemiluminescence study of UVA-induced oxidative stress in human skin in vivo. J. Investig. Dermatol. 2004, 122, 1020–1029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanson, K.M.; Simon, J.D. Epidermal trans-urocanic acid and the UV-A-induced photoaging of the skin. Proc. Natl. Acad. Sci. USA 1998, 95, 10576–10578. [Google Scholar] [CrossRef] [Green Version]
- Krutmann, J. Ultraviolet A radiation-induced biological effects in human skin: Relevance for photoaging and photodermatosis. J. Dermatol. Sci. 2000, 23, S22–S26. [Google Scholar] [CrossRef]
- Šnyrychova, I.; Kos, P.B.; Hideg, E. Hydroxyl radicals are not the protagonists of UV-B-induced damage in isolated thylakoid membranes. Funct. Plant Biol. 2007, 34, 1112–1121. [Google Scholar] [CrossRef]
- Halliwell, B.; Gutteridge, J.M.C. Free Radicals in Biology and Medicine, 5th ed.; Oxford University Press: Oxford, UK, 2015. [Google Scholar]
- Galano, A.; Tan, D.X.; Reiter, R.J. Melatonin as a natural ally against oxidative stress: A physicochemical examination. J. Pineal Res. 2011, 51, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Repine, J.E.; Eaton, J.W.; Anders, M.W.; Hoidal, J.R.; Fox, R.B. Generation of hydroxyl radical by enzymes, chemicals, and human phagocytes in vitro. Detection with the anti-inflammatory agent, dimethyl sulfoxide. J. Clin. Investig. 1979, 64, 1642–1651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snezhkina, A.V.; Kudryavtseva, A.V.; Kardymon, O.L.; Savvateeva, M.V.; Melnikova, N.V.; Krasnov, G.S.; Dmitriev, A.A. ROS generation and antioxidant defense systems in normal and malignant cells. Oxidative Med. Cell. Longev. 2019, 2019. [Google Scholar] [CrossRef]
- Krieg, P.; Fürstenberger, G. The role of lipoxygenases in epidermis. Biochim. Biophys. Acta 2014, 1841, 390–400. [Google Scholar] [CrossRef]
- Dang, P.M.-C.; Rolas, L.; El-Benna, J. The dual role of reactive oxygen species-generating nicotinamide adenine dinucleotide phosphate oxidases in gastrointestinal inflammation and therapeutic perspectives. Antioxid. Redox Signal. 2020, 33, 354–373. [Google Scholar] [CrossRef]
- Zuo, L.; Christofi, F.L.; Wright, V.P.; Bao, S.; Clanton, T.L. Lipoxygenase-dependent superoxide release in skeletal muscle. J. Appl. Physiol. 2004, 97, 661–668. [Google Scholar] [CrossRef] [Green Version]
- Lambeth, J.D.; Neish, A.S. Nox enzymes and new thinking on reactive oxygen: A double-edged sword revisited. Annu. Rev. Pathol. Mech. Dis. 2014, 9, 119–145. [Google Scholar] [CrossRef]
- Poljšak, B.; Dahmane, R. Free radicals and extrinsic skin aging. Dermatol. Res. Pract. 2012, 2012. [Google Scholar] [CrossRef] [Green Version]
- Dreher, F.; Denig, N.; Gabard, B.; Schwindt, D.A.; Maibach, H.I. Effect of topical antioxidants on UV-induced erythema formation when administered after exposure. Dermatology 1999, 198, 52–55. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.-H.; Lin, J.-Y.; Gupta, R.D.; Tournas, J.A.; Burch, J.A.; Selim, M.A.; Monteiro-Riviere, N.A.; Grichnik, J.M.; Zielinski, J.; Pinnell, S.R. Ferulic acid stabilizes a solution of vitamins C and E and doubles its photoprotection of skin. J. Investig. Dermatol. 2005, 125, 826–832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Z.; Moghadasian, M.H. Chemistry, natural sources, dietary intake and pharmacokinetic properties of ferulic acid: A review. Food Chem. 2008, 109, 691–702. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Basak, P.; Dutta, S.; Chowdhury, S.; Sil, P.C. New insights into the ameliorative effects of ferulic acid in pathophysiological conditions. Food Chem. Toxicol. 2017, 103, 41–55. [Google Scholar] [CrossRef]
- Zdunska, K.; Dana, A.; Kolodziejczak, A.; Rotsztejn, H. Antioxidant properties of ferulic acid and its possible application. Skin Pharmacol. Physiol. 2018, 31, 332–336. [Google Scholar] [CrossRef]
- Ou, S.; Kwok, K.-C. Ferulic acid: Pharmaceutical functions, preparation and applications in foods. J. Sci. Food Agric. 2004, 84, 1261–1269. [Google Scholar] [CrossRef]
- Saija, A.; Tomaino, A.; Trombetta, D.; De Pasquale, A.; Uccella, N.; Barbuzzi, T.; Paolino, D.; Bonina, F. In vitro and in vivo evaluation of caffeic and ferulic acids as topical photoprotective agents. Int. J. Pharm. 2000, 199, 39–47. [Google Scholar] [CrossRef]
- Harwansh, R.K.; Mukherjee, P.K.; Bahadur, S.; Biswas, R. Enhanced permeability of ferulic acid loaded nanoemulsion based gel through skin against UVA mediated oxidative stress. Life Sci. 2015, 141, 202–211. [Google Scholar] [CrossRef]
- Mancuso, C.; Santangelo, R. Ferulic acid: Pharmacological and toxicological aspects. Food Chem. Toxicol. 2014, 65, 185–195. [Google Scholar] [CrossRef]
- Zhang, L.-W.; Al-Suwayeh, S.A.; Hsieh, P.-W.; Fang, J.-Y. A comparison of skin delivery of ferulic acid and its derivatives: Evaluation of their efficacy and safety. Int. J. Pharm. 2010, 399, 44–51. [Google Scholar] [CrossRef]
- de Paiva, L.B.; Goldbeck, R.; dos Santos, W.D.; Squina, F.M. Ferulic acid and derivatives: Molecules with potential application in the pharmaceutical field. Braz. J. Pharm. Sci. 2013, 9, 395–411. [Google Scholar] [CrossRef] [Green Version]
- Graf, E. Antioxidant potential of ferulic acid. Free Radic. Biol. Med. 1992, 13, 435–448. [Google Scholar] [CrossRef]
- Clifford, M.N. Chlorogenic acids and other cinnamates—Nature, occurrence, dietary burden, absorption and metabolism. J. Sci. Food Agric. 2000, 80, 1033–1043. [Google Scholar] [CrossRef]
- Faria, A.; Fernandes, I.; Norberto, S.; Mateus, N.; Calhau, C. Interplay between anthocyanins and gut microbiota. J. Agric. Food Chem. 2014, 62, 6898–6902. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Sarrias, A.; Espin, J.C.; Tomas-Barberan, F.A. Non-extractable polyphenols produce gut microbiota metabolites that persist in circulation and show anti-inflammatory and free radical scavenging effects. Trends Food Sci. Technol. 2017, 69, 281–288. [Google Scholar] [CrossRef]
- Halliwell, B.; Rafter, J.; Jenner, A. Health promotion by flavonoids, tocopherols, tocotrienols, and other phenols: Direct or indirect effects? Antioxidant or not? Am. J. Clin. Nutr. 2005, 81, 268S–276S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez-Mateos, A.; Vauzour, D.; Krueger, C.G.; Shanmuganayagam, D.; Reed, J.; Calani, L.; Mena, P.; Del Rio, D.; Crozier, A. Bioavailability, bioactivity and impact on health of dietary flavonoids and related compounds: An update. Arch. Toxicol. 2014, 88, 1803–1853. [Google Scholar] [CrossRef]
- Halliwell, B.; Whiteman, M. Measuring reactive species and oxidative damage in vivo and in cell culture: How should you do it and what do the results mean? Br. J. Pharmacol. 2004, 142, 231–255. [Google Scholar] [CrossRef] [Green Version]
- Dangles, O.; Dufour, C.; Tonnele, C.; Trouillas, P. The physical chemistry of polyphenols: Insights into the activity of polyphenols in humans at the molecular level. In Recent Advances in Polyphenol Research; Yoshida, K., Cheynier, V., Quiedau, S., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2017; pp. 1–35. [Google Scholar]
- Košinova, P.; Berka, K.; Wykes, M.; Otyepka, M.; Trouillas, P. Positioning of antioxidant quercetin and its metabolites in lipid bilayer membranes: Implication for their lipid-peroxidation inhibition. J. Phys. Chem. B 2012, 116, 1309–1318. [Google Scholar] [CrossRef]
- Galano, A.; Mazzone, G.; Alvarez-Diduk, R.; Marino, T.; Alvarez-Idaboy, J.R.; Russo, N. Food antioxidants: Chemical insights at the molecular level. Annu. Rev. Food Sci. Technol. 2016, 7, 335–352. [Google Scholar] [CrossRef]
- Amić, A.; Marković, Z.; Dimitrić Marković, J.M.; Milenković, D.; Stepanić, V. Antioxidative potential of ferulic acid phenoxyl radical. Phytochemistry 2020, 170, 112218. [Google Scholar] [CrossRef] [PubMed]
- Sies, H. Strategies of antioxidant defense. Eur. J. Biochem. 1993, 215, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Galleano, M.; Verstraeten, S.V.; Oteiza, P.I.; Fraga, C.G. Antioxidant actions of flavonoids: Thermodynamic and kinetic analysis. Arch. Biochem. Biophys. 2010, 501, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Forman, H.J.; Davies, K.J.A.; Ursini, F. How do nutritional antioxidants really work: Nucleophilic tone and para-hormesis versus free radical scavenging in vivo. Free Radic. Biol. Med. 2014, 66, 24–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leopoldini, M.; Russo, N.; Chiodo, S.; Toscano, M. Iron chelation by the powerful antioxidant flavonoid quercetin. J. Agric. Food Chem. 2006, 54, 6343–6351. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Zhao, Y.; Schultz, N.E.; Truhlar, D.G. Design of density functionals by combining the method of constraint satisfaction with parametrization for thermochemistry, thermochemical kinetics, and noncovalent interactions. J. Chem. Theory Comput. 2006, 2, 364–382. [Google Scholar] [CrossRef]
- Mardirossian, N.; Head-Gordon, M. How accurate are the Minnesota density functionals for noncovalent interactions, isomerization energies, thermochemistry, and barrier heights involving molecules composed of main-group elements? J. Chem. Theory Comput. 2016, 12, 4303–4325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dzib, E.; Cabellos, J.L.; Ortiz-Chi, F.; Pan, S.; Galano, A.; Merino, G. Eyringpy: A program for computing rate constants in the gas phase and in solution. Int. J. Quantum Chem. 2019, 119, e25686. [Google Scholar] [CrossRef] [Green Version]
- Eckart, C. The penetration of a potential barrier by electrons. Phys. Rev. 1930, 35, 1303–1309. [Google Scholar] [CrossRef]
- Alvarez-Idaboy, J.R.; Mora-Diez, N.; Boyd, R.J.; Vivier-Bunge, A. On the importance of prereactive complexes in molecule−radical reactions: Hydrogen abstraction from aldehydes by OH. J. Am. Chem. Soc. 2001, 123, 2018–2024. [Google Scholar] [CrossRef]
- Galano, A. Free radicals induced oxidative stress at a molecular level: The current status, challenges and perspectives of computational chemistry based protocols. J. Mex. Chem. Soc. 2015, 59, 231–262. [Google Scholar] [CrossRef] [Green Version]
- Mayer, J.M.; Hrovat, D.A.; Thomas, J.L.; Borden, W.T. Proton-coupled electron transfer versus hydrogen atom transfer in benzyl/toluene, methoxyl/methanol, and phenoxyl/phenol self-exchange reactions. J. Am. Chem. Soc. 2002, 124, 11142–11147. [Google Scholar] [CrossRef] [PubMed]
- Keith, T.A. AIMAll; Version 17.11.14; TS Gristmill Software: Overland Park, KS, USA, 2017; Available online: http://aim.tkgristmill.com (accessed on 18 May 2020).
- Kaur, N.; Kumari, I.; Gupta, S.; Goel, N. Spin inversion phenomenon and two-state reactivity mechanism for direct benzene hydroxylation by V4O10 cluster. J. Phys. Chem. A 2016, 120, 9588–9597. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Guerra Pedregal, J.; Funes-Ardoiz, I.; Maseras, F. EasyMECP: Quick Setup of MECP Calculations with Gaussian. 2018. Available online: https://github.com/jaimergp/easymecp (accessed on 14 April 2020).
- Zhao, Y.; Truhlar, D.G. Density functionals with broad applicability in chemistry. Acc. Chem. Res. 2008, 41, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- Skrzypczak-Jankun, E.; Bross, R.A.; Carroll, R.T.; Dunham, W.R.; Funk, M.O., Jr. Three-dimensional structure of a purple lipoxygenase. J. Am. Chem. Soc. 2001, 113, 10814–10820. [Google Scholar] [CrossRef] [PubMed]
- BIOVIA, Dassault Systèmes. Discovery Studio Modeling Environment; Dassault Systèmes: San Diego, CA, USA, 2017. [Google Scholar]
- Ingold, K.U.; Pratt, D.A. Advances in radical-trapping antioxidant chemistry in the 21st century: A kinetics and mechanisms perspective. Chem. Rev. 2014, 114, 9022–9046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tureček, F.; Syrstad, E.A. Mechanism and energetics of intramolecular hydrogen transfer in amide and peptide radicals and cation-radicals. J. Am. Chem. Soc. 2003, 125, 3353–3369. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; O’Malley, P.J.; Popelier, P.L.A. Mechanistic aspects of hydrogen abstraction for phenolic antioxidants. Electronic structure and topological electron density analysis. Phys. Chem. Chem. Phys. 2005, 7, 614–619. [Google Scholar] [CrossRef]
- Galano, A. On the direct scavenging activity of melatonin towards hydroxyl and a series of peroxyl radicals. Phys. Chem. Chem. Phys. 2011, 13, 7178–7188. [Google Scholar] [CrossRef]
- Schröder, D.; Shaik, S.; Schwarz, H. Two-state reactivity as a new concept in organometallic chemistry. Acc. Chem. Res. 2000, 33, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Harvey, J.N. Spin-forbidden reactions: Computational insight into mechanisms and kinetics. WIREs Comput. Mol. Sci. 2014, 4, 1–14. [Google Scholar] [CrossRef]
- Harvey, J.N.; Aschi, M.; Schwarz, H.; Koch, W. The singlet and triplet states of phenyl cation. A hybrid approach for locating minimum energy crossing points between non-interacting potential energy surfaces. Theor. Chem. Acc. 1998, 99, 95–99. [Google Scholar] [CrossRef]
- Harvey, J.N.; Aschi, M. Modelling spin-forbidden reactions: Recombination of carbon monoxide with iron tetracarbonyl. Faraday Discuss. 2003, 124, 129–143. [Google Scholar] [CrossRef]
- Lykhin, A.O.; Kaliakin, D.S.; de Polo, G.E.; Kuzubov, A.A.; Varganov, S.A. Nonadiabatic transition state theory: Application to intersystem crossings in the active sites of metal-sulfur proteins. Int. J. Quantum Chem. 2016, 116, 750–761. [Google Scholar] [CrossRef] [Green Version]
- Tošović, J.; Marković, S. Antioxidative activity of chlorogenic acid relative to trolox in aqueous solution—DFT study. Food Chem. 2019, 278, 469–475. [Google Scholar] [CrossRef] [PubMed]
- Neta, P.; Grodkowski, J. Rate constants for reactions of phenoxyl radicals in solution. J. Phys. Chem. Ref. Data 2005, 34, 109–199. [Google Scholar] [CrossRef] [Green Version]
- Schwartz, M.; Marshall, P.; Berry, R.J.; Ehlers, C.J.; Petersson, G.A. Computational study of the kinetics of hydrogen abstraction from fluoromethanes by the hydroxyl radical. J. Phys. Chem. A 1998, 102, 10074–10081. [Google Scholar] [CrossRef] [Green Version]
- Bors, W.; Heller, W.; Michel, C.; Saran, M. Flavonoids as antioxidants: Determination of radical-scavenging efficiencies. In Methods in Enzymology; Packer, L., Glazer, A.N., Eds.; Academic Press: San Diego, CA, USA, 1990; pp. 343–355. [Google Scholar]
- Casier, B.; Sisourat, N.; Carniato, S.; Capron, N. Keto–enol tautomerism in micro-hydrated acetylacetone: An atoms-in-molecules study. Theor. Chem. Acc. 2018, 138, 107. [Google Scholar] [CrossRef]
- Yamabe, S.; Tsuchida, N.; Miyajima, K. Reaction paths of keto-enol tautomerization of β-diketones. J. Phys. Chem. A 2004, 108, 2750–2757. [Google Scholar] [CrossRef]
- Ohashi, H.; Yamamoto, E.; Lewis, N.G.; Towers, G.H.N. 5-Hydroxyferulic acid in Zea Mays and Hordeum Vulgare cell walls. Phytochemistry 1987, 26, 1915–1916. [Google Scholar]
- Kylli, P.; Nousiainen, P.; Biely, P.; Sipilä, J.; Tenkanen, M. Heinonen, M. Antioxidant potential of hydroxycinnamic acid glycoside esters. J. Agric. Food Chem. 2008, 56, 4797–4805. [Google Scholar] [CrossRef]
- Erdemgil, F.Z.; Şanli, S.; Şanli, N.; Özkan, G.; Barbosa, J.; Guiteras, J.; Beltrán, J.L. Determination of pKa values of some hydroxylated benzoic acids in methanol–water binary mixtures by LC methodology and potentiometry. Talanta 2007, 72, 489–496. [Google Scholar] [CrossRef]
- ACD/Percepta. ACD/Labs Release 2020.2.0. Available online: https://www.acdlabs.com/products/percepta/predictors/pka/ (accessed on 9 May 2021).
- Truong, D.H.; Nhung, N.T.A.; Dao, D.Q. Iron ions chelation-based antioxidant potential vs. pro-oxidant risk of ferulic acid: A DFT study in aqueous phase. Comput. Theor. Chem. 2020, 1185, 112905. [Google Scholar] [CrossRef]
- Holtomo, O.; Nsangou, M.; Fifen, J.J.; Motapon, O. Antioxidative potency and UV–Vis spectra features of the compounds resulting from the chelation of Fe2+ by caffeic acid phenethyl ester and two of its derivatives. Comput. Theor. Chem. 2015, 1067, 135–147. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Path | ΔrG | ΔG≠ | ν | kTST | κEck | kTST/Eck |
|---|---|---|---|---|---|---|
| C1 | −41.5 | 14.1 | −256 | 2.71 × 102 | 1.1 | 2.89 × 102 |
| C3 | −48.2 | 11.1 | −254 | 4.70 × 104 | 1.1 | 5.00 × 104 |
| C5 | −47.6 | 12.9 | −363 | 2.23 × 103 | 1.1 | 2.53 × 103 |
| C8 | −50.4 | 8.6 | −306 | 3.31 × 106 | 0.9 | 2.98 × 106 |
| = | 3.03 × 106 | |||||
| n | ΔrG | ΔG≠ | ν | kTST | κEck | kTST/Eck |
|---|---|---|---|---|---|---|
| 0 | −28.2 | 72.9 | −1551 | 2.3 × 10−41 | 1282.7 | 3.0 × 10−38 |
| 1 | −24.3 | 23.9 | −1384 | 1.9 × 10−5 | 10.5 | 2.0 × 10−4 |
| 2 | −27.1 | 11.8 | −966 | 1.4 × 104 | 2.8 | 4.1 × 104 |
| 3 | −27.2 | 10.1 | −838 | 2.3 × 105 | 2.1 | 4.8 × 105 |
| Conformation | ΔGbind | Ki (µM) | ΔGinter | ΔGvdw+hbond+desolv | ΔGelec | ΔGtotal | ΔGtor | ΔGunb |
|---|---|---|---|---|---|---|---|---|
| LOX-FA | −8.29 | 0.84 | −9.78 | −3.37 | −6.41 | −0.25 | 1.49 | −0.25 |
| LOX-5OHFA | −7.25 | 4.82 | −9.04 | −3.75 | −5.30 | −1.72 | 1.79 | −1.72 |
| 1:1 Fe2+−FA Complexes | 1:1 Fe2+−5OHFA Complexes |
|---|---|
Fe2+−FA−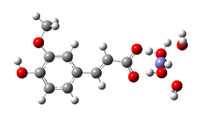 ΔrG = −9.2476 kcal/mol | Fe2+−5OHFA− ΔrG = −9.8757 kcal/mol |
Fe2+−FA2−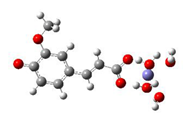 ΔrG = −12.3638 kcal/mol | Fe2+−5OHFA2−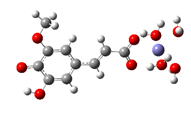 ΔrG = −11.2186 kcal/mol |
Fe2+−FA−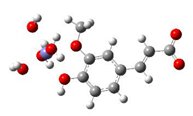 ΔrG = −2.5853 kcal/mol | Fe2+−5OHFA− ΔrG = −1.8091 kcal/mol |
Fe2+−FA2− ΔrG = −16.4640 kcal/mol | Fe2+−FA2− ΔrG = −16.2287 kcal/mol |
| 1:2 Fe2+−FA complexes | 1:2 Fe2+−5OHFA complexes |
Fe2+−(FA−)2 ΔrG = −20.9369 kcal/mol | Fe2+−(5OHFA−)2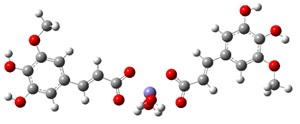 ΔrG = −21.7620 kcal/mol |
Fe2+−(FA2−)2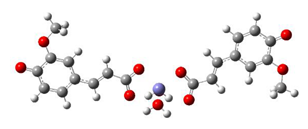 ΔrG = −23.9257 kcal/mol | Fe2+−(5OHFA2−)2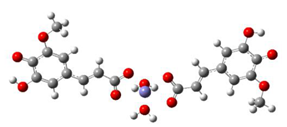 ΔrG = −25.6670 kcal/mol |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amić, A.; Dimitrić Marković, J.M.; Marković, Z.; Milenković, D.; Milanović, Ž.; Antonijević, M.; Mastiľák Cagardová, D.; Rodríguez-Guerra Pedregal, J. Theoretical Study of Radical Inactivation, LOX Inhibition, and Iron Chelation: The Role of Ferulic Acid in Skin Protection against UVA Induced Oxidative Stress. Antioxidants 2021, 10, 1303. https://doi.org/10.3390/antiox10081303
Amić A, Dimitrić Marković JM, Marković Z, Milenković D, Milanović Ž, Antonijević M, Mastiľák Cagardová D, Rodríguez-Guerra Pedregal J. Theoretical Study of Radical Inactivation, LOX Inhibition, and Iron Chelation: The Role of Ferulic Acid in Skin Protection against UVA Induced Oxidative Stress. Antioxidants. 2021; 10(8):1303. https://doi.org/10.3390/antiox10081303
Chicago/Turabian StyleAmić, Ana, Jasmina M. Dimitrić Marković, Zoran Marković, Dejan Milenković, Žiko Milanović, Marko Antonijević, Denisa Mastiľák Cagardová, and Jaime Rodríguez-Guerra Pedregal. 2021. "Theoretical Study of Radical Inactivation, LOX Inhibition, and Iron Chelation: The Role of Ferulic Acid in Skin Protection against UVA Induced Oxidative Stress" Antioxidants 10, no. 8: 1303. https://doi.org/10.3390/antiox10081303