

Antioxidant Supplementation in Oxidative Stress-Related Diseases: What Have We Learned from Studies on Alpha-Tocopherol?
 , , and
, , and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Process of Oxidative Damage in Health and Disease
2.1. Generation of Reactive Oxygen Species (ROS)
2.2. Implications of the ROS Balance in Maintenance of Health and Disease
2.2.1. Physiological Range of ROS: Normal Cellular Functioning
2.2.2. Pushing the Boundaries of the ROS Balance
2.2.3. The Role of Excessive ROS in Cardiovascular Diseases
2.2.4. The Role of Excessive ROS in Neurodegenerative Diseases
2.2.5. The Role of ROS in Cancer Pathogenesis
3. Role of Antioxidants in ROS Elimination
3.1. Working Mechanisms of Antioxidants
3.2. Antioxidant Supplementation in Age-Related Diseases
3.3. Antioxidant Supplementation in α-Tocopherol-Deficiency
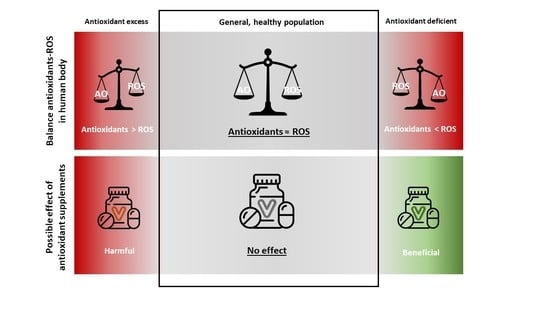
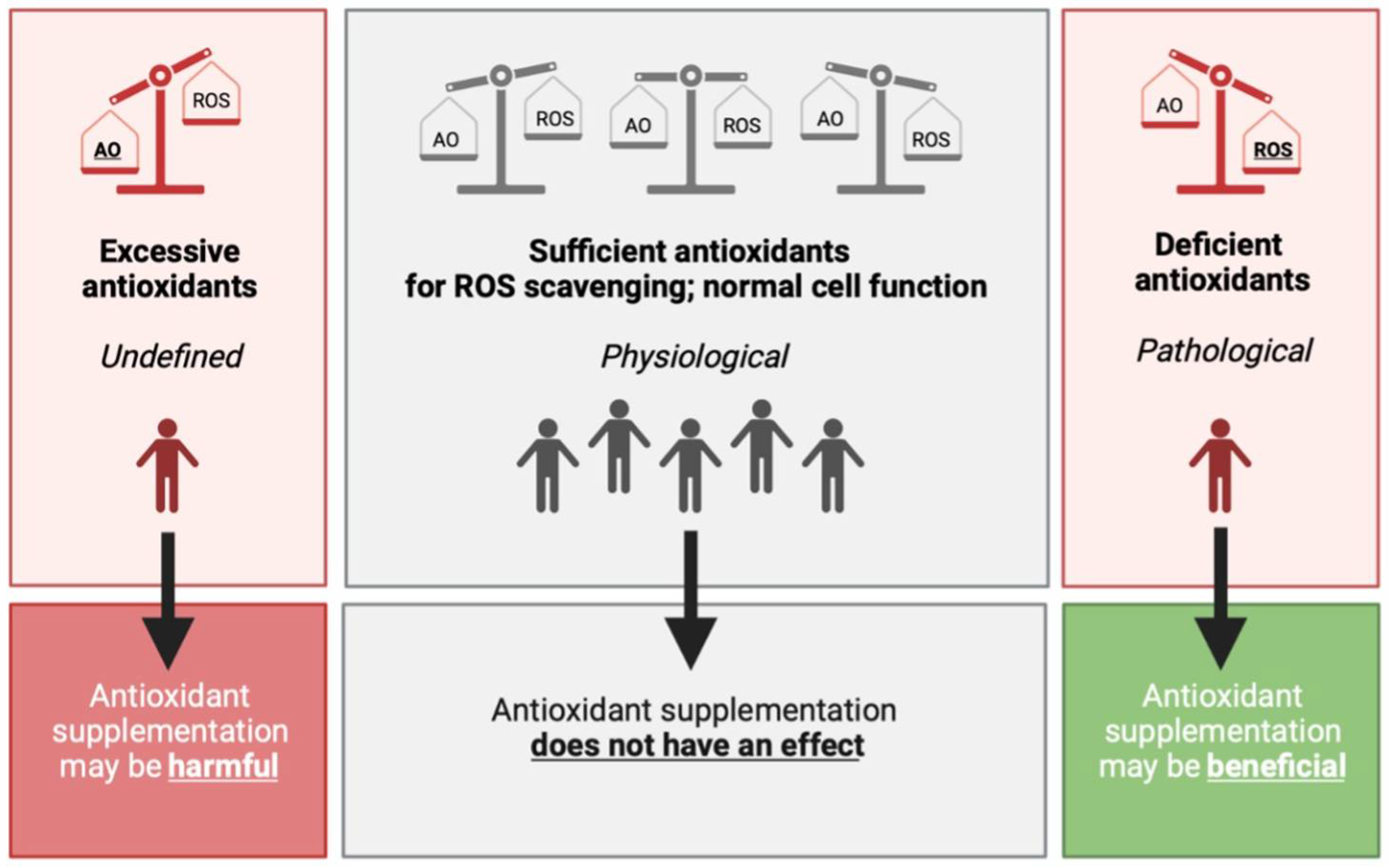
3.4. Antioxidant Supplementation in the General, Healthy Population
3.5. Antioxidant Circulating Levels Versus Antioxidative Capacity
4. Discussion and Concluding Remarks
4.1. Antioxidant Supplements: Is There Really Any Benefit?
4.2. Final Remarks and Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Kiefte-de Jong, J.C.; Mathers, J.C.; Franco, O.H. Nutrition and healthy ageing: The key ingredients. Proc. Nutr. Soc. 2014, 73, 249–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Zhang, Z.; Ren, Y.; Wang, Y.; Fang, J.; Yue, H.; Ma, S.; Guan, F. Aging and age-related diseases: From mechanisms to therapeutic strategies. Biogerontology 2021, 22, 165–187. [Google Scholar] [CrossRef]
- Guarente, L.; Kenyon, C. Genetic pathways that regulate ageing in model organisms. Nature 2000, 408, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Melzer, D.; Pilling, L.C.; Ferrucci, L. The genetics of human ageing. Nat. Rev. Genet. 2020, 21, 88–101. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, Y.; Liu, C.; Lu, M.; Dong, Q.; Wang, Z.; Wang, Z.; Xiong, W.; Zhang, N.; Zhou, J.; Liu, Q.; et al. Calorie restriction is the most reasonable anti-ageing intervention: A meta-analysis of survival curves. Sci. Rep. 2018, 8, 5779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erbaba, B.; Arslan-Ergul, A.; Adams, M.M. Effects of caloric restriction on the antagonistic and integrative hallmarks of aging. Ageing Res. Rev. 2021, 66, 101228. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef]
- Phaniendra, A.; Jestadi, D.B.; Periyasamy, L. Free radicals: Properties, sources, targets, and their implication in various diseases. Indian J. Clin. Biochem. 2015, 30, 11–26. [Google Scholar] [CrossRef]
- Sies, H. Oxidative stress: A concept in redox biology and medicine. Redox Biol. 2015, 4, 180–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizvi, S.; Raza, S.T.; Ahmed, F.; Ahmad, A.; Abbas, S.; Mahdi, F. The role of vitamin e in human health and some diseases. Sultan Qaboos Univ. Med. J. 2014, 14, e157–e165. [Google Scholar] [PubMed]
- Wright, M.E.; Lawson, K.A.; Weinstein, S.J.; Pietinen, P.; Taylor, P.R.; Virtamo, J.; Albanes, D. Higher baseline serum concentrations of vitamin E are associated with lower total and cause-specific mortality in the Alpha-Tocopherol, Beta-Carotene Cancer Prevention Study. Am. J. Clin. Nutr. 2006, 84, 1200–1207. [Google Scholar] [CrossRef] [PubMed]
- Stolzenberg-Solomon, R.Z.; Sheffler-Collins, S.; Weinstein, S.; Garabrant, D.H.; Mannisto, S.; Taylor, P.; Virtamo, J.; Albanes, D. Vitamin E intake, alpha-tocopherol status, and pancreatic cancer in a cohort of male smokers. Am. J. Clin. Nutr. 2009, 89, 584–591. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Weinstein, S.J.; Yu, K.; Männistö, S.; Albanes, D. Relationship Between Serum Alpha-Tocopherol and Overall and Cause-Specific Mortality. Circ. Res. 2019, 125, 29–40. [Google Scholar] [CrossRef]
- Wang, L.; Sesso, H.D.; Glynn, R.J.; Christen, W.G.; Bubes, V.; Manson, J.E.; Buring, J.E.; Gaziano, J.M. Vitamin E and C supplementation and risk of cancer in men: Posttrial follow-up in the Physicians’ Health Study II randomized trial. Am. J. Clin. Nutr. 2014, 100, 915–923. [Google Scholar] [CrossRef] [Green Version]
- Sesso, H.D.; Buring, J.E.; Christen, W.G.; Kurth, T.; Belanger, C.; MacFadyen, J.; Bubes, V.; Manson, J.E.; Glynn, R.J.; Gaziano, J.M. Vitamins E and C in the prevention of cardiovascular disease in men: The Physicians’ Health Study II randomized controlled trial. JAMA 2008, 300, 2123–2133. [Google Scholar] [CrossRef] [Green Version]
- Lee, I.M.; Cook, N.R.; Gaziano, J.M.; Gordon, D.; Ridker, P.M.; Manson, J.E.; Hennekens, C.H.; Buring, J.E. Vitamin E in the primary prevention of cardiovascular disease and cancer: The Women’s Health Study: A randomized controlled trial. JAMA 2005, 294, 56–65. [Google Scholar] [CrossRef]
- Myung, S.K.; Ju, W.; Cho, B.; Oh, S.W.; Park, S.M.; Koo, B.K.; Park, B.; Korean Meta-Analysis Study Group. Efficacy of vitamin and antioxidant supplements in prevention of cardiovascular disease: Systematic review and meta-analysis of randomised controlled trials. Bmj 2013, 346, f10. [Google Scholar] [CrossRef] [Green Version]
- Di Meo, S.; Reed, T.T.; Venditti, P.; Victor, V.M. Role of ROS and RNS Sources in Physiological and Pathological Conditions. Oxid. Med. Cell. Longev. 2016, 2016, 1245049. [Google Scholar] [CrossRef]
- Kalyanaraman, B.; Cheng, G.; Hardy, M.; Ouari, O.; Bennett, B.; Zielonka, J. Teaching the basics of reactive oxygen species and their relevance to cancer biology: Mitochondrial reactive oxygen species detection, redox signaling, and targeted therapies. Redox Biol. 2018, 15, 347–362. [Google Scholar] [CrossRef]
- Luo, J.; Mills, K.; le Cessie, S.; Noordam, R.; van Heemst, D. Ageing, age-related diseases and oxidative stress: What to do next? Ageing Res. Rev. 2020, 57, 100982. [Google Scholar] [CrossRef] [PubMed]
- Higdon, A.; Diers, A.R.; Oh, J.Y.; Landar, A.; Darley-Usmar, V.M. Cell signalling by reactive lipid species: New concepts and molecular mechanisms. Biochem. J. 2012, 442, 453–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prins, J.R.; Schoots, M.H.; Wessels, J.I.; Campmans-Kuijpers, M.J.E.; Navis, G.J.; van Goor, H.; Robertson, S.A.; van der Beek, E.N.; Sobrevia, L.; Gordijn, S.J. The influence of the dietary exposome on oxidative stress in pregnancy complications. Mol. Aspects Med. 2022, 87, 101098. [Google Scholar] [CrossRef]
- Galaris, D.; Barbouti, A.; Pantopoulos, K. Iron homeostasis and oxidative stress: An intimate relationship. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 118535. [Google Scholar] [CrossRef] [PubMed]
- Bartosz, G. Reactive oxygen species: Destroyers or messengers? Biochem. Pharmacol. 2009, 77, 1303–1315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrer-Sueta, G.; Campolo, N.; Trujillo, M.; Bartesaghi, S.; Carballal, S.; Romero, N.; Alvarez, B.; Radi, R. Biochemistry of Peroxynitrite and Protein Tyrosine Nitration. Chem. Rev. 2018, 118, 1338–1408. [Google Scholar] [CrossRef] [PubMed]
- Torrens-Mas, M.; Pons, D.G.; Sastre-Serra, J.; Oliver, J.; Roca, P. Sexual hormones regulate the redox status and mitochondrial function in the brain. Pathological implications. Redox Biol. 2020, 31, 101505. [Google Scholar] [CrossRef]
- Dennis, K.K.; Go, Y.M.; Jones, D.P. Redox Systems Biology of Nutrition and Oxidative Stress. J. Nutr. 2019, 149, 553–565. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, X.; Vikash, V.; Ye, Q.; Wu, D.; Liu, Y.; Dong, W. ROS and ROS-Mediated Cellular Signaling. Oxid. Med. Cell. Longev. 2016, 2016, 4350965. [Google Scholar] [CrossRef]
- Cross, J.V.; Templeton, D.J. Thiol oxidation of cell signaling proteins: Controlling an apoptotic equilibrium. J. Cell. Biochem. 2004, 93, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Reynaert, N.L.; van der Vliet, A.; Guala, A.S.; McGovern, T.; Hristova, M.; Pantano, C.; Heintz, N.H.; Heim, J.; Ho, Y.; Matthews, D.E. Dynamic redox control of NF-kappaB through glutaredoxin-regulated S-glutathionylation of inhibitory kappaB kinase beta. Proc. Natl. Acad. Sci. USA 2006, 103, 13086–13091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Courtois, G.; Fauvarque, M.O. The Many Roles of Ubiquitin in NF-κB Signaling. Biomedicines 2018, 6, 43. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Liu, H.T. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002, 12, 9–18. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, K.; Shi, Y.; Shao, C. The tango of ROS and p53 in tissue stem cells. Cell Death Differ. 2018, 25, 639–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ristow, M.; Schmeisser, S. Extending life span by increasing oxidative stress. Free Radic. Biol. Med. 2011, 51, 327–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brieger, K.; Schiavone, S.; Miller, F.J., Jr.; Krause, K.H. Reactive oxygen species: From health to disease. Swiss Med. Wkly. 2012, 142, w13659. [Google Scholar] [CrossRef]
- Urbonaviciute, V.; Luo, H.; Sjöwall, C.; Bengtsson, A.; Holmdahl, R. Low Production of Reactive Oxygen Species Drives Systemic Lupus Erythematosus. Trends Mol. Med. 2019, 25, 826–835. [Google Scholar] [CrossRef]
- O’Neill, S.; Brault, J.; Stasia, M.J.; Knaus, U.G. Genetic disorders coupled to ROS deficiency. Redox Biol. 2015, 6, 135–156. [Google Scholar] [CrossRef] [Green Version]
- Aggarwal, V.; Tuli, H.S.; Varol, A.; Thakral, F.; Yerer, M.B.; Sak, K.; Varol, M.; Jain, A.; Khan, M.A.; Sethi, G. Role of Reactive Oxygen Species in Cancer Progression: Molecular Mechanisms and Recent Advancements. Biomolecules 2019, 9, 735. [Google Scholar] [CrossRef]
- Yang, S.; Lian, G. ROS and diseases: Role in metabolism and energy supply. Mol. Cell. Biochem. 2020, 467, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Ago, T.; Liu, T.; Zhai, P.; Chen, W.; Li, H.; Molkentin, J.D.; Vatner, S.F.; Sadoshima, J. A redox-dependent pathway for regulating class II HDACs and cardiac hypertrophy. Cell 2008, 133, 978–993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gniwotta, C.; Morrow, J.D.; Roberts, L.J., 2nd; Kühn, H. Prostaglandin F2-like compounds, F2-isoprostanes, are present in increased amounts in human atherosclerotic lesions. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 3236–3241. [Google Scholar] [CrossRef]
- Dai, D.F.; Johnson, S.C.; Villarin, J.J.; Chin, M.T.; Nieves-Cintrón, M.; Chen, T.; Marcinek, D.J.; Dorn, G.W., 2nd; Kang, Y.J.; Prolla, T.A.; et al. Mitochondrial oxidative stress mediates angiotensin II-induced cardiac hypertrophy and Galphaq overexpression-induced heart failure. Circ. Res. 2011, 108, 837–846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.; Prosser, B.L.; Bamboye, M.A.; Gondim, A.N.S.; Santos, C.X.; Martin, D.; Ghigo, A.; Perino, A.; Brewer, A.C.; Ward, C.W.; et al. Contractile Function During Angiotensin-II Activation: Increased Nox2 Activity Modulates Cardiac Calcium Handling via Phospholamban Phosphorylation. J. Am. Coll. Cardiol. 2015, 66, 261–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cervantes Gracia, K.; Llanas-Cornejo, D.; Husi, H. CVD and Oxidative Stress. J. Clin. Med. 2017, 6, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayala, A.; Muñoz, M.F.; Argüelles, S. Lipid Peroxidation: Production, Metabolism, and Signaling Mechanisms of Malondialdehyde and 4-Hydroxy-2-Nonenal. Oxidative Med. Cell. Longev. 2014, 2014, 360438. [Google Scholar] [CrossRef] [Green Version]
- Csala, M.; Kardon, T.; Legeza, B.; Lizák, B.; Mandl, J.; Margittai, É.; Puskás, F.; Száraz, P.; Szelényi, P.; Bánhegyi, G. On the role of 4-hydroxynonenal in health and disease. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2015, 1852, 826–838. [Google Scholar] [CrossRef] [Green Version]
- Stocker, R.; Keaney, J.F., Jr. Role of oxidative modifications in atherosclerosis. Physiol. Rev. 2004, 84, 1381–1478. [Google Scholar] [CrossRef]
- Kattoor, A.J.; Pothineni, N.V.K.; Palagiri, D.; Mehta, J.L. Oxidative Stress in Atherosclerosis. Curr. Atheroscler. Rep. 2017, 19, 42. [Google Scholar] [CrossRef]
- Girotti, A.W.; Korytowski, W. Cholesterol Peroxidation as a Special Type of Lipid Oxidation in Photodynamic Systems. Photochem. Photobiol. 2019, 95, 73–82. [Google Scholar] [CrossRef] [Green Version]
- Olkkonen, V.M.; Béaslas, O.; Nissilä, E. Oxysterols and their cellular effectors. Biomolecules 2012, 2, 76–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poli, G.; Biasi, F.; Leonarduzzi, G. Oxysterols in the pathogenesis of major chronic diseases. Redox Biol. 2013, 1, 125–130. [Google Scholar] [CrossRef] [Green Version]
- De Medina, P.; Silvente-Poirot, S.; Poirot, M. Oxysterols are potential physiological regulators of ageing. Ageing Res. Rev. 2022, 77, 101615. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Noordam, R.; Jukema, J.W.; van Dijk, K.W.; Hägg, S.; Grassmann, F.; le Cessie, S.; van Heemst, D. Low mitochondrial copy number drives atherogenic cardiovascular disease: Evidence from prospective cohort analyses in the UK Biobank combined with Mendelian Randomization. medRxiv 2021. [Google Scholar] [CrossRef]
- Kim, G.H.; Kim, J.E.; Rhie, S.J.; Yoon, S. The Role of Oxidative Stress in Neurodegenerative Diseases. Exp. Neurobiol. 2015, 24, 325–340. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.H.; Cha, M.; Lee, B.H. Neuroprotective Effect of Antioxidants in the Brain. Int. J. Mol. Sci. 2020, 21, 7152. [Google Scholar] [CrossRef]
- Wang, X.; Wang, W.; Li, L.; Perry, G.; Lee, H.G.; Zhu, X. Oxidative stress and mitochondrial dysfunction in Alzheimer’s disease. Biochim. Biophys. Acta 2014, 1842, 1240–1247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilkins, H.M.; Swerdlow, R.H. Mitochondrial links between brain aging and Alzheimer’s disease. Transl. Neurodegener. 2021, 10, 33. [Google Scholar] [CrossRef]
- González-Rodríguez, P.; Zampese, E.; Stout, K.A.; Guzman, J.N.; Ilijic, E.; Yang, B.; Tkatch, T.; Stavarache, M.A.; Wokosin, D.L.; Gao, L.; et al. Disruption of mitochondrial complex I induces progressive parkinsonism. Nature 2021, 599, 650–656. [Google Scholar] [CrossRef] [PubMed]
- Moloney, J.N.; Cotter, T.G. ROS signalling in the biology of cancer. Semin. Cell Dev. Biol. 2018, 80, 50–64. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kim, J.; Bae, J.S. ROS homeostasis and metabolism: A critical liaison for cancer therapy. Exp. Mol. Med. 2016, 48, e269. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Villani, R.M.; Wang, H.; Simpson, M.J.; Roberts, M.S.; Tang, M.; Liang, X. The role of cellular reactive oxygen species in cancer chemotherapy. J. Exp. Clin. Cancer Res. 2018, 37, 266. [Google Scholar] [CrossRef] [Green Version]
- Assi, M. The differential role of reactive oxygen species in early and late stages of cancer. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2017, 313, R646–R653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenwood, H.E.; McCormick, P.N.; Gendron, T.; Glaser, M.; Pereira, R.; Maddocks, O.D.K.; Sander, K.; Zhang, T.; Koglin, N.; Lythgoe, M.F.; et al. Measurement of Tumor Antioxidant Capacity and Prediction of Chemotherapy Resistance in Preclinical Models of Ovarian Cancer by Positron Emission Tomography. Clin. Cancer Res. 2019, 25, 2471–2482. [Google Scholar] [CrossRef] [Green Version]
- Allen, B.G.; Bodeker, K.L.; Smith, M.C.; Monga, V.; Sandhu, S.; Hohl, R.; Carlisle, T.; Brown, H.; Hollenbeck, N.; Vollstedt, S.; et al. First-in-Human Phase I Clinical Trial of Pharmacologic Ascorbate Combined with Radiation and Temozolomide for Newly Diagnosed Glioblastoma. Clin. Cancer Res. 2019, 25, 6590–6597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoenfeld, J.D.; Alexander, M.S.; Waldron, T.J.; Sibenaller, Z.A.; Spitz, D.R.; Buettner, G.R.; Allen, B.G.; Cullen, J.J. Pharmacological Ascorbate as a Means of Sensitizing Cancer Cells to Radio-Chemotherapy While Protecting Normal Tissue. Semin. Radiat. Oncol. 2019, 29, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Flieger, J.; Flieger, W.; Baj, J.; Maciejewski, R. Antioxidants: Classification, Natural Sources, Activity/Capacity Measurements, and Usefulness for the Synthesis of Nanoparticles. Materials 2021, 14, 4135. [Google Scholar] [CrossRef]
- Niki, E. Role of vitamin E as a lipid-soluble peroxyl radical scavenger: In vitro and in vivo evidence. Free Radic. Biol. Med. 2014, 66, 3–12. [Google Scholar] [CrossRef]
- Ricciarelli, R.; Zingg, J.M.; Azzi, A. The 80th anniversary of vitamin E: Beyond its antioxidant properties. Biol. Chem. 2002, 383, 457–465. [Google Scholar] [CrossRef]
- Ighodaro, O.M.; Akinloye, O.A. First line defence antioxidants-superoxide dismutase (SOD), catalase (CAT) and glutathione peroxidase (GPX): Their fundamental role in the entire antioxidant defence grid. Alex. J. Med. 2018, 54, 287–293. [Google Scholar] [CrossRef] [Green Version]
- Lubos, E.; Loscalzo, J.; Handy, D.E. Glutathione peroxidase-1 in health and disease: From molecular mechanisms to therapeutic opportunities. Antioxid. Redox Signal. 2011, 15, 1957–1997. [Google Scholar] [CrossRef] [Green Version]
- Tatone, C.; Carbone, M.C.; Falone, S.; Aimola, P.; Giardinelli, A.; Caserta, D.; Marci, R.; Pandolfi, A.; Ragnelli, A.M.; Amicarelli, F. Age-dependent changes in the expression of superoxide dismutases and catalase are associated with ultrastructural modifications in human granulosa cells. Mol. Hum. Reprod. 2006, 12, 655–660. [Google Scholar] [CrossRef] [Green Version]
- Kozakiewicz, M.; Kornatowski, M.; Krzywińska, O.; Kędziora-Kornatowska, K. Changes in the blood antioxidant defense of advanced age people. Clin. Interv. Aging 2019, 14, 763–771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siti, H.N.; Kamisah, Y.; Kamsiah, J. The role of oxidative stress, antioxidants and vascular inflammation in cardiovascular disease (a review). Vasc. Pharmacol. 2015, 71, 40–56. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.; Song, H. Antioxidant vitamins intake and the risk of coronary heart disease: Meta-analysis of cohort studies. Eur. J. Cardiovasc. Prev. Rehabil. 2008, 15, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Kubota, Y.; Iso, H.; Date, C.; Kikuchi, S.; Watanabe, Y.; Wada, Y.; Inaba, Y.; Tamakoshi, A.; JACC Study Group. Dietary intakes of antioxidant vitamins and mortality from cardiovascular disease: The Japan Collaborative Cohort Study (JACC) study. Stroke 2011, 42, 1665–1672. [Google Scholar] [CrossRef] [PubMed]
- Engelhart, M.J.; Geerlings, M.I.; Ruitenberg, A.; van Swieten, J.C.; Hofman, A.; Witteman, J.C.; Breteler, M.M.B. Dietary intake of antioxidants and risk of Alzheimer disease. JAMA 2002, 287, 3223–3229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zandi, P.P.; Anthony, J.C.; Khachaturian, A.S.; Stone, S.V.; Gustafson, D.; Tschanz, J.T.; Norton, M.C.; Welsh-Bohmer, K.A.; Breitner, J.C.; Cache County Study Group. Reduced risk of Alzheimer disease in users of antioxidant vitamin supplements: The Cache County Study. Arch. Neurol. 2004, 61, 82–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Etminan, M.; Gill, S.S.; Samii, A. Intake of vitamin E, vitamin C, and carotenoids and the risk of Parkinson’s disease: A meta-analysis. Lancet Neurol. 2005, 4, 362–365. [Google Scholar] [CrossRef]
- Wang, H.; O’Reilly, É.J.; Weisskopf, M.G.; Logroscino, G.; McCullough, M.L.; Schatzkin, A.; Kolonel, L.N.; Ascherio, A. Vitamin E intake and risk of amyotrophic lateral sclerosis: A pooled analysis of data from 5 prospective cohort studies. Am. J. Epidemiol. 2011, 173, 595–602. [Google Scholar] [CrossRef] [PubMed]
- Veldink, J.H.; Kalmijn, S.; Groeneveld, G.J.; Wunderink, W.; Koster, A.; de Vries, J.H.; van der Luyt, J.; Wokke, J.H.J.; van den Berg, L.H. Intake of polyunsaturated fatty acids and vitamin E reduces the risk of developing amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 2007, 78, 367–371. [Google Scholar] [CrossRef] [Green Version]
- Gaziano, J.M.; Glynn, R.J.; Christen, W.G.; Kurth, T.; Belanger, C.; MacFadyen, J.; Bubes, V.; Manson, J.E.; Sesso, H.D.; Buring, J.E. Vitamins E and C in the prevention of prostate and total cancer in men: The Physicians’ Health Study II randomized controlled trial. JAMA 2009, 301, 52–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, J.; le Cessie, S.; van Heemst, D.; Noordam, R. Diet-Derived Circulating Antioxidants and Risk of Coronary Heart Disease: A Mendelian Randomization Study. J. Am. Coll. Cardiol. 2021, 77, 45–54. [Google Scholar] [CrossRef]
- Martens, L.G.; Luo, J.; Dijk, K.W.V.; Jukema, J.W.; Noordam, R.; Heemst, D.V. Diet-derived antioxidants do not decrease the risk of ischemic stroke: A Mendelian Randomization Study in over 1 million participants. medRxiv 2021. [Google Scholar]
- Williams, D.M.; Hägg, S.; Pedersen, N.L. Circulating antioxidants and Alzheimer disease prevention: A Mendelian randomization study. Am. J. Clin. Nutr. 2019, 109, 90–98. [Google Scholar] [CrossRef] [Green Version]
- Khadangi, F.; Azzi, A. Vitamin E—The Next 100 Years. IUBMB Life 2019, 71, 411–415. [Google Scholar] [CrossRef] [PubMed]
- Gabsi, S.; Gouider-Khouja, N.; Belal, S.; Fki, M.; Kefi, M.; Turki, I.; Hamida, M.B.; Kayden, H.; Mebazaa, R.; Hentati, F. Effect of vitamin E supplementation in patients with ataxia with vitamin E deficiency. Eur. J. Neurol. 2001, 8, 477–481. [Google Scholar] [CrossRef]
- Ouahchi, K.; Arita, M.; Kayden, H.; Hentati, F.; Ben Hamida, M.; Sokol, R.; Arai, H.; Inoue, K.; Mandel, J.L.; Koenig, M. Ataxia with isolated vitamin E deficiency is caused by mutations in the alpha-tocopherol transfer protein. Nat. Genet. 1995, 9, 141–145. [Google Scholar] [CrossRef]
- Maxfield, L.; Crane, J.S. Vitamin C Deficiency; StatPearls Publishing: Treasure Island, FL, USA, 2022. Available online: https://www.ncbi.nlm.nih.gov/books/NBK493187/ (accessed on 1 September 2021).
- De Mutsert, R.; den Heijer, M.; Rabelink, T.J.; Smit, J.W.; Romijn, J.A.; Jukema, J.W.; de Roos, A.; Cobbaert, C.M.; Kloppenburg, M.; le Cessie, S.; et al. The Netherlands Epidemiology of Obesity (NEO) study: Study design and data collection. Eur. J. Epidemiol. 2013, 28, 513–523. [Google Scholar] [CrossRef]
- Martens, L.G.; Luo, J.; Meulmeester, F.L.; Ashrafi, N.; van Eekelen, E.W.; de Mutsert, R.; Mook-Kanamori, D.O.; Rosendaal, F.R.; van Dijk, K.W.; Mills, K.; et al. Associations between Lifestyle Factors and Vitamin E Metabolites in the General Population. Antioxidants 2020, 9, 1280. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Meulmeester, F.L.; Martens, L.G.; Ashrafi, N.; de Mutsert, R.; Mook-Kanamori, D.O.; Rosendaal, F.R.; Willems van Dijk, K.; le Cessie, S.; Mills, K.; et al. Urinary oxidized, but not enzymatic vitamin E metabolites are inversely associated with measures of glucose homeostasis in middle-aged healthy individuals. Clin. Nutr. 2021, 40, 4192–4200. [Google Scholar] [CrossRef]
- Meulmeester, F.L.; Luo, J.; Martens, L.G.; Ashrafi, N.; de Mutsert, R.; Mook-Kanamori, D.O.; Lamb, H.J.; Rosendaal, F.R.; Willems van Dijk, K.; Mills, K.; et al. Association of measures of body fat with serum alpha-tocopherol and its metabolites in middle-aged individuals. Nutr. Metab. Cardiovasc. Dis. 2021, 31, 2407–2415. [Google Scholar] [CrossRef]
- Luo, J.; Hashimoto, Y.; Martens, L.G.; Meulmeester, F.L.; Ashrafi, N.; Mook-Kanamori, D.O.; Rosendaal, F.R.; Jukema, J.W.; van Dijk, K.W.; van Heemst, D.; et al. Associations of metabolomic profiles with circulating vitamin E and urinary vitamin E metabolites in middle-aged individuals. Nutrition 2022, 93, 111440. [Google Scholar] [CrossRef] [PubMed]
- Lodge, J.K. Vitamin E bioavailability in humans. J. Plant Physiol. 2005, 162, 790–796. [Google Scholar] [CrossRef] [PubMed]
- Rubio, C.P.; Hernández-Ruiz, J.; Martinez-Subiela, S.; Tvarijonaviciute, A.; Ceron, J.J. Spectrophotometric assays for total antioxidant capacity (TAC) in dog serum: An update. BMC Vet. Res. 2016, 12, 166. [Google Scholar] [CrossRef] [Green Version]
- Schmölz, L.; Birringer, M.; Lorkowski, S.; Wallert, M. Complexity of vitamin E metabolism. World J. Biol. Chem. 2016, 7, 14–43. [Google Scholar] [CrossRef]
- Sies, H.; Berndt, C.; Jones, D.P. Oxidative Stress. Annu. Rev. Biochem. 2017, 86, 715–748. [Google Scholar] [CrossRef]
- Putchala, M.C.; Ramani, P.; Sherlin, H.J.; Premkumar, P.; Natesan, A. Ascorbic acid and its pro-oxidant activity as a therapy for tumours of oral cavity—A systematic review. Arch. Oral Biol. 2013, 58, 563–574. [Google Scholar] [CrossRef]
- Pearson, P.; Lewis, S.A.; Britton, J.; Young, I.S.; Fogarty, A. The pro-oxidant activity of high-dose vitamin E supplements in vivo. BioDrugs 2006, 20, 271–273. [Google Scholar] [CrossRef]
- Traber, M.G.; Stevens, J.F. Vitamins C and E: Beneficial effects from a mechanistic perspective. Free Radic. Biol. Med. 2011, 51, 1000–1013. [Google Scholar] [CrossRef] [PubMed]
- Mesdaghinia, E.; Mohammad-Ebrahimi, B.; Foroozanfard, F.; Banafshe, H.R. The effect of vitamin E and aspirin on the uterine artery blood flow in women with recurrent abortion: A single-blind randomized controlled trial. Int. J. Reprod. Biomed. 2017, 15, 635–640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katerji, M.; Filippova, M.; Duerksen-Hughes, P. Approaches and Methods to Measure Oxidative Stress in Clinical Samples: Research Applications in the Cancer Field. Oxid. Med. Cell. Longev. 2019, 2019, 1279250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frei, B. Efficacy of Dietary Antioxidants to Prevent Oxidative Damage and Inhibit Chronic Disease. J. Nutr. 2004, 134, 3196S–3198S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, C.; Ding, P.; Xie, C.; Ye, C.; Ye, M.; Pan, C.; Cao, X.; Zhang, S.; Zheng, S. Potential application of the oxidative nucleic acid damage biomarkers in detection of diseases. Oncotarget 2017, 8, 75767–75777. [Google Scholar] [CrossRef] [Green Version]
- Dalle-Donne, I.; Rossi, R.; Giustarini, D.; Milzani, A.; Colombo, R. Protein carbonyl groups as biomarkers of oxidative stress. Clin. Chim. Acta 2003, 329, 23–38. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meulmeester, F.L.; Luo, J.; Martens, L.G.; Mills, K.; van Heemst, D.; Noordam, R. Antioxidant Supplementation in Oxidative Stress-Related Diseases: What Have We Learned from Studies on Alpha-Tocopherol? Antioxidants 2022, 11, 2322. https://doi.org/10.3390/antiox11122322
Meulmeester FL, Luo J, Martens LG, Mills K, van Heemst D, Noordam R. Antioxidant Supplementation in Oxidative Stress-Related Diseases: What Have We Learned from Studies on Alpha-Tocopherol? Antioxidants. 2022; 11(12):2322. https://doi.org/10.3390/antiox11122322
Chicago/Turabian StyleMeulmeester, Fleur L., Jiao Luo, Leon G. Martens, Kevin Mills, Diana van Heemst, and Raymond Noordam. 2022. "Antioxidant Supplementation in Oxidative Stress-Related Diseases: What Have We Learned from Studies on Alpha-Tocopherol?" Antioxidants 11, no. 12: 2322. https://doi.org/10.3390/antiox11122322