
Inhibition of Cell Proliferation and Cell Viability by Sinecatechins in Cutaneous SCC Cells Is Related to an Imbalance of ROS and Loss of Mitochondrial Membrane Potential


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells and Treatment
2.2. Cell Proliferation
2.3. Apoptosis Induction and Cell Viability
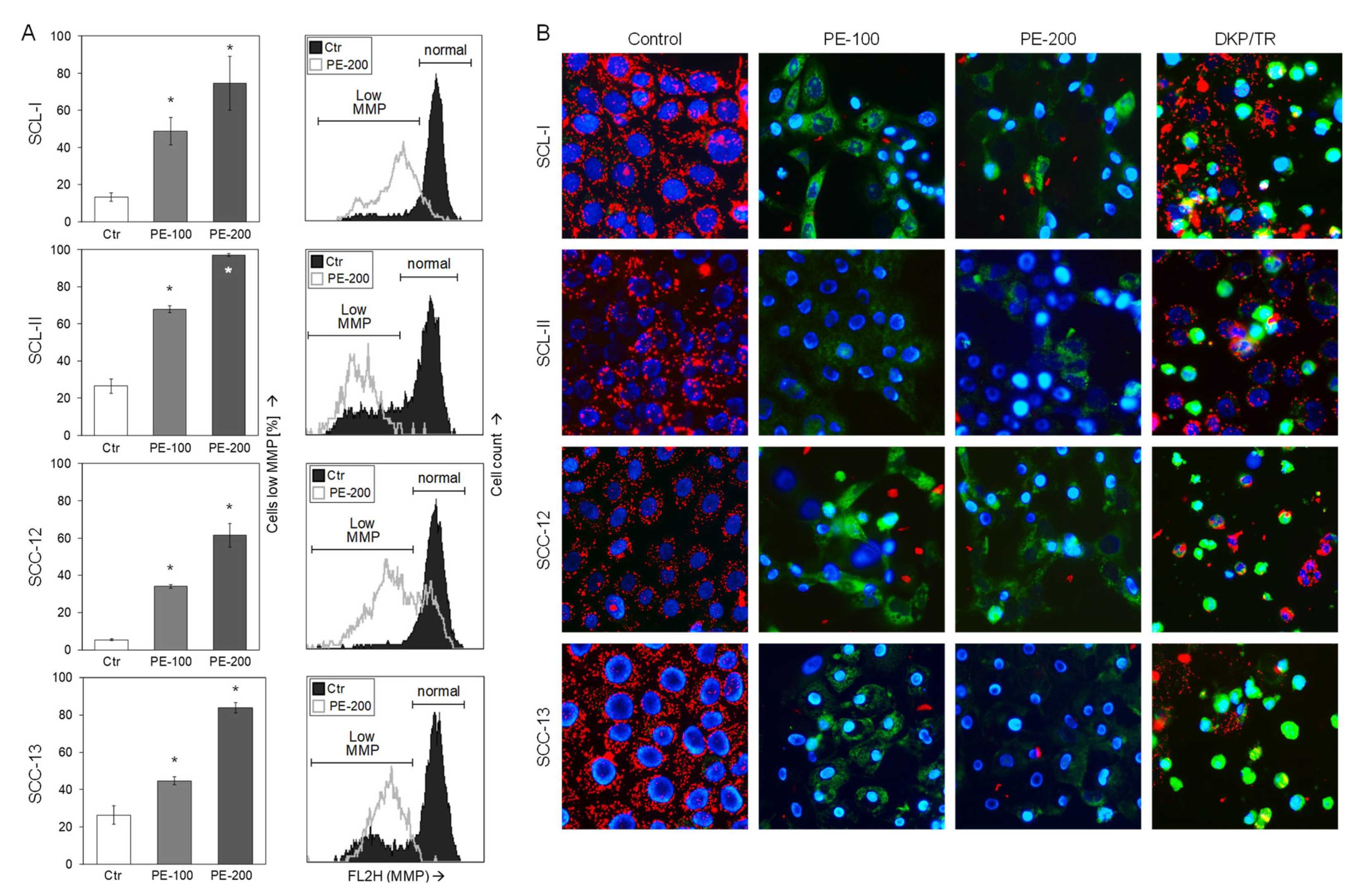
2.4. Mitochondrial Membrane Potential
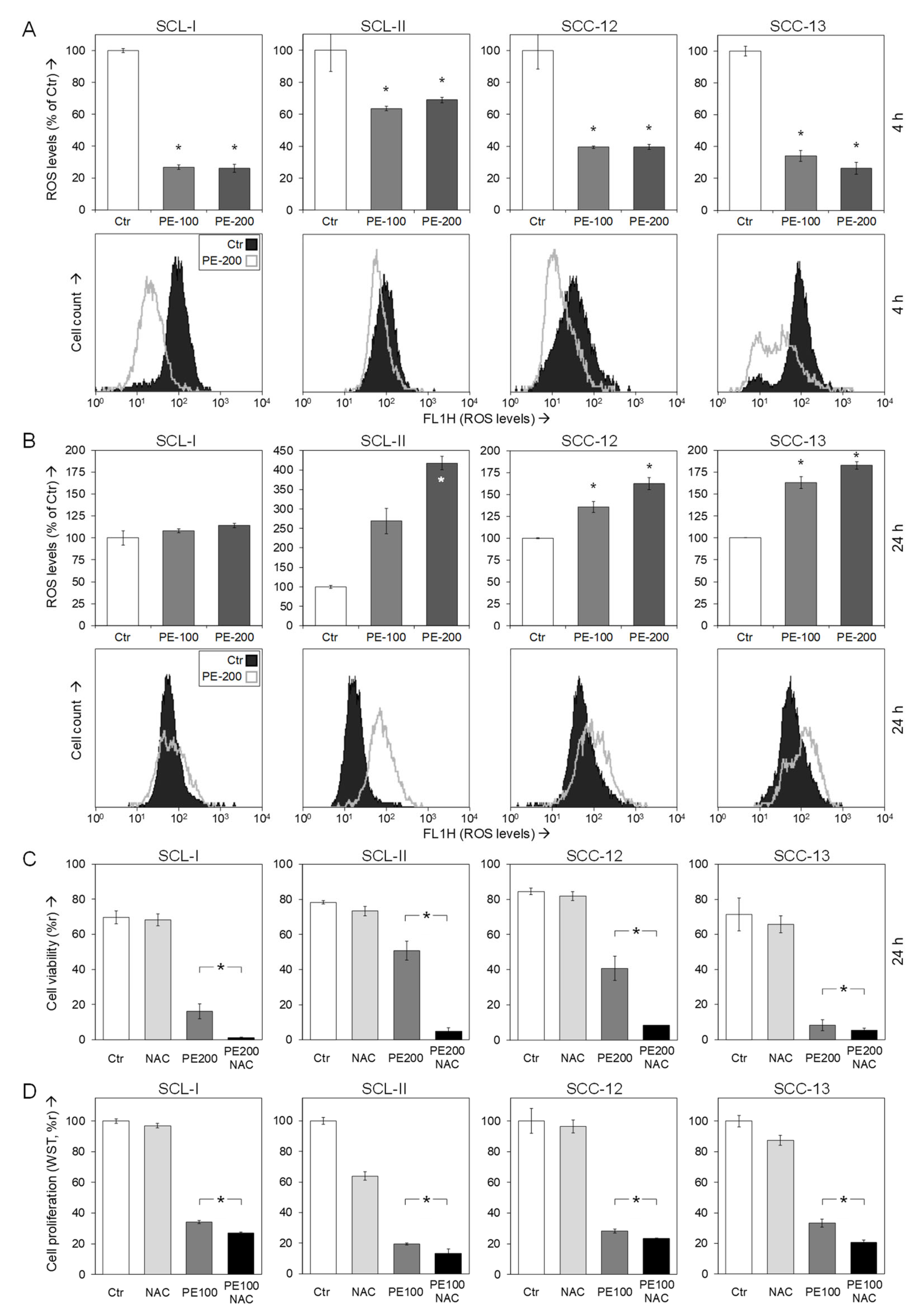
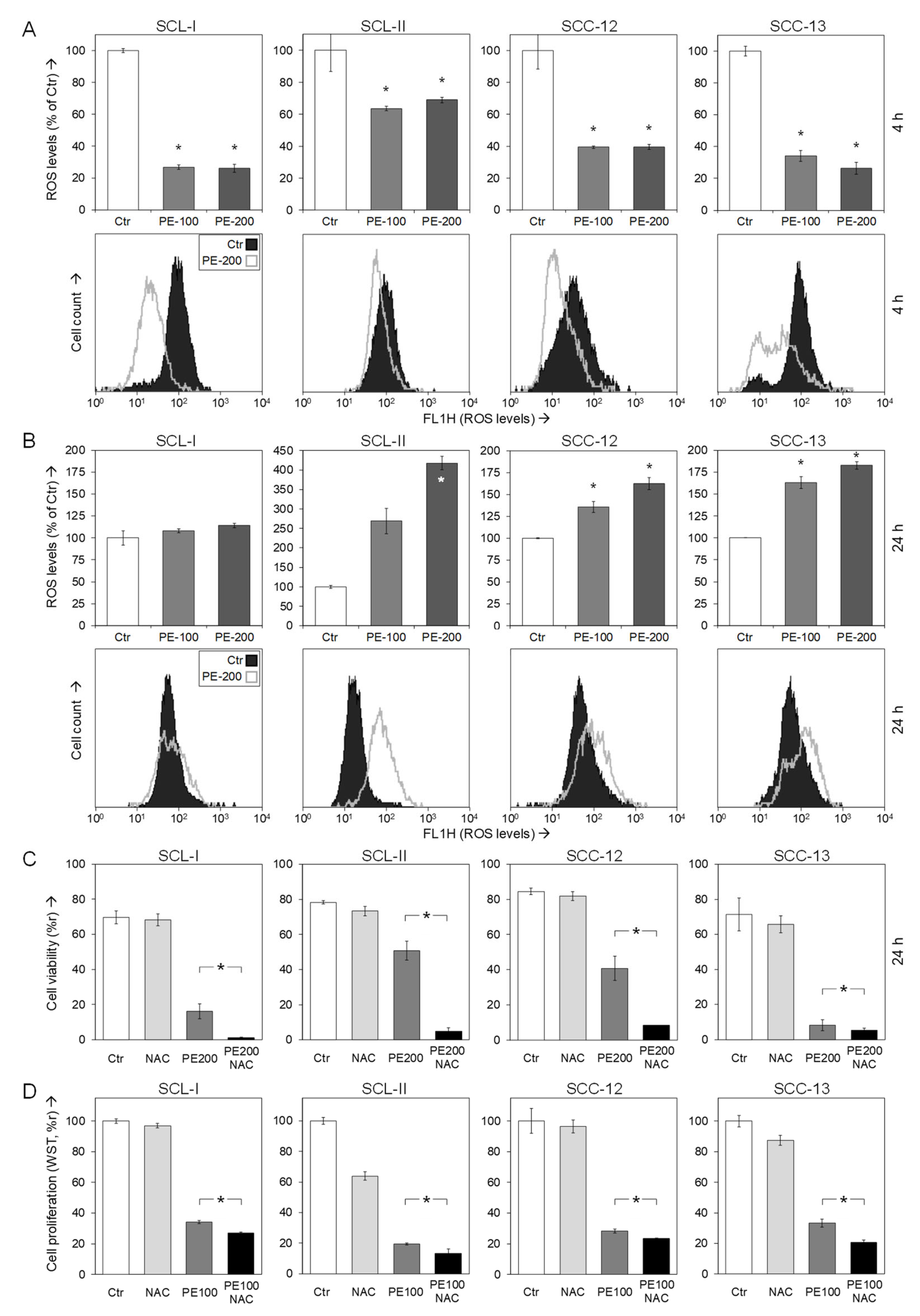
2.5. Analysis of Reactive Oxygen Species
2.6. Western Blotting
2.7. Statistical Analyses
3. Results
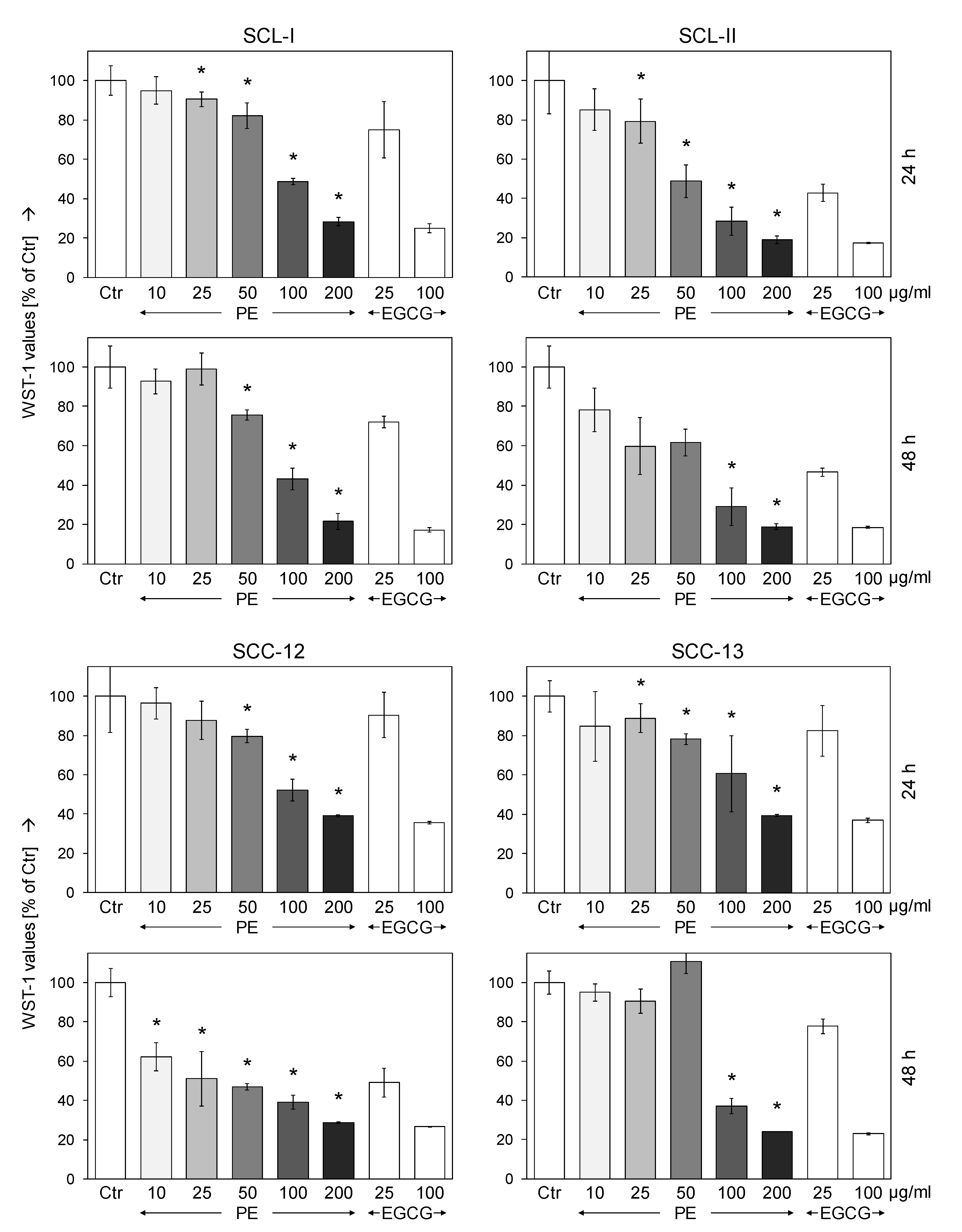
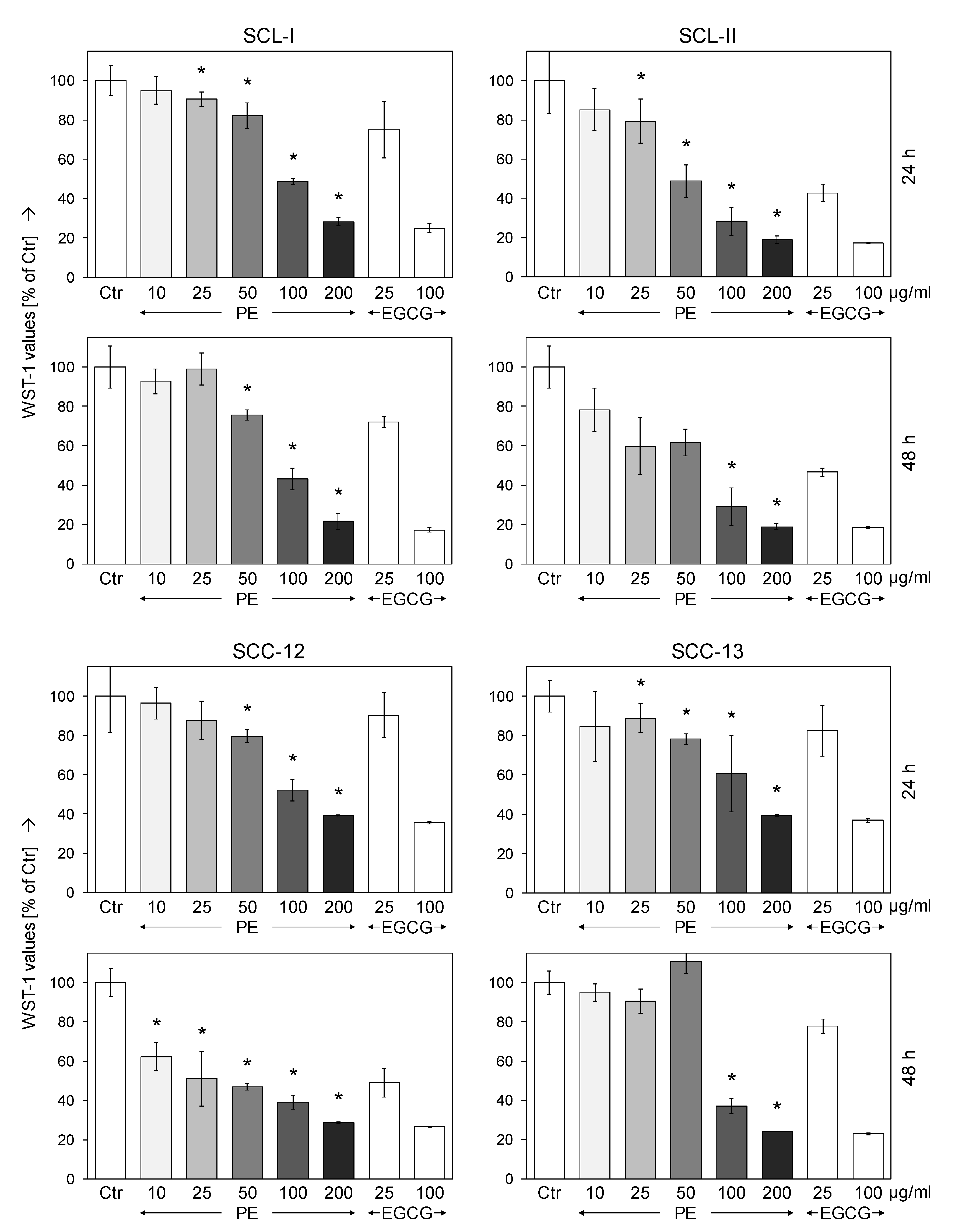
3.1. Dose-Dependent Inhibition of Cell Proliferation by Sinecatechins (PE)
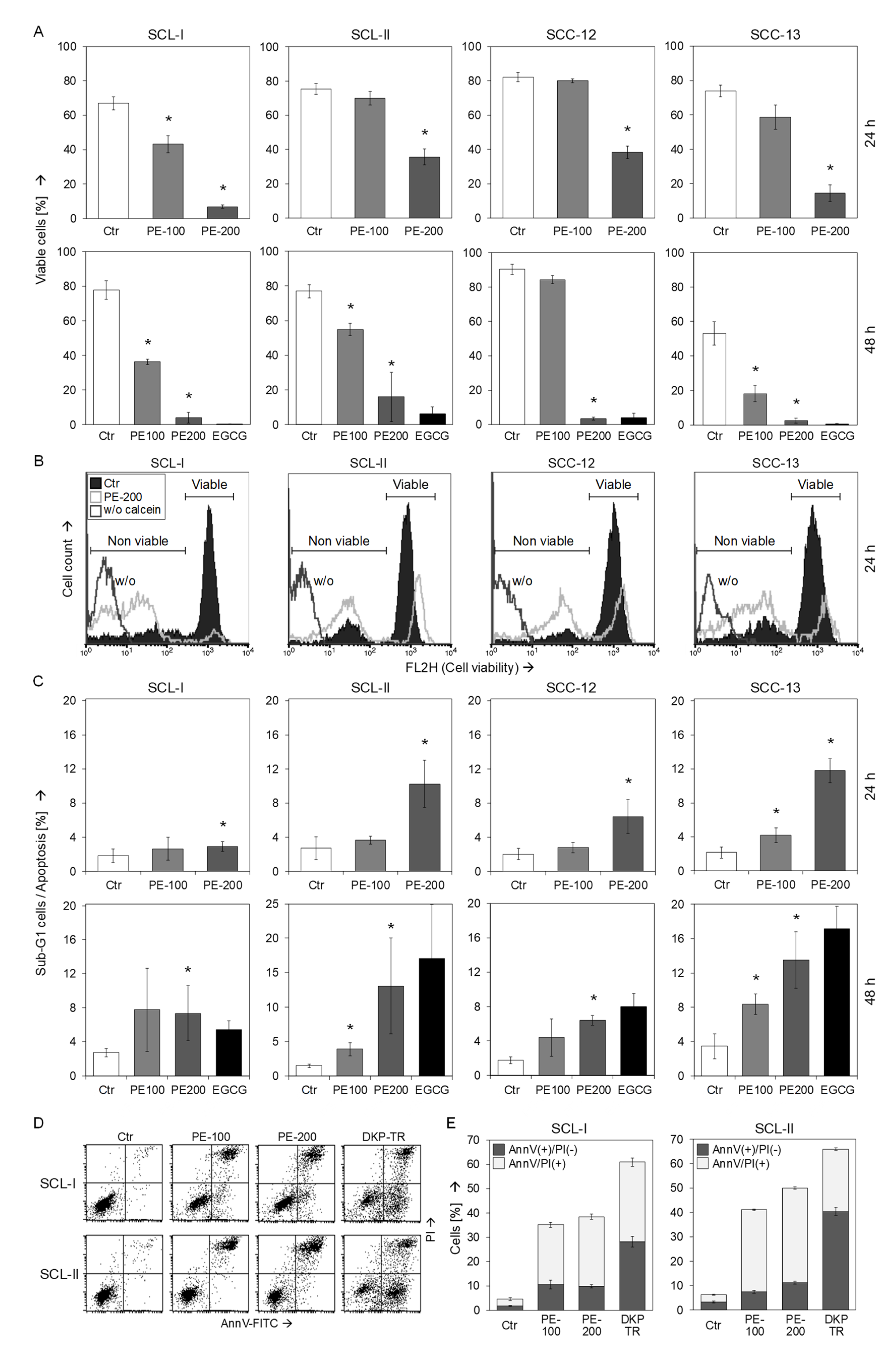
3.2. Strongly Reduced Cell Viability and Moderate Induction of Early Apoptosis
3.3. Loss of Mitochondrial Membrane Potential
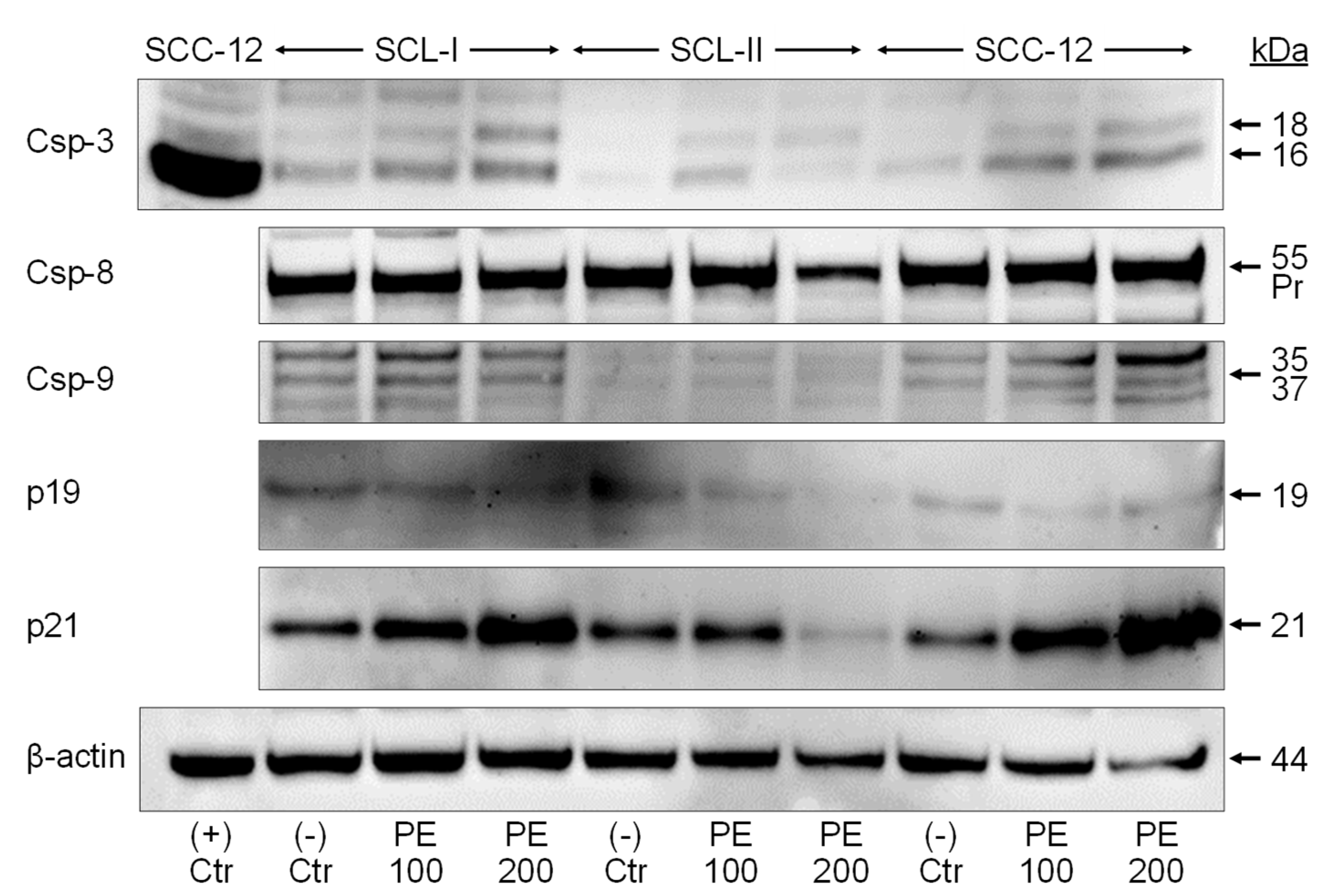
3.4. Caspase Activation and Upregulation of p21
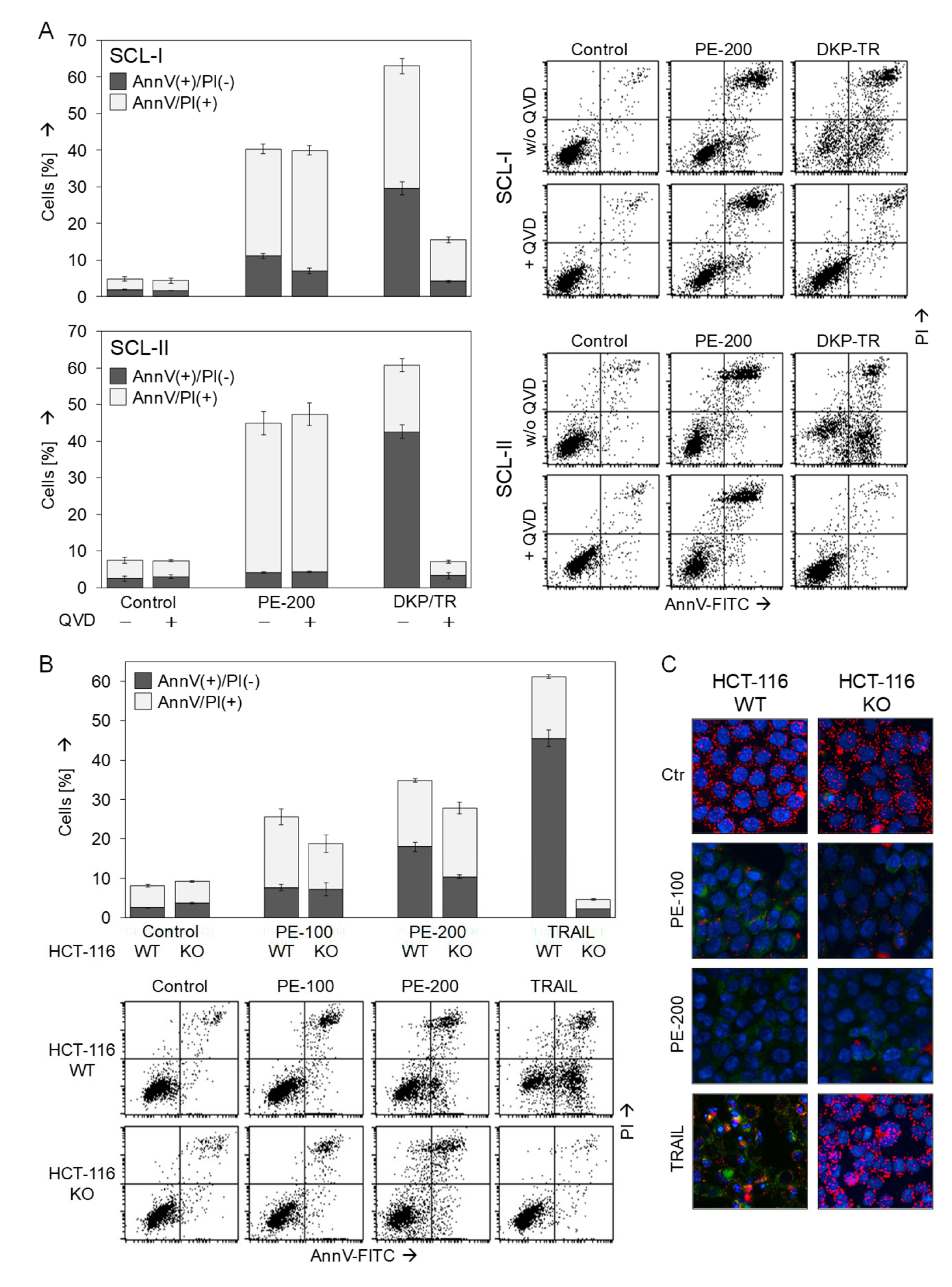
3.5. No Dependence on Caspases or the Proapoptotic Bcl-2 Proteins Bax and Bak
3.6. Role of Reactive Oxygen Species
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alam, M.; Ratner, D. Cutaneous squamous-cell carcinoma. N. Engl. J. Med. 2001, 344, 975–983. [Google Scholar] [CrossRef] [PubMed]
- Szewczyk, M.; Pazdrowski, J.; Golusiński, P.; Dańczak-Pazdrowska, A.; Marszałek, S.; Golusiński, W. Analysis of selected risk factors for nodal metastases in head and neck cutaneous squamous cell carcinoma. Eur. Arch. Otorhinolaryngol. 2015, 272, 3007–3012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agbai, O.N.; Buster, K.; Sanchez, M.; Hernandez, C.; Kundu, R.V.; Chiu, M.; Roberts, W.E.; Draelos, Z.D.; Bhushan, R.; Taylor, S.C.; et al. Skin cancer and photoprotection in people of color: A review and recommendations for physicians and the public. J. Am. Acad. Dermatol. 2014, 70, 748–762. [Google Scholar] [CrossRef] [PubMed]
- Amaral, T.; Osewold, M.; Presser, D.; Meiwes, A.; Garbe, C.; Leiter, U. Advanced cutaneous squamous cell carcinoma: Real world data of patient profiles and treatment patterns. J. Eur. Acad. Dermatol. Venereol. 2019, 33 (Suppl. 8), 44–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stockfleth, E. The importance of treating the field in actinic keratosis. J. Eur. Acad. Dermatol. Venereol. 2017, 31 (Suppl. 2), 8–11. [Google Scholar] [CrossRef] [Green Version]
- Cramer, P.; Stockfleth, E. Actinic keratosis: Where do we stand and where is the future going to take us? Expert Opin. Emerg. Drugs 2020, 25, 49–58. [Google Scholar] [CrossRef]
- Stockfleth, E.; Meyer, T. Sinecatechins (Polyphenon E) ointment for treatment of external genital warts and possible future indications. Expert. Opin. Biol. Ther. 2014, 14, 1033–1043. [Google Scholar] [CrossRef]
- Bobyr, I.; Campanati, A.; Offidani, A. “SENECA” Sinecatechins 10% ointment: A green tea extract for the treatment of actinic keratosis. Case series. Dermatol. Ther. 2018, 31, e12634. [Google Scholar] [CrossRef]
- Khan, N.; Mukhtar, H. Tea Polyphenols in Promotion of Human Health. Nutrients 2018, 11, 39. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.Y.; Han, Z.; Li, X.; Xie, H.H.; Zhu, S.S. Mechanism of EGCG promoting apoptosis of MCF-7 cell line in human breast cancer. Oncol. Lett. 2017, 14, 3623–3627. [Google Scholar] [CrossRef] [Green Version]
- Gu, J.J.; Qiao, K.S.; Sun, P.; Chen, P.; Li, Q. Study of EGCG induced apoptosis in lung cancer cells by inhibiting PI3K/Akt signaling pathway. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 4557–4563. [Google Scholar] [CrossRef]
- Fu, J.D.; Yao, J.J.; Wang, H.; Cui, W.G.; Leng, J.; Ding, L.Y.; Fan, K.Y. Effects of EGCG on proliferation and apoptosis of gastric cancer SGC7901 cells via down-regulation of HIF-1α and VEGF under a hypoxic state. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 155–161. [Google Scholar] [CrossRef]
- Roufayel, R.; Mezher, R.; Storey, K.B. The Role of Retinoblastoma Protein in Cell Cycle Regulation: An Updated Review. Curr. Mol. Med. 2021, 21, 620–629. [Google Scholar] [CrossRef]
- Roy, A.; Banerjee, S. p27 and leukemia: Cell cycle and beyond. J. Cell. Physiol. 2015, 230, 504–509. [Google Scholar] [CrossRef]
- Li, J.; Poi, M.J.; Tsai, M.D. Regulatory mechanisms of tumor suppressor P16(INK4A) and their relevance to cancer. Biochemistry 2011, 50, 5566–5582. [Google Scholar] [CrossRef] [Green Version]
- Eberle, J. Countering TRAIL Resistance in Melanoma. Cancers 2019, 11, 656. [Google Scholar] [CrossRef] [Green Version]
- Chipuk, J.E.; Moldoveanu, T.; Llambi, F.; Parsons, M.J.; Green, D.R. The BCL-2 family reunion. Mol. Cell 2010, 37, 299–310. [Google Scholar] [CrossRef]
- Fischer, U.; Janicke, R.U.; Schulze-Osthoff, K. Many cuts to ruin: A comprehensive update of caspase substrates. Cell Death Differ. 2003, 10, 76–100. [Google Scholar] [CrossRef]
- Wang, C.; Youle, R.J. Predominant requirement of Bax for apoptosis in HCT116 cells is determined by Mcl-1’s inhibitory effect on Bak. Oncogene 2012, 31, 3177–3189. [Google Scholar] [CrossRef] [Green Version]
- Gillissen, B.; Richter, A.; Richter, A.; Overkamp, T.; Essmann, F.; Hemmati, P.G.; Preissner, R.; Belka, C.; Daniel, P.T. Targeted therapy of the XIAP/proteasome pathway overcomes TRAIL-resistance in carcinoma by switching apoptosis signaling to a Bax/Bak-independent ‘type I’ mode. Cell Death Dis. 2013, 4, e643. [Google Scholar] [CrossRef] [Green Version]
- Gillissen, B.; Richter, A.; Richter, A.; Preissner, R.; Schulze-Osthoff, K.; Essmann, F.; Daniel, P.T. Bax/Bak-independent mitochondrial depolarization and reactive oxygen species induction by sorafenib overcome resistance to apoptosis in renal cell carcinoma. J. Biol. Chem. 2017, 292, 6478–6492. [Google Scholar] [CrossRef] [Green Version]
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef]
- Zhu, J.; May, S.; Ulrich, C.; Stockfleth, E.; Eberle, J. High ROS Production by Celecoxib and Enhanced Sensitivity for Death Ligand-Induced Apoptosis in Cutaneous SCC Cell Lines. Int. J. Mol. Sci. 2021, 22, 3622. [Google Scholar] [CrossRef]
- Zhu, J.; Langer, P.; Ulrich, C.; Eberle, J. Crucial Role of Reactive Oxygen Species (ROS) for the Proapoptotic Effects of Indirubin Derivatives in Cutaneous SCC Cells. Antioxidants 2021, 10, 1514. [Google Scholar] [CrossRef]
- Rheinwald, J.G.; Beckett, M.A. Tumorigenic keratinocyte lines requiring anchorage and fibroblast support cultured from human squamous cell carcinomas. Cancer Res. 1981, 41, 1657–1663. [Google Scholar]
- Tilgen, W.; Boukamp, P.; Breitkreutz, D.; Dzarlieva, R.T.; Engstner, M.; Haag, D.; Fusenig, N.E. Preservation of morphological, functional, and karyotypic traits during long-term culture and in vivo passage of two human skin squamous cell carcinomas. Cancer Res. 1983, 43, 5995–6011. [Google Scholar]
- Fecker, L.F.; Stockfleth, E.; Braun, F.K.; Rodust, P.M.; Schwarz, C.; Köhler, A.; Leverkus, M.; Eberle, J. Enhanced death ligand-induced apoptosis in cutaneous SCC cells by treatment with diclofenac/hyaluronic acid correlates with downregulation of c-FLIP. J. Investig. Dermatol. 2010, 130, 2098–2109. [Google Scholar] [CrossRef] [Green Version]
- Manach, C.; Scalbert, A.; Morand, C.; Rémésy, C.; Jiménez, L. Polyphenols: Food sources and bioavailability. Am. J. Clin. Nutr. 2004, 79, 727–747. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.; Wan, S.B.; Yang, H.; Yuan, J.; Chan, T.H.; Dou, Q.P. EGCG, green tea polyphenols and their synthetic analogs and prodrugs for human cancer prevention and treatment. Adv. Clin. Chem. 2011, 53, 155–177. [Google Scholar] [CrossRef] [Green Version]
- Stockfleth, E.; Meyer, T. The use of sinecatechins (polyphenon E) ointment for treatment of external genital warts. Expert Opin. Biol. Ther. 2012, 12, 783–793. [Google Scholar] [CrossRef]
- Gross, G.; Meyer, K.G.; Pres, H.; Thielert, C.; Tawfik, H.; Mescheder, A. A randomized, double-blind, four-arm parallel-group, placebo-controlled Phase II/III study to investigate the clinical efficacy of two galenic formulations of Polyphenon E in the treatment of external genital warts. J. Eur. Acad. Dermatol. Venereol. 2007, 21, 1404–1412. [Google Scholar] [CrossRef] [PubMed]
- Prasanth, M.I.; Sivamaruthi, B.S.; Chaiyasut, C.; Tencomnao, T. A Review of the Role of Green Tea (Camellia sinensis) in Antiphotoaging, Stress Resistance, Neuroprotection, and Autophagy. Nutrients 2019, 11, 474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, A.M.; Baliga, M.S.; Katiyar, S.K. Epigallocatechin-3-gallate induces apoptosis in estrogen receptor-negative human breast carcinoma cells via modulation in protein expression of p53 and Bax and caspase-3 activation. Mol. Cancer Ther. 2005, 4, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Luo, K.W.; Xia, J.; Cheng, B.H.; Gao, H.C.; Fu, L.W.; Luo, X.L. Tea polyphenol EGCG inhibited colorectal-cancer-cell proliferation and migration via downregulation of STAT3. Gastroenterol. Rep. 2021, 9, 59–70. [Google Scholar] [CrossRef]
- Mohan, K.V.; Gunasekaran, P.; Varalakshmi, E.; Hara, Y.; Nagini, S. In vitro evaluation of the anticancer effect of lactoferrin and tea polyphenol combination on oral carcinoma cells. Cell Biol. Int. 2007, 31, 599–608. [Google Scholar] [CrossRef]
- Shimizu, M.; Deguchi, A.; Lim, J.T.; Moriwaki, H.; Kopelovich, L.; Weinstein, I.B. (−)-Epigallocatechin gallate and polyphenon E inhibit growth and activation of the epidermal growth factor receptor and human epidermal growth factor receptor-2 signaling pathways in human colon cancer cells. Clin. Cancer Res. 2005, 11, 2735–2746. [Google Scholar] [CrossRef] [Green Version]
- Xiong, Y.; Hannon, G.J.; Zhang, H.; Casso, D.; Kobayashi, R.; Beach, D. p21 is a universal inhibitor of cyclin kinases. Nature 1993, 366, 701–704. [Google Scholar] [CrossRef]
- Abbas, T.; Dutta, A. p21 in cancer: Intricate networks and multiple activities. Nat. Rev. Cancer 2009, 9, 400–414. [Google Scholar] [CrossRef]
- Liang, Y.C.; Lin-Shiau, S.Y.; Chen, C.F.; Lin, J.K. Inhibition of cyclin-dependent kinases 2 and 4 activities as well as induction of Cdk inhibitors p21 and p27 during growth arrest of human breast carcinoma cells by (−)-epigallocatechin-3-gallate. J. Cell. Biochem. 1999, 75, 1–12. [Google Scholar] [CrossRef]
- Gupta, S.; Hussain, T.; Mukhtar, H. Molecular pathway for (−)-epigallocatechin-3-gallate-induced cell cycle arrest and apoptosis of human prostate carcinoma cells. Arch. Biochem. Biophys. 2003, 410, 177–185. [Google Scholar] [CrossRef]
- Balasubramanian, S.; Adhikary, G.; Eckert, R.L. The Bmi-1 polycomb protein antagonizes the (−)-epigallocatechin-3-gallate-dependent suppression of skin cancer cell survival. Carcinogenesis 2010, 31, 496–503. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Zhang, D.Y.; Zhang, W.; Zhao, X.; Yuan, C.; Ye, F. The effect of green tea extract and EGCG on the signaling network in squamous cell carcinoma. Nutr. Cancer 2011, 63, 466–475. [Google Scholar] [CrossRef]
- Luo, K.W.; Lung, W.Y.; Chun, X.; Luo, X.L.; Huang, W.R. EGCG inhibited bladder cancer T24 and 5637 cell proliferation and migration via PI3K/AKT pathway. Oncotarget 2018, 9, 12261–12272. [Google Scholar] [CrossRef] [Green Version]
- Zhu, W.; Li, M.C.; Wang, F.R.; Mackenzie, G.G.; Oteiza, P.I. The inhibitory effect of ECG and EGCG dimeric procyanidins on colorectal cancer cells growth is associated with their actions at lipid rafts and the inhibition of the epidermal growth factor receptor signaling. Biochem. Pharmacol. 2020, 175, 113923. [Google Scholar] [CrossRef]
- Singh, M.; Singh, R.; Bhui, K.; Tyagi, S.; Mahmood, Z.; Shukla, Y. Tea polyphenols induce apoptosis through mitochondrial pathway and by inhibiting nuclear factor-kappaB and Akt activation in human cervical cancer cells. Oncol. Res. 2011, 19, 245–257. [Google Scholar] [CrossRef]
- Chakrabarty, S.; Nag, D.; Ganguli, A.; Das, A.; Ghosh Dastidar, D.; Chakrabarti, G. Theaflavin and epigallocatechin-3-gallate synergistically induce apoptosis through inhibition of PI3K/Akt signaling upon depolymerizing microtubules in HeLa cells. J. Cell. Biochem. 2019, 120, 5987–6003. [Google Scholar] [CrossRef]
- Tipoe, G.L.; Leung, T.M.; Hung, M.W.; Fung, M.L. Green tea polyphenols as an anti-oxidant and anti-inflammatory agent for cardiovascular protection. Cardiovasc. Hematol. Disord. Drug Targets 2007, 7, 135–144. [Google Scholar] [CrossRef]
- Singh, N.A.; Mandal, A.K.; Khan, Z.A. Potential neuroprotective properties of epigallocatechin-3-gallate (EGCG). Nutr. J. 2016, 15, 60. [Google Scholar] [CrossRef] [Green Version]
- Avadhani, K.S.; Manikkath, J.; Tiwari, M.; Chandrasekhar, M.; Godavarthi, A.; Vidya, S.M.; Hariharapura, R.C.; Kalthur, G.; Udupa, N.; Mutalik, S. Skin delivery of epigallocatechin-3-gallate (EGCG) and hyaluronic acid loaded nano-transfersomes for antioxidant and anti-aging effects in UV radiation induced skin damage. Drug Deliv. 2017, 24, 61–74. [Google Scholar] [CrossRef] [Green Version]
- Franke, J.C.; Plotz, M.; Prokop, A.; Geilen, C.C.; Schmalz, H.G.; Eberle, J. New caspase-independent but ROS-dependent apoptosis pathways are targeted in melanoma cells by an iron-containing cytosine analogue. Biochem. Pharmacol. 2010, 79, 575–586. [Google Scholar] [CrossRef] [Green Version]
- Soltan, M.Y.; Sumarni, U.; Assaf, C.; Langer, P.; Reidel, U.; Eberle, J. Key Role of Reactive Oxygen Species (ROS) in Indirubin Derivative-Induced Cell Death in Cutaneous T-Cell Lymphoma Cells. Int. J. Mol. Sci. 2019, 20, 1158. [Google Scholar] [CrossRef] [Green Version]
- Zhivkova, V.; Kiecker, F.; Langer, P.; Eberle, J. Crucial role of reactive oxygen species (ROS) for the proapoptotic effects of indirubin derivative DKP-073 in melanoma cells. Mol. Carcinog. 2019, 58, 258–269. [Google Scholar] [CrossRef]
- Quast, S.A.; Berger, A.; Eberle, J. ROS-dependent phosphorylation of Bax by wortmannin sensitizes melanoma cells for TRAIL-induced apoptosis. Cell Death Dis. 2013, 4, e839. [Google Scholar] [CrossRef]
- Liu, P.; Yang, W.T.; Shi, L.; Zhang, H.Y.; Xu, Y.; Wang, P.R.; Zhang, G.L.; Chen, W.R.; Zhang, B.B.; Wang, X.L. Concurrent photothermal therapy and photodynamic therapy for cutaneous squamous cell carcinoma by gold nanoclusters under a single NIR laser irradiation. J. Mater. Chem. B 2019, 7, 6924–6933. [Google Scholar] [CrossRef]
- Austin, E.; Koo, E.; Jagdeo, J. Thermal photodynamic therapy increases apoptosis and reactive oxygen species generation in cutaneous and mucosal squamous cell carcinoma cells. Sci. Rep. 2018, 8, 12599. [Google Scholar] [CrossRef]
- Niu, T.H.; Tian, Y.; Wang, G.Y.; Guo, G.J.; Tong, Y.; Shi, Y. Inhibition of ROS-NF-kappa B-dependent autophagy enhances Hypocrellin A united LED red light-induced apoptosis in squamous carcinoma A431 cells. Cell. Signal. 2020, 69, 109550. [Google Scholar] [CrossRef]
- Khiewkamrop, P.; Phunsomboon, P.; Richert, L.; Pekthong, D.; Srisawang, P. Epistructured catechins, EGCG and EC facilitate apoptosis induction through targeting de novo lipogenesis pathway in HepG2 cells. Cancer Cell Int. 2018, 18, 46. [Google Scholar] [CrossRef]
- Tsai, C.Y.; Chen, C.Y.; Chiou, Y.H.; Shyu, H.W.; Lin, K.H.; Chou, M.C.; Huang, M.H.; Wang, Y.F. Epigallocatechin-3-Gallate Suppresses Human Herpesvirus 8 Replication and Induces ROS Leading to Apoptosis and Autophagy in Primary Effusion Lymphoma Cells. Int. J. Mol. Sci. 2017, 19, 16. [Google Scholar] [CrossRef] [Green Version]
- Ranzato, E.; Martinotti, S.; Magnelli, V.; Murer, B.; Biffo, S.; Mutti, L.; Burlando, B. Epigallocatechin-3-gallate induces mesothelioma cell death via H2O2-dependent T-type Ca2+ channel opening. J. Cell. Mol. Med. 2012, 16, 2667–2678. [Google Scholar] [CrossRef]
- Kaźmierczak-Barańska, J.; Boguszewska, K.; Adamus-Grabicka, A.; Karwowski, B.T. Two Faces of Vitamin C-Antioxidative and Pro-Oxidative Agent. Nutrients 2020, 12, 1501. [Google Scholar] [CrossRef]
- Chobot, V.; Hadacek, F.; Bachmann, G.; Weckwerth, W.; Kubicova, L. In Vitro Evaluation of Pro- and Antioxidant Effects of Flavonoid Tricetin in Comparison to Myricetin. Molecules 2020, 25, 5850. [Google Scholar] [CrossRef] [PubMed]
- do Carmo, M.A.V.; Granato, D.; Azevedo, L. Antioxidant/pro-oxidant and antiproliferative activities of phenolic-rich foods and extracts: A cell-based point of view. Adv. Food Nutr. Res. 2021, 98, 253–280. [Google Scholar] [CrossRef] [PubMed]
- Moloney, J.N.; Cotter, T.G. ROS signalling in the biology of cancer. Semin. Cell Dev. Biol. 2018, 80, 50–64. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, J.; Gillissen, B.; Dang Tran, D.L.; May, S.; Ulrich, C.; Stockfleth, E.; Eberle, J. Inhibition of Cell Proliferation and Cell Viability by Sinecatechins in Cutaneous SCC Cells Is Related to an Imbalance of ROS and Loss of Mitochondrial Membrane Potential. Antioxidants 2022, 11, 1416. https://doi.org/10.3390/antiox11071416
Zhu J, Gillissen B, Dang Tran DL, May S, Ulrich C, Stockfleth E, Eberle J. Inhibition of Cell Proliferation and Cell Viability by Sinecatechins in Cutaneous SCC Cells Is Related to an Imbalance of ROS and Loss of Mitochondrial Membrane Potential. Antioxidants. 2022; 11(7):1416. https://doi.org/10.3390/antiox11071416
Chicago/Turabian StyleZhu, Jiaqi, Bernd Gillissen, Dieu Linh Dang Tran, Stefanie May, Claas Ulrich, Eggert Stockfleth, and Jürgen Eberle. 2022. "Inhibition of Cell Proliferation and Cell Viability by Sinecatechins in Cutaneous SCC Cells Is Related to an Imbalance of ROS and Loss of Mitochondrial Membrane Potential" Antioxidants 11, no. 7: 1416. https://doi.org/10.3390/antiox11071416