
Formyl-Peptide Receptor 2 Signaling Redirects Glucose and Glutamine into Anabolic Pathways in Metabolic Reprogramming of Lung Cancer Cells

 ,
, 
 , ,
, ,  and
and 
Abstract
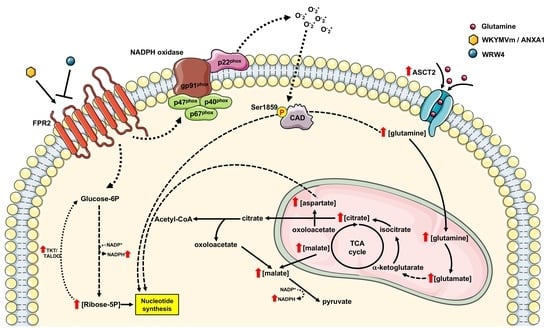
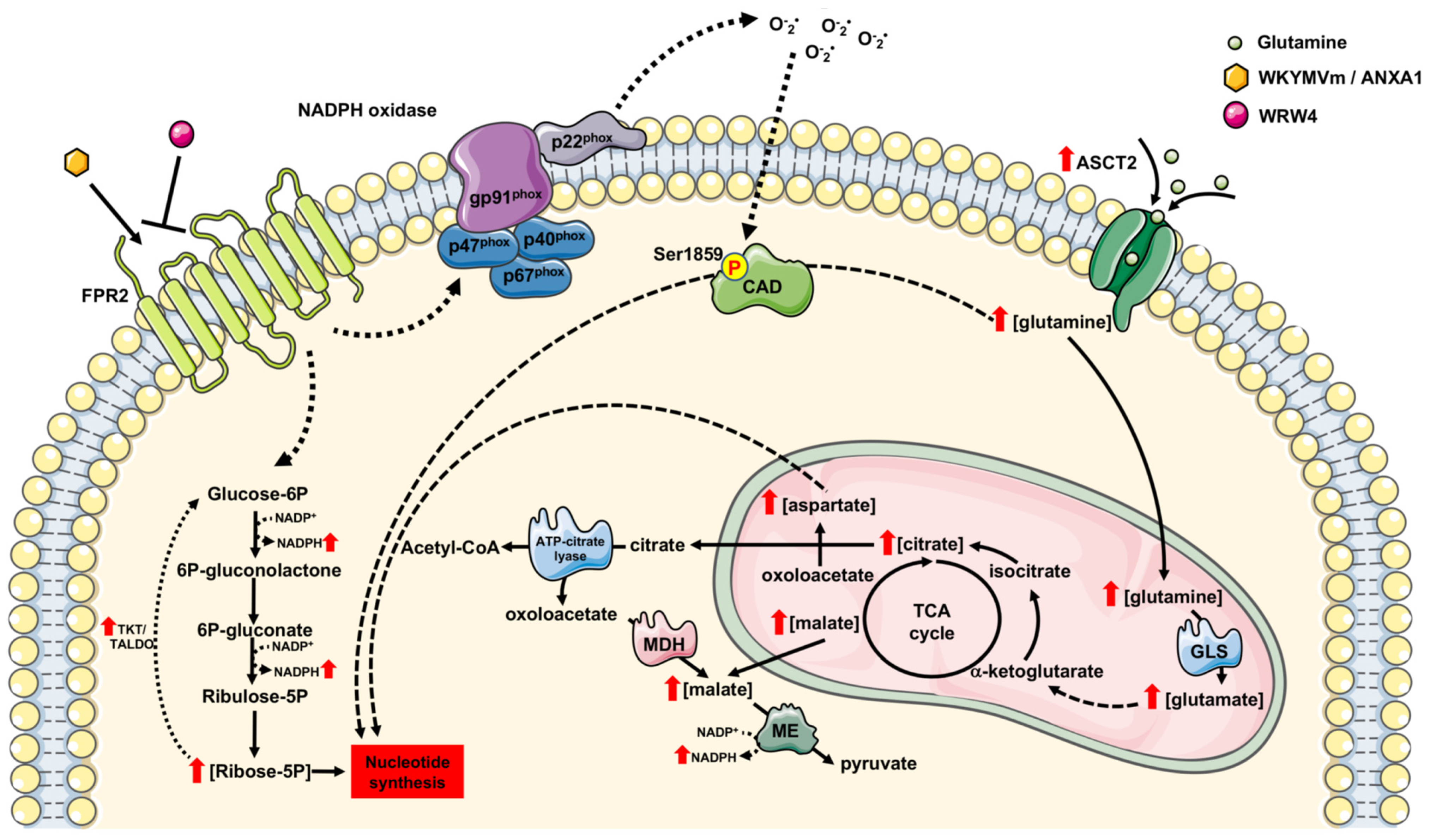
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Reagents
2.2. Metabolomic Analysis by LC-MS
2.3. p22phoxCrispr/Cas9 Double-Nickase CaLu-6 Cells
2.4. Protein Extraction and Western Blot
2.5. Mitochondrial Membrane Potential Assay
2.6. NADP+/NADPH Assay
2.7. Transketolase Activity Assay
2.8. Statistical Analysis
3. Results and Discussion
3.1. FPR2 Stimulation Triggers PPP
3.2. NADPH Production and the Non-Oxidative Phase of PPP Are Regulated by FPR2
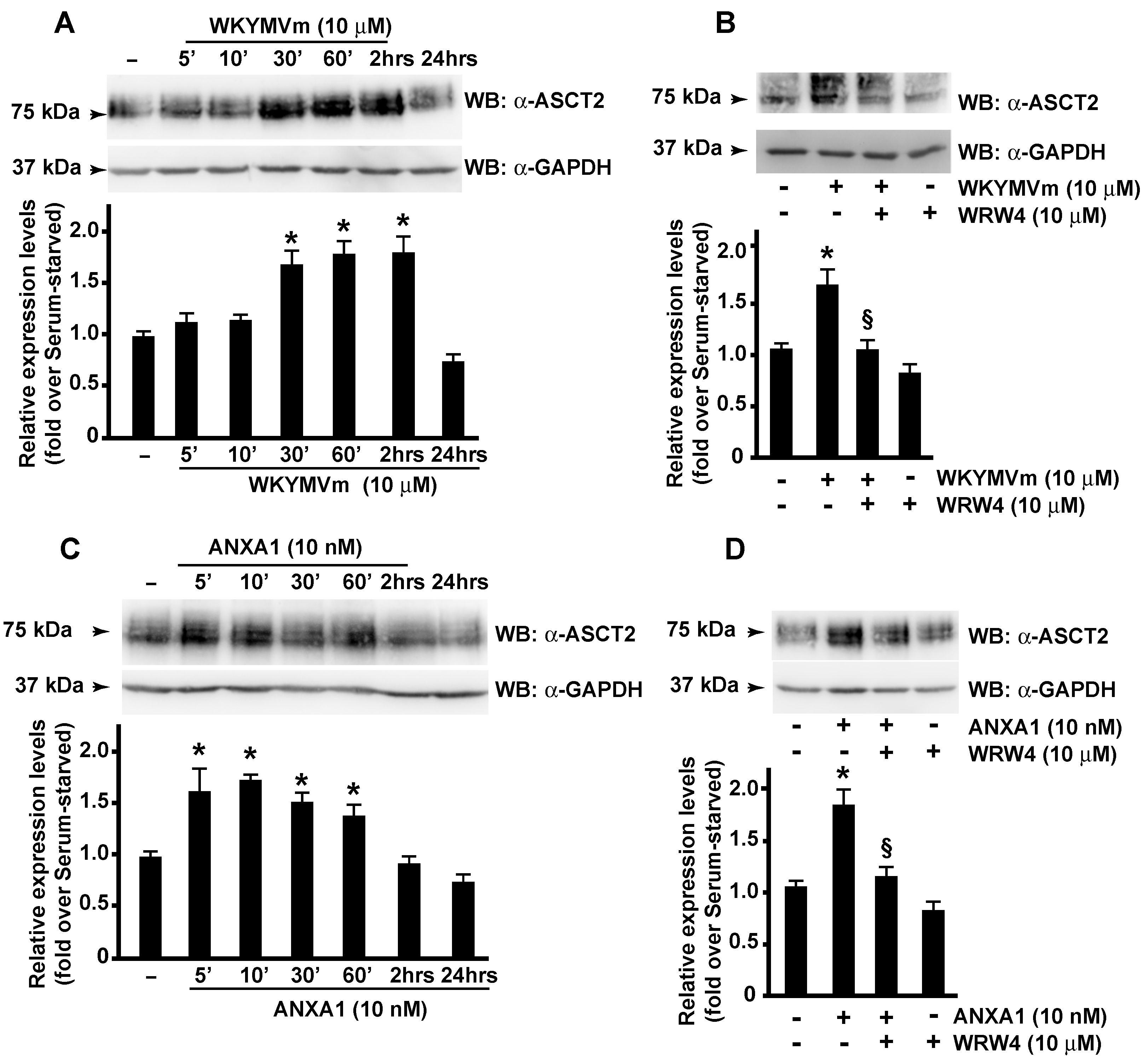
3.3. FPR2 Signaling Modulates ASCT2 Expression
3.4. FPR2 Signaling Regulates De Novo Synthesis of Pyrimidine Nucleotides
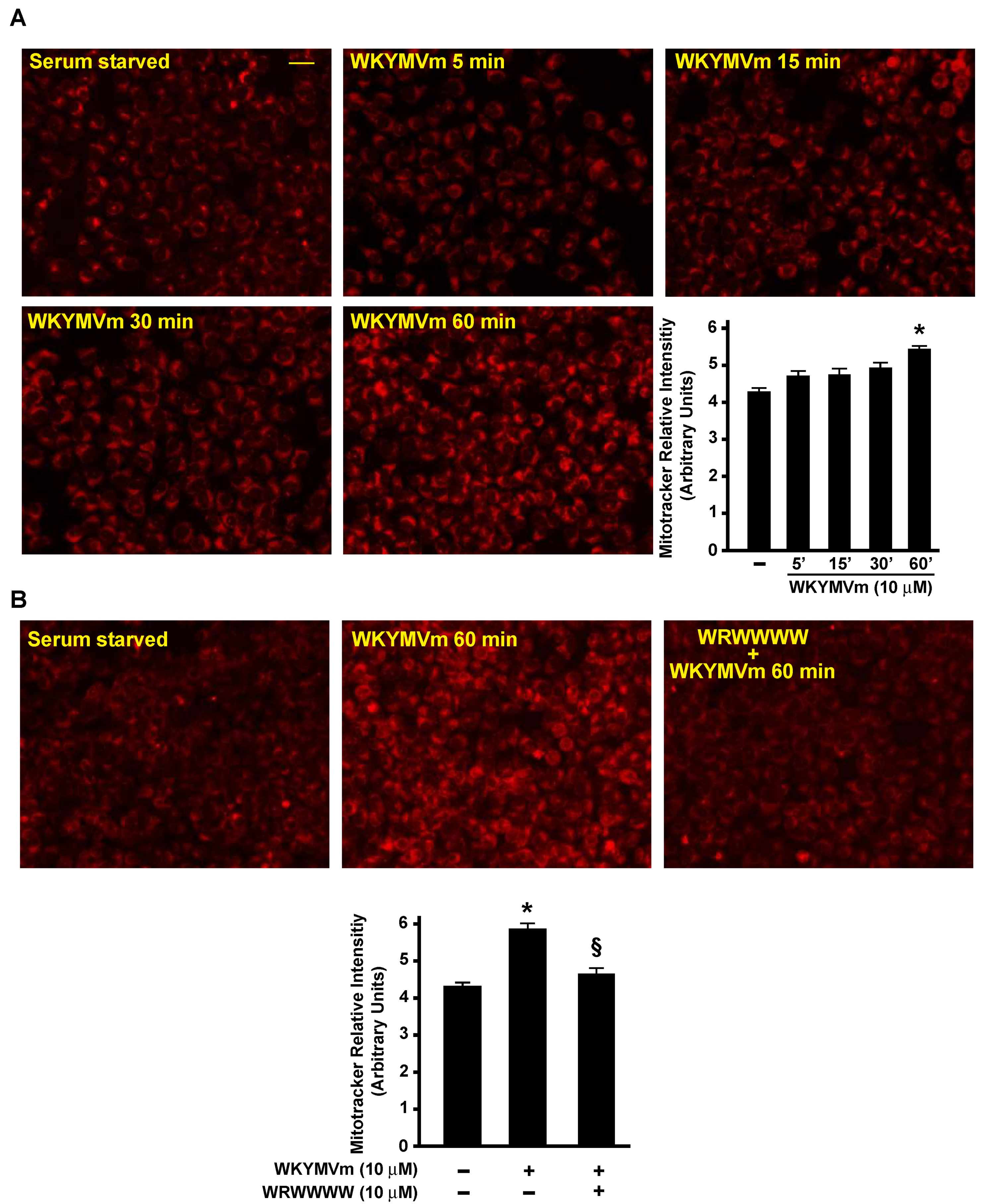
3.5. FPR2 Signaling Regulates TCA
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Abdel-Wahab, A.F.; Mahmoud, W.; Al-Harizy, R.M. Targeting glucose metabolism to suppress cancer progression: Prospective of anti-glycolytic cancer therapy. Pharmacol. Res. 2019, 150, 104511. [Google Scholar] [CrossRef] [PubMed]
- Koppenol, W.H.; Bounds, P.L.; Dang, C.V. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat. Rev. Cancer 2011, 11, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Epstein, T.; Gatenby, R.A.; Brown, J.S. The Warburg effect as an adaptation of cancer cells to rapid fluctuations in energy demand. PLoS ONE 2017, 12, e0185085. [Google Scholar] [CrossRef]
- Wei, Z.; Liu, X.; Cheng, C.; Yu, W.; Yi, P. Metabolism of Amino Acids in Cancer. Front. Cell Dev. Biol. 2020, 8, 603837. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.L.; Nokin, M.J.; Teres, S.; Tome, M.; Bodineau, C.; Galmar, O.; Pasquet, J.M.; Rousseau, B.; van Liempd, S.; Falcon-Perez, J.M.; et al. Downregulation of Glutamine Synthetase, not glutaminolysis, is responsible for glutamine addiction in Notch1-driven acute lymphoblastic leukemia. Mol. Oncol. 2021, 15, 1412–1431. [Google Scholar] [CrossRef]
- Ahn, C.S.; Metallo, C.M. Mitochondria as biosynthetic factories for cancer proliferation. Cancer Metab. 2015, 3, 1. [Google Scholar] [CrossRef]
- Wellen, K.E.; Lu, C.; Mancuso, A.; Lemons, J.M.; Ryczko, M.; Dennis, J.W.; Rabinowitz, J.D.; Coller, H.A.; Thompson, C.B. The hexosamine biosynthetic pathway couples growth factor-induced glutamine uptake to glucose metabolism. Genes Dev. 2010, 24, 2784–2799. [Google Scholar] [CrossRef]
- Fan, J.; Kang, H.B.; Shan, C.; Elf, S.; Lin, R.; Xie, J.; Gu, T.L.; Aguiar, M.; Lonning, S.; Chung, T.W.; et al. Tyr-301 phosphorylation inhibits pyruvate dehydrogenase by blocking substrate binding and promotes the Warburg effect. J. Biol. Chem. 2014, 289, 26533–26541. [Google Scholar] [CrossRef]
- Han, J.; Zhang, Y.; Xu, J.; Zhang, T.; Wang, H.; Wang, Z.; Jiang, Y.; Zhou, L.; Yang, M.; Hua, Y.; et al. Her4 promotes cancer metabolic reprogramming via the c-Myc-dependent signaling axis. Cancer Lett. 2021, 496, 57–71. [Google Scholar] [CrossRef]
- Balic, J.J.; Albargy, H.; Luu, K.; Kirby, F.J.; Jayasekara, W.S.N.; Mansell, F.; Garama, D.J.; De Nardo, D.; Baschuk, N.; Louis, C.; et al. STAT3 serine phosphorylation is required for TLR4 metabolic reprogramming and IL-1beta expression. Nat. Commun. 2020, 11, 3816. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhang, X.; Chen, K.; Cheng, Y.; Liu, S.; Xia, M.; Chen, Y.; Zhu, H.; Li, Z.; Cao, X. CCR7 Chemokine Receptor-Inducible lnc-Dpf3 Restrains Dendritic Cell Migration by Inhibiting HIF-1alpha-Mediated Glycolysis. Immunity 2019, 50, 600–615.e15. [Google Scholar] [CrossRef] [PubMed]
- Thorne, J.L.; Campbell, M.J. Nuclear receptors and the Warburg effect in cancer. Int. J. Cancer 2015, 137, 1519–1527. [Google Scholar] [CrossRef]
- Busch, L.; Vieten, S.; Brodel, S.; Endres, K.; Bufe, B. Emerging contributions of formyl peptide receptors to neurodegenerative diseases. Biol. Chem. 2022, 403, 27–41. [Google Scholar] [CrossRef] [PubMed]
- Cattaneo, F.; Guerra, G.; Ammendola, R. Expression and signaling of formyl-peptide receptors in the brain. Neurochem Res. 2010, 35, 2018–2026. [Google Scholar] [CrossRef]
- Weiss, E.; Kretschmer, D. Formyl-Peptide Receptors in Infection, Inflammation, and Cancer. Trends Immunol. 2018, 39, 815–829. [Google Scholar] [CrossRef]
- Xiang, Y.; Yao, X.; Chen, K.; Wang, X.; Zhou, J.; Gong, W.; Yoshimura, T.; Huang, J.; Wang, R.; Wu, Y.; et al. The G-protein coupled chemoattractant receptor FPR2 promotes malignant phenotype of human colon cancer cells. Am. J. Cancer Res. 2016, 6, 2599–2610. [Google Scholar]
- Snapkov, I.; Oqvist, C.O.; Figenschau, Y.; Kogner, P.; Johnsen, J.I.; Sveinbjornsson, B. The role of formyl peptide receptor 1 (FPR1) in neuroblastoma tumorigenesis. BMC Cancer 2016, 16, 490. [Google Scholar] [CrossRef]
- Perretti, M.; Godson, C. Formyl peptide receptor type 2 agonists to kick-start resolution pharmacology. Br. J. Pharmacol. 2020, 177, 4595–4600. [Google Scholar] [CrossRef]
- Ammendola, R.; Parisi, M.; Esposito, G.; Cattaneo, F. Pro-Resolving FPR2 Agonists Regulate NADPH Oxidase-Dependent Phosphorylation of HSP27, OSR1, and MARCKS and Activation of the Respective Upstream Kinases. Antioxidants 2021, 10, 134. [Google Scholar] [CrossRef]
- Ammendola, R.; Russo, L.; De Felice, C.; Esposito, F.; Russo, T.; Cimino, F. Low-affinity receptor-mediated induction of superoxide by N-formyl-methionyl-leucyl-phenylalanine and WKYMVm in IMR90 human fibroblasts. Free Radic. Biol. Med. 2004, 36, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Le, Y.; Murphy, P.M.; Wang, J.M. Formyl-peptide receptors revisited. Trends Immunol. 2002, 23, 541–548. [Google Scholar] [CrossRef]
- Cattaneo, F.; Parisi, M.; Fioretti, T.; Sarnataro, D.; Esposito, G.; Ammendola, R. Nuclear localization of Formyl-Peptide Receptor 2 in human cancer cells. Arch. Biochem. Biophys. 2016, 603, 10–19. [Google Scholar] [CrossRef] [PubMed]
- He, H.Q.; Ye, R.D. The Formyl Peptide Receptors: Diversity of Ligands and Mechanism for Recognition. Molecules 2017, 22, 455. [Google Scholar] [CrossRef]
- Perretti, M.; Chiang, N.; La, M.; Fierro, I.M.; Marullo, S.; Getting, S.J.; Solito, E.; Serhan, C.N. Endogenous lipid- and peptide-derived anti-inflammatory pathways generated with glucocorticoid and aspirin treatment activate the lipoxin A4 receptor. Nat. Med. 2002, 8, 1296–1302. [Google Scholar] [CrossRef]
- Kim, Y.E.; Park, W.S.; Ahn, S.Y.; Sung, D.K.; Sung, S.I.; Kim, J.H.; Chang, Y.S. WKYMVm hexapeptide, a strong formyl peptide receptor 2 agonist, attenuates hyperoxia-induced lung injuries in newborn mice. Sci. Rep. 2019, 9, 6815. [Google Scholar] [CrossRef]
- Galvao, I.; de Carvalho, R.V.H.; Vago, J.P.; Silva, A.L.N.; Carvalho, T.G.; Antunes, M.M.; Ribeiro, F.M.; Menezes, G.B.; Zamboni, D.S.; Sousa, L.P.; et al. The role of annexin A1 in the modulation of the NLRP3 inflammasome. Immunology 2020, 160, 78–89. [Google Scholar] [CrossRef]
- Ye, R.D.; Sun, L. Emerging functions of serum amyloid A in inflammation. J. Leukoc. Biol. 2015, 98, 923–929. [Google Scholar] [CrossRef]
- Cooray, S.N.; Gobbetti, T.; Montero-Melendez, T.; McArthur, S.; Thompson, D.; Clark, A.J.; Flower, R.J.; Perretti, M. Ligand-specific conformational change of the G-protein-coupled receptor ALX/FPR2 determines proresolving functional responses. Proc. Natl. Acad. Sci. USA 2013, 110, 18232–18237. [Google Scholar] [CrossRef]
- Cattaneo, F.; Russo, R.; Castaldo, M.; Chambery, A.; Zollo, C.; Esposito, G.; Pedone, P.V.; Ammendola, R. Phosphoproteomic analysis sheds light on intracellular signaling cascades triggered by Formyl-Peptide Receptor 2. Sci. Rep. 2019, 9, 17894. [Google Scholar] [CrossRef]
- Iaccio, A.; Cattaneo, F.; Mauro, M.; Ammendola, R. FPRL1-mediated induction of superoxide in LL-37-stimulated IMR90 human fibroblast. Arch. Biochem. Biophys. 2009, 481, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Cattaneo, F.; Iaccio, A.; Guerra, G.; Montagnani, S.; Ammendola, R. NADPH-oxidase-dependent reactive oxygen species mediate EGFR transactivation by FPRL1 in WKYMVm-stimulated human lung cancer cells. Free Radic. Biol. Med. 2011, 51, 1126–1136. [Google Scholar] [CrossRef] [PubMed]
- Tonks, N.K. Redox redux: Revisiting PTPs and the control of cell signaling. Cell 2005, 121, 667–670. [Google Scholar] [CrossRef] [PubMed]
- Chiarugi, P.; Cirri, P. Redox regulation of protein tyrosine phosphatases during receptor tyrosine kinase signal transduction. Trends Biochem. Sci. 2003, 28, 509–514. [Google Scholar] [CrossRef]
- Annunziata, M.C.; Parisi, M.; Esposito, G.; Fabbrocini, G.; Ammendola, R.; Cattaneo, F. Phosphorylation Sites in Protein Kinases and Phosphatases Regulated by Formyl Peptide Receptor 2 Signaling. Int. J. Mol. Sci. 2020, 21, 3818. [Google Scholar] [CrossRef]
- Cattaneo, F.; Guerra, G.; Parisi, M.; De Marinis, M.; Tafuri, D.; Cinelli, M.; Ammendola, R. Cell-surface receptors transactivation mediated by g protein-coupled receptors. Int. J. Mol. Sci. 2014, 15, 19700–19728. [Google Scholar] [CrossRef]
- Castaldo, M.; Zollo, C.; Esposito, G.; Ammendola, R.; Cattaneo, F. NOX2-Dependent Reactive Oxygen Species Regulate Formyl-Peptide Receptor 1-Mediated TrkA Transactivation in SH-SY5Y Cells. Oxidative Med. Cell. Longev. 2019, 2019, 2051235. [Google Scholar] [CrossRef]
- Cattaneo, F.; Castaldo, M.; Parisi, M.; Faraonio, R.; Esposito, G.; Ammendola, R. Formyl Peptide Receptor 1 Modulates Endothelial Cell Functions by NADPH Oxidase-Dependent VEGFR2 Transactivation. Oxidative Med. Cell. Longev. 2018, 2018, 2609847. [Google Scholar] [CrossRef]
- Cattaneo, F.; Parisi, M.; Ammendola, R. WKYMVm-induced cross-talk between FPR2 and HGF receptor in human prostate epithelial cell line PNT1A. FEBS Lett. 2013, 587, 1536–1542. [Google Scholar] [CrossRef]
- Cattaneo, F.; Parisi, M.; Ammendola, R. Distinct signaling cascades elicited by different formyl peptide receptor 2 (FPR2) agonists. Int. J. Mol. Sci. 2013, 14, 7193–7230. [Google Scholar] [CrossRef]
- Sommella, E.; Badolati, N.; Riccio, G.; Salviati, E.; Bottone, S.; Dentice, M.; Campiglia, P.; Tenore, G.C.; Stornaiuolo, M.; Novellino, E. A Boost in Mitochondrial Activity Underpins the Cholesterol-Lowering Effect of Annurca Apple Polyphenols on Hepatic Cells. Nutrients 2019, 11, 163. [Google Scholar] [CrossRef] [Green Version]
- Riccio, G.; Sommella, E.; Badolati, N.; Salviati, E.; Bottone, S.; Campiglia, P.; Dentice, M.; Tenore, G.C.; Stornaiuolo, M.; Novellino, E. Annurca Apple Polyphenols Protect Murine Hair Follicles from Taxane Induced Dystrophy and Hijacks Polyunsaturated Fatty Acid Metabolism toward beta-Oxidation. Nutrients 2018, 10, 1808. [Google Scholar] [CrossRef]
- Badolati, N.; Sommella, E.; Riccio, G.; Salviati, E.; Heintz, D.; Bottone, S.; Di Cicco, E.; Dentice, M.; Tenore, G.; Campiglia, P.; et al. Annurca Apple Polyphenols Ignite Keratin Production in Hair Follicles by Inhibiting the Pentose Phosphate Pathway and Amino Acid Oxidation. Nutrients 2018, 10, 1406. [Google Scholar] [CrossRef]
- Schiattarella, G.G.; Cattaneo, F.; Carrizzo, A.; Paolillo, R.; Boccella, N.; Ambrosio, M.; Damato, A.; Pironti, G.; Franzone, A.; Russo, G.; et al. Akap1 Regulates Vascular Function and Endothelial Cells Behavior. Hypertension 2018, 71, 507–517. [Google Scholar] [CrossRef] [PubMed]
- Pavone, L.M.; Cattaneo, F.; Rea, S.; De Pasquale, V.; Spina, A.; Sauchelli, E.; Mastellone, V.; Ammendola, R. Intracellular signaling cascades triggered by the NK1 fragment of hepatocyte growth factor in human prostate epithelial cell line PNT1A. Cell. Signal. 2011, 23, 1961–1971. [Google Scholar] [CrossRef]
- Patra, K.C.; Hay, N. The pentose phosphate pathway and cancer. Trends Biochem. Sci. 2014, 39, 347–354. [Google Scholar] [CrossRef]
- Lane, A.N.; Fan, T.W. Regulation of mammalian nucleotide metabolism and biosynthesis. Nucleic Acids Res. 2015, 43, 2466–2485. [Google Scholar] [CrossRef]
- Christophe, T.; Karlsson, A.; Dugave, C.; Rabiet, M.J.; Boulay, F.; Dahlgren, C. The synthetic peptide Trp-Lys-Tyr-Met-Val-Met-NH2 specifically activates neutrophils through FPRL1/lipoxin A4 receptors and is an agonist for the orphan monocyte-expressed chemoattractant receptor FPRL2. J. Biol. Chem. 2001, 276, 21585–21593. [Google Scholar] [CrossRef]
- Leoni, G.; Alam, A.; Neumann, P.A.; Lambeth, J.D.; Cheng, G.; McCoy, J.; Hilgarth, R.S.; Kundu, K.; Murthy, N.; Kusters, D.; et al. Annexin A1, formyl peptide receptor, and NOX1 orchestrate epithelial repair. J. Clin. Investig. 2013, 123, 443–454. [Google Scholar] [CrossRef]
- Iaccio, A.; Collinet, C.; Gesualdi, N.M.; Ammendola, R. Protein kinase C-alpha and -delta are required for NADPH oxidase activation in WKYMVm-stimulated IMR90 human fibroblasts. Arch. Biochem. Biophys. 2007, 459, 288–294. [Google Scholar] [CrossRef]
- Esposito, G.; Carsana, A. Metabolic Alterations in Cardiomyocytes of Patients with Duchenne and Becker Muscular Dystrophies. J. Clin. Med. 2019, 8, 2151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nogueira, V.; Hay, N. Molecular pathways: Reactive oxygen species homeostasis in cancer cells and implications for cancer therapy. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2013, 19, 4309–4314. [Google Scholar] [CrossRef]
- Liu, H.; Huang, D.; McArthur, D.L.; Boros, L.G.; Nissen, N.; Heaney, A.P. Fructose induces transketolase flux to promote pancreatic cancer growth. Cancer Res. 2010, 70, 6368–6376. [Google Scholar] [CrossRef]
- Scalise, M.; Console, L.; Rovella, F.; Galluccio, M.; Pochini, L.; Indiveri, C. Membrane Transporters for Amino Acids as Players of Cancer Metabolic Rewiring. Cells 2020, 9, 2028. [Google Scholar] [CrossRef] [PubMed]
- Bergstrom, J.; Furst, P.; Noree, L.O.; Vinnars, E. Intracellular free amino acid concentration in human muscle tissue. J. Appl. Physiol. 1974, 36, 693–697. [Google Scholar] [CrossRef]
- Altman, B.J.; Stine, Z.E.; Dang, C.V. From Krebs to clinic: Glutamine metabolism to cancer therapy. Nat. Rev. Cancer 2016, 16, 619–634. [Google Scholar] [CrossRef] [PubMed]
- Bhutia, Y.D.; Babu, E.; Ramachandran, S.; Ganapathy, V. Amino Acid transporters in cancer and their relevance to “glutamine addiction”: Novel targets for the design of a new class of anticancer drugs. Cancer Res. 2015, 75, 1782–1788. [Google Scholar] [CrossRef]
- Wise, D.R.; Thompson, C.B. Glutamine addiction: A new therapeutic target in cancer. Trends Biochem. Sci. 2010, 35, 427–433. [Google Scholar] [CrossRef]
- Nicklin, P.; Bergman, P.; Zhang, B.; Triantafellow, E.; Wang, H.; Nyfeler, B.; Yang, H.; Hild, M.; Kung, C.; Wilson, C.; et al. Bidirectional transport of amino acids regulates mTOR and autophagy. Cell 2009, 136, 521–534. [Google Scholar] [CrossRef]
- Marshall, A.D.; van Geldermalsen, M.; Otte, N.J.; Lum, T.; Vellozzi, M.; Thoeng, A.; Pang, A.; Nagarajah, R.; Zhang, B.; Wang, Q.; et al. ASCT2 regulates glutamine uptake and cell growth in endometrial carcinoma. Oncogenesis 2017, 6, e367. [Google Scholar] [CrossRef]
- Wang, V.M.; Ferreira, R.M.M.; Almagro, J.; Evan, T.; Legrave, N.; Zaw Thin, M.; Frith, D.; Carvalho, J.; Barry, D.J.; Snijders, A.P.; et al. CD9 identifies pancreatic cancer stem cells and modulates glutamine metabolism to fuel tumour growth. Nat. Cell Biol. 2019, 21, 1425–1435. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Xiao, D.; Zhao, Z.; Ren, P.; Li, C.; Hu, Y.; Shi, J.; Su, H.; Wang, L.; Liu, H.; et al. Epigenetic silencing of microRNA-137 enhances ASCT2 expression and tumor glutamine metabolism. Oncogenesis 2017, 6, e356. [Google Scholar] [CrossRef] [PubMed]
- Jeon, Y.J.; Khelifa, S.; Ratnikov, B.; Scott, D.A.; Feng, Y.; Parisi, F.; Ruller, C.; Lau, E.; Kim, H.; Brill, L.M.; et al. Regulation of glutamine carrier proteins by RNF5 determines breast cancer response to ER stress-inducing chemotherapies. Cancer Cell 2015, 27, 354–369. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Han, M.; Zhang, X.; He, Y.; Yuan, C.; Xiong, Y.; Li, X.; Zeng, C.; Lu, K.; Zhu, H.; et al. Discoidin domain receptor 1 promotes hepatocellular carcinoma progression through modulation of SLC1A5 and the mTORC1 signaling pathway. Cell. Oncol. 2022, 45, 163–178. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhao, T.; Li, Z.; Wang, L.; Yuan, S.; Sun, L. The role of ASCT2 in cancer: A review. Eur. J. Pharmacol. 2018, 837, 81–87. [Google Scholar] [CrossRef]
- Korangath, P.; Teo, W.W.; Sadik, H.; Han, L.; Mori, N.; Huijts, C.M.; Wildes, F.; Bharti, S.; Zhang, Z.; Santa-Maria, C.A.; et al. Targeting Glutamine Metabolism in Breast Cancer with Aminooxyacetate. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2015, 21, 3263–3273. [Google Scholar] [CrossRef]
- Liu, P.; Ge, M.; Hu, J.; Li, X.; Che, L.; Sun, K.; Cheng, L.; Huang, Y.; Pilo, M.G.; Cigliano, A.; et al. A functional mammalian target of rapamycin complex 1 signaling is indispensable for c-Myc-driven hepatocarcinogenesis. Hepatology 2017, 66, 167–181. [Google Scholar] [CrossRef]
- Seo, H.R.; Kim, J.; Bae, S.; Soh, J.W.; Lee, Y.S. Cdk5-mediated phosphorylation of c-Myc on Ser-62 is essential in transcriptional activation of cyclin B1 by cyclin G1. J. Biol. Chem. 2008, 283, 15601–15610. [Google Scholar] [CrossRef]
- Li, G.; Li, D.; Wang, T.; He, S. Pyrimidine Biosynthetic Enzyme CAD: Its Function, Regulation, and Diagnostic Potential. Int. J. Mol. Sci. 2021, 22, 253. [Google Scholar] [CrossRef]
- Sigoillot, F.D.; Kotsis, D.H.; Serre, V.; Sigoillot, S.M.; Evans, D.R.; Guy, H.I. Nuclear localization and mitogen-activated protein kinase phosphorylation of the multifunctional protein CAD. J. Biol. Chem. 2005, 280, 25611–25620. [Google Scholar] [CrossRef]
- Ben-Sahra, I.; Howell, J.J.; Asara, J.M.; Manning, B.D. Stimulation of de novo pyrimidine synthesis by growth signaling through mTOR and S6K1. Science 2013, 339, 1323–1328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robitaille, A.M.; Christen, S.; Shimobayashi, M.; Cornu, M.; Fava, L.L.; Moes, S.; Prescianotto-Baschong, C.; Sauer, U.; Jenoe, P.; Hall, M.N. Quantitative phosphoproteomics reveal mTORC1 activates de novo pyrimidine synthesis. Science 2013, 339, 1320–1323. [Google Scholar] [CrossRef] [PubMed]
- Sarbassov, D.D.; Sabatini, D.M. Redox regulation of the nutrient-sensitive raptor-mTOR pathway and complex. J. Biol. Chem. 2005, 280, 39505–39509. [Google Scholar] [CrossRef] [PubMed]
- Martensson, J.; Sundqvist, M.; Manandhar, A.; Ieremias, L.; Zhang, L.; Ulven, T.; Xie, X.; Bjorkman, L.; Forsman, H. The Two Formyl Peptide Receptors Differently Regulate GPR84-Mediated Neutrophil NADPH Oxidase Activity. J. Innate Immun. 2021, 13, 242–256. [Google Scholar] [CrossRef]
- Peshavariya, H.M.; Taylor, C.J.; Goh, C.; Liu, G.S.; Jiang, F.; Chan, E.C.; Dusting, G.J. Annexin peptide Ac2-26 suppresses TNFalpha-induced inflammatory responses via inhibition of Rac1-dependent NADPH oxidase in human endothelial cells. PLoS ONE 2013, 8, e60790. [Google Scholar] [CrossRef]
- Filina, Y.; Gabdoulkhakova, A.; Rizvanov, A.; Safronova, V. MAP kinases in regulation of NOX activity stimulated through two types of formyl peptide receptors in murine bone marrow granulocytes. Cell. Signal. 2022, 90, 110205. [Google Scholar] [CrossRef]
- Yoo, H.C.; Park, S.J.; Nam, M.; Kang, J.; Kim, K.; Yeo, J.H.; Kim, J.K.; Heo, Y.; Lee, H.S.; Lee, M.Y.; et al. A Variant of SLC1A5 Is a Mitochondrial Glutamine Transporter for Metabolic Reprogramming in Cancer Cells. Cell Metab. 2020, 31, 267–283.e12. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pecchillo Cimmino, T.; Pagano, E.; Stornaiuolo, M.; Esposito, G.; Ammendola, R.; Cattaneo, F. Formyl-Peptide Receptor 2 Signaling Redirects Glucose and Glutamine into Anabolic Pathways in Metabolic Reprogramming of Lung Cancer Cells. Antioxidants 2022, 11, 1692. https://doi.org/10.3390/antiox11091692
Pecchillo Cimmino T, Pagano E, Stornaiuolo M, Esposito G, Ammendola R, Cattaneo F. Formyl-Peptide Receptor 2 Signaling Redirects Glucose and Glutamine into Anabolic Pathways in Metabolic Reprogramming of Lung Cancer Cells. Antioxidants. 2022; 11(9):1692. https://doi.org/10.3390/antiox11091692
Chicago/Turabian StylePecchillo Cimmino, Tiziana, Ester Pagano, Mariano Stornaiuolo, Gabriella Esposito, Rosario Ammendola, and Fabio Cattaneo. 2022. "Formyl-Peptide Receptor 2 Signaling Redirects Glucose and Glutamine into Anabolic Pathways in Metabolic Reprogramming of Lung Cancer Cells" Antioxidants 11, no. 9: 1692. https://doi.org/10.3390/antiox11091692
APA StylePecchillo Cimmino, T., Pagano, E., Stornaiuolo, M., Esposito, G., Ammendola, R., & Cattaneo, F. (2022). Formyl-Peptide Receptor 2 Signaling Redirects Glucose and Glutamine into Anabolic Pathways in Metabolic Reprogramming of Lung Cancer Cells. Antioxidants, 11(9), 1692. https://doi.org/10.3390/antiox11091692