NOX2-Induced Activation of Arginase and Diabetes-Induced Retinal Endothelial Cell Senescence


 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mouse Model
2.2. Endothelial Cell Culture
2.3. High Glucose or Hydrogen Peroxide Treatment of Endothelial Cells
2.4. Nitric Oxide Assays
2.5. Superoxide Assay
2.6. Arginase Activity Assay
2.7. Senescence Associated β-Galactosidase (SA-β-Gal) Activity Assay
2.8. Western Blot
2.9. Statistical Analysis
3. Results
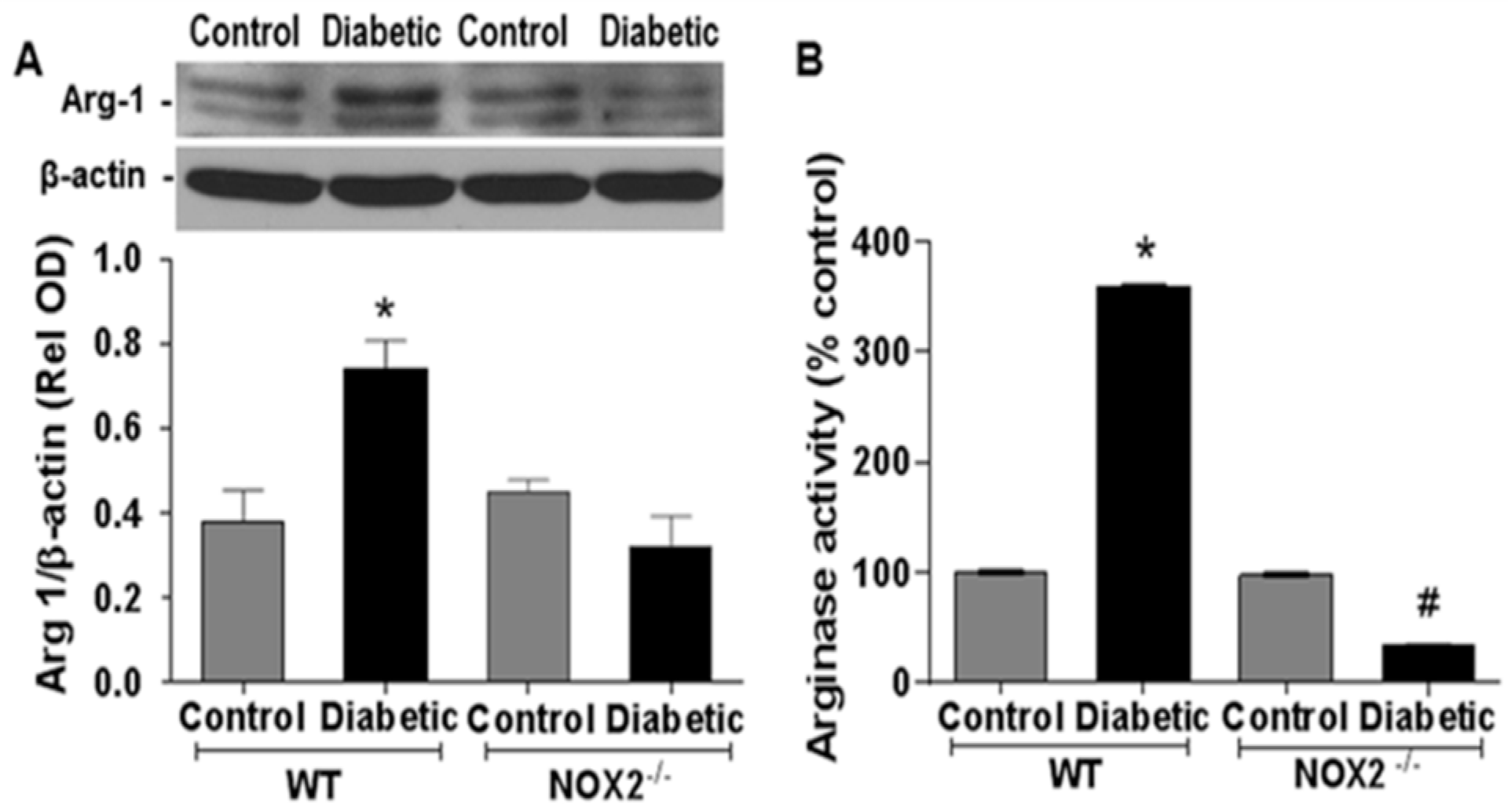
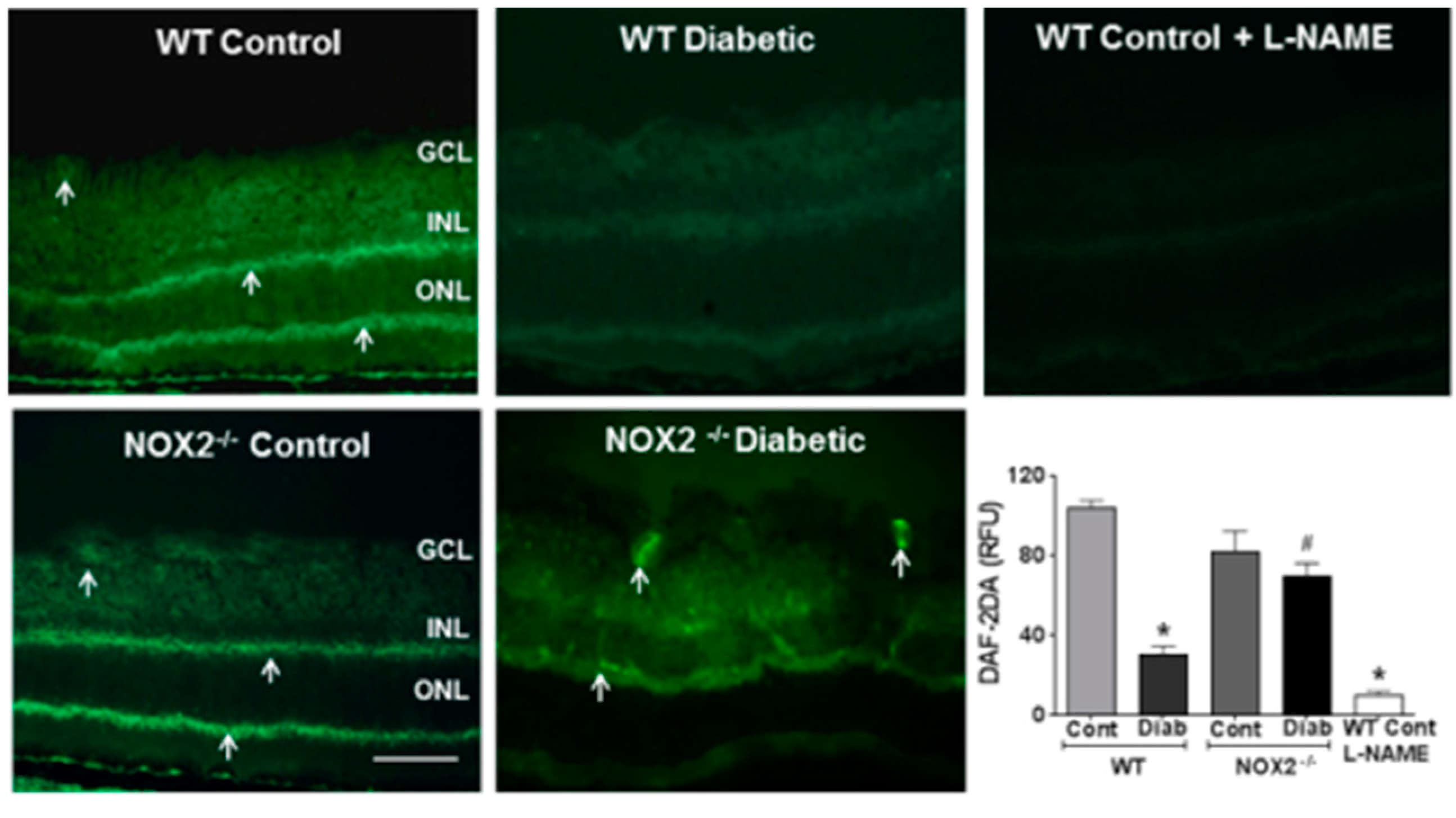
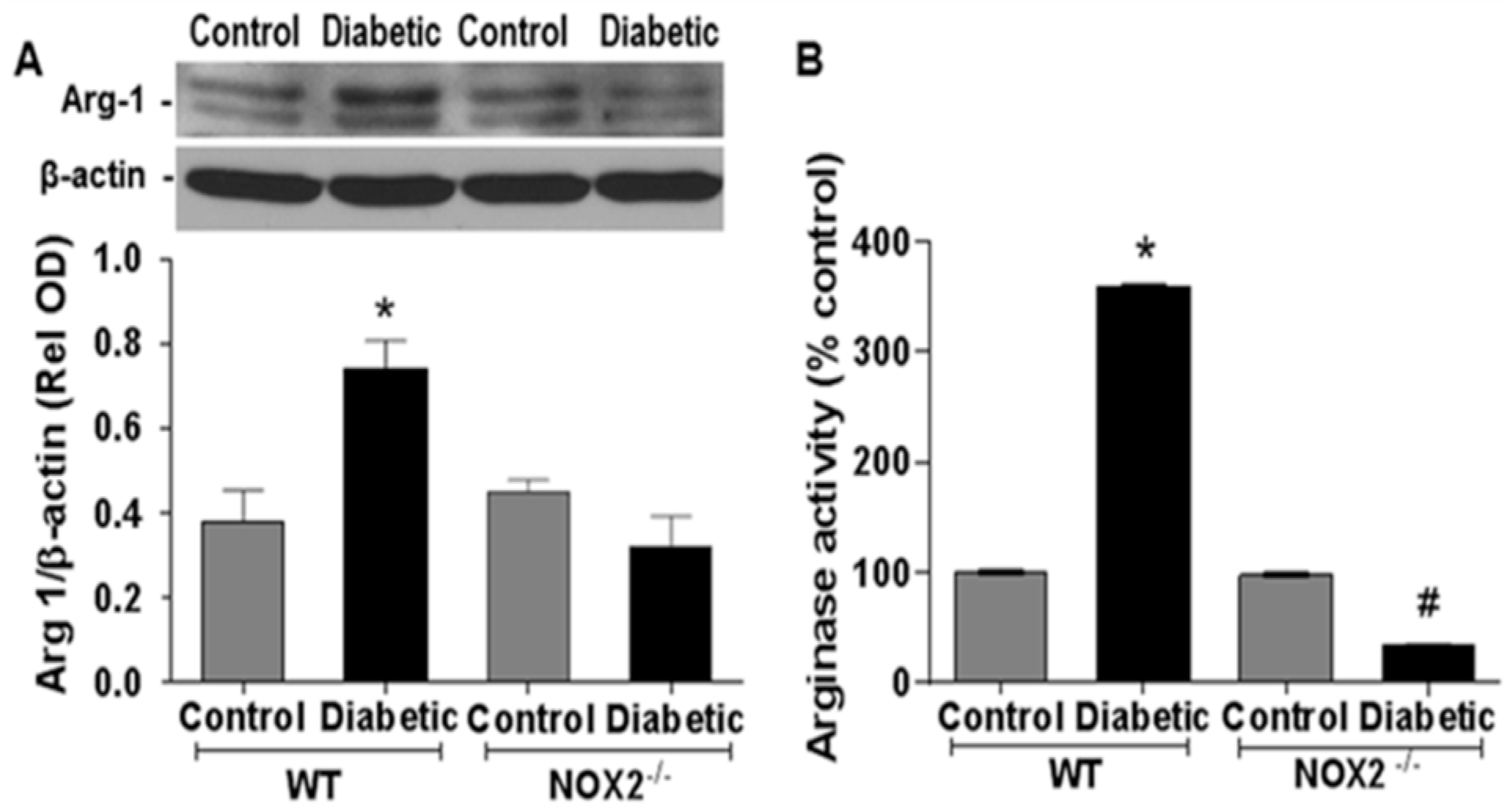
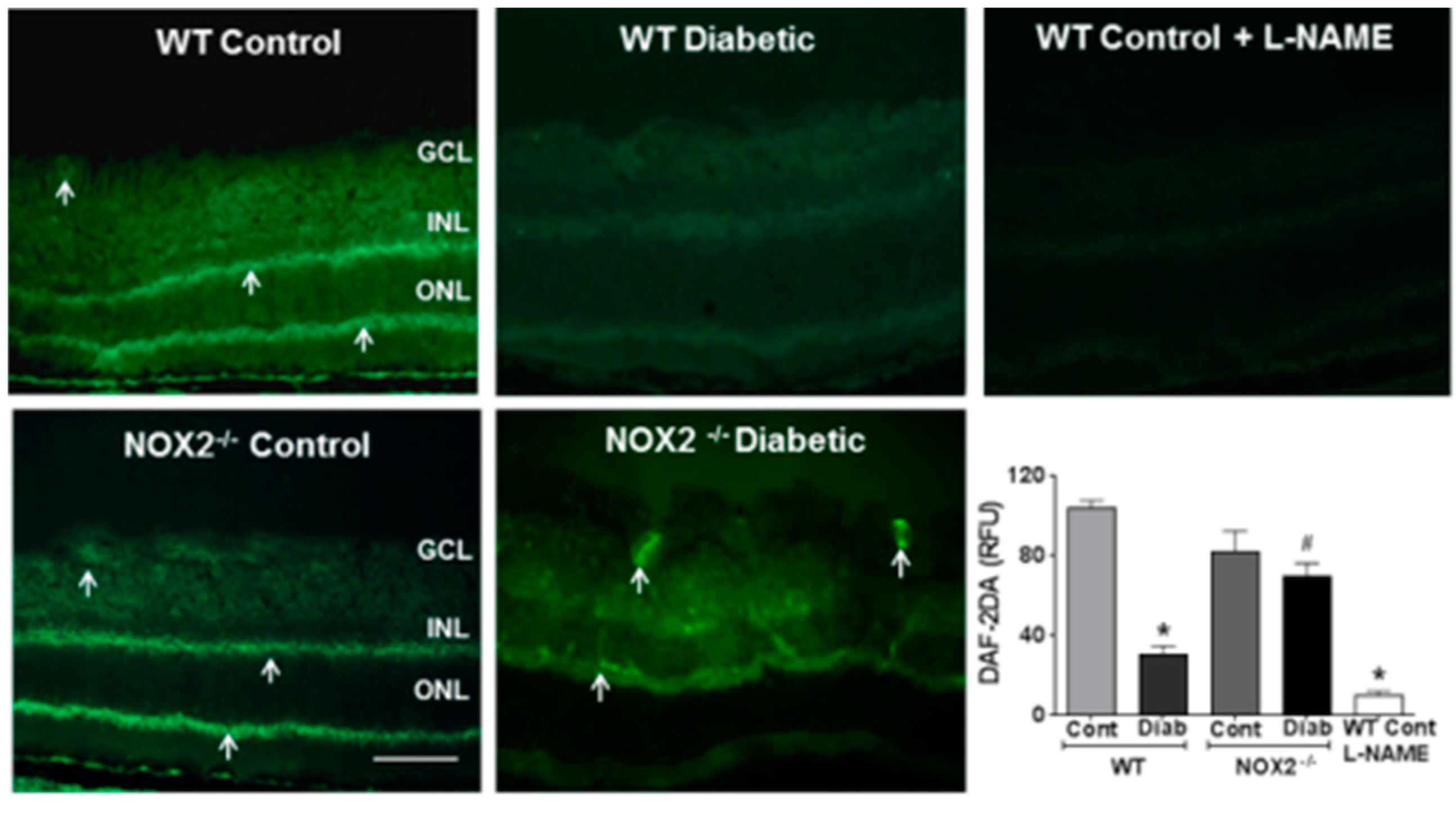
3.1. Effects of NOX2 Deletion on Arginase Expression/Activity and NO Formation
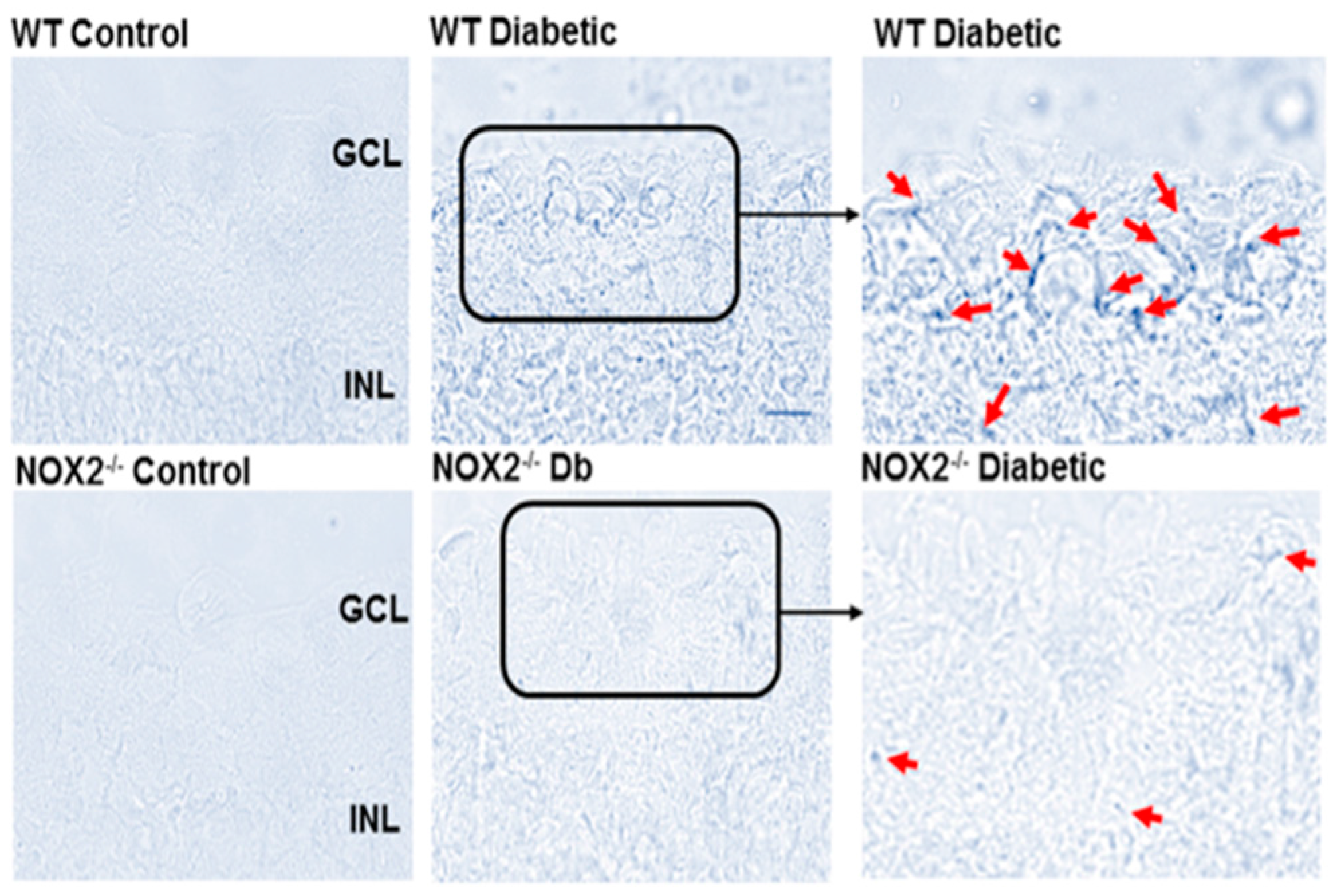
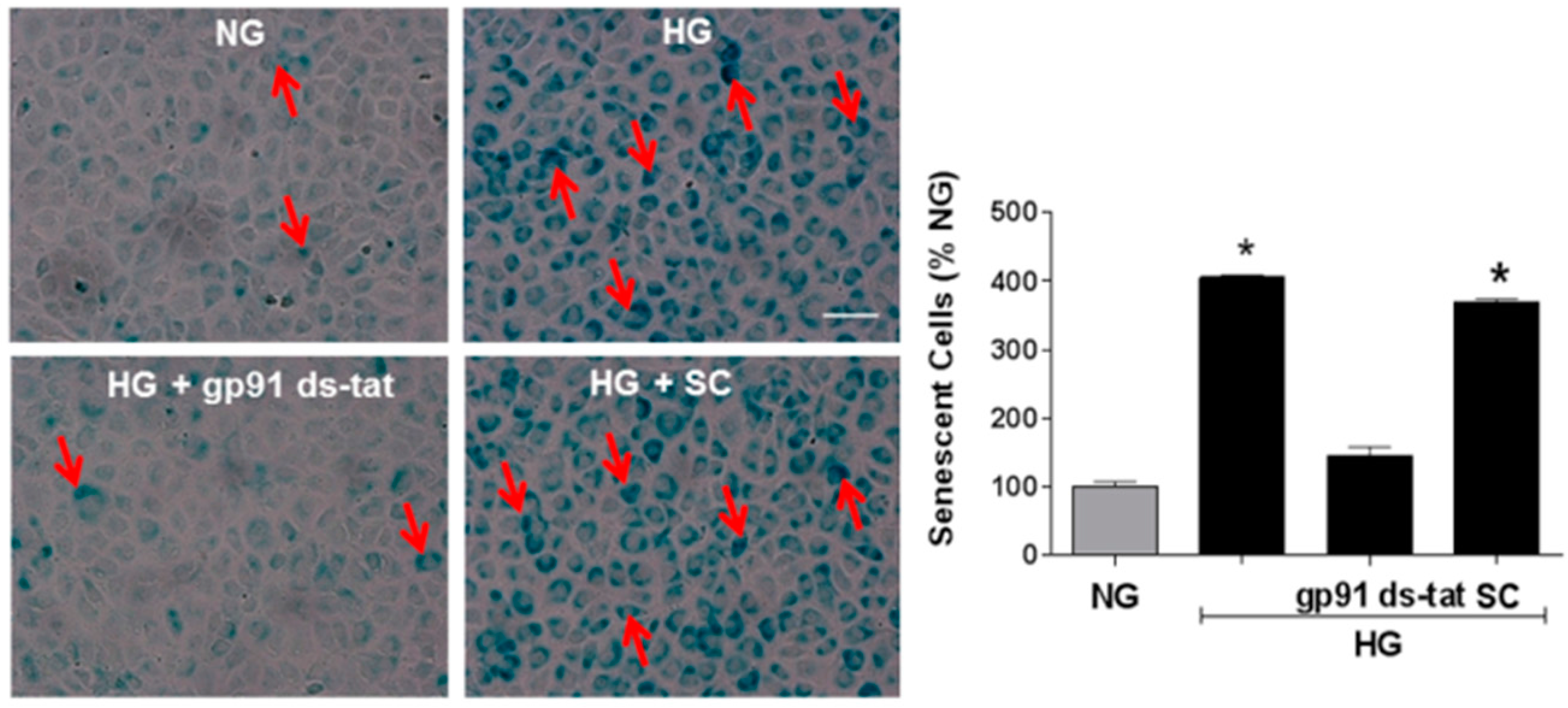
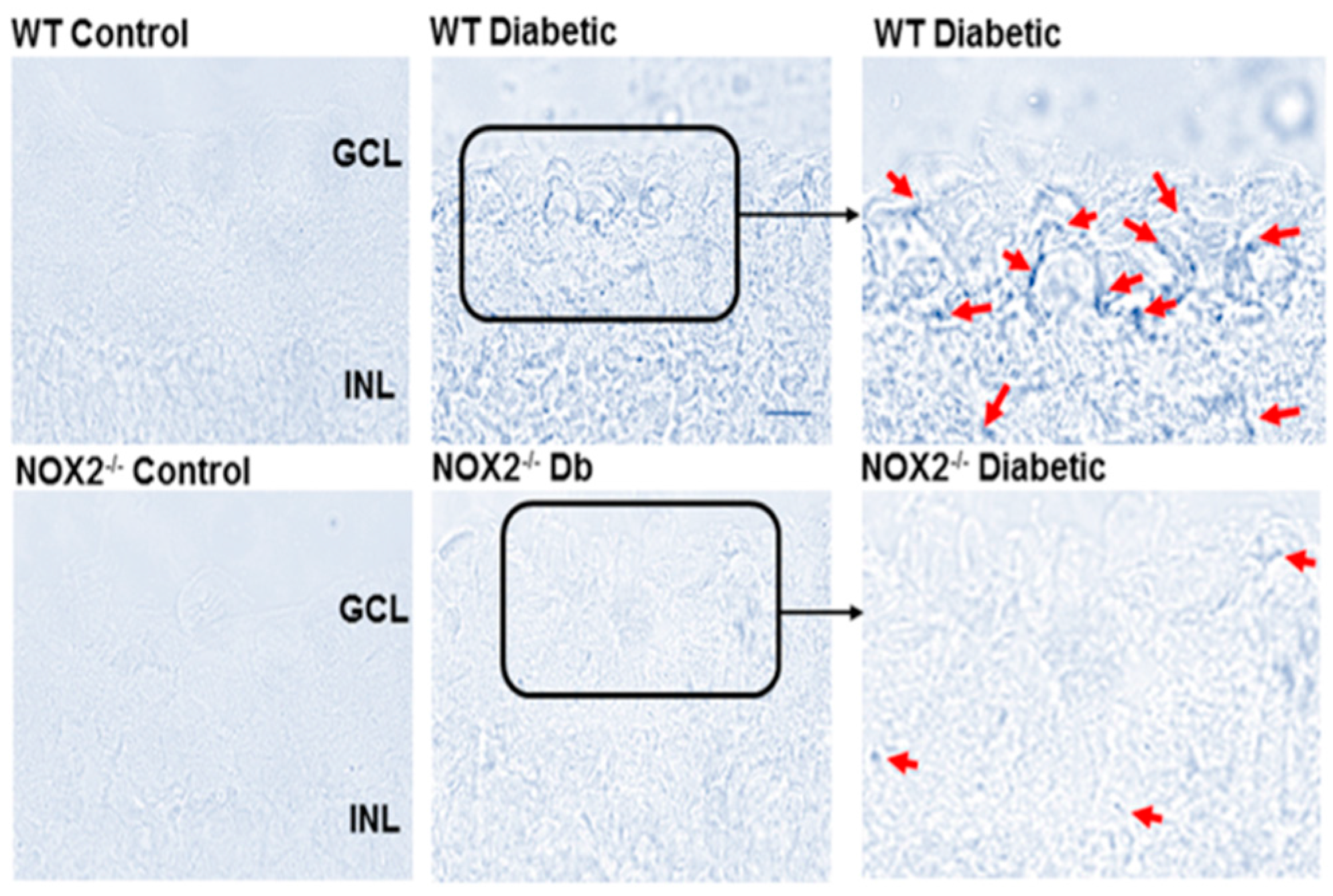
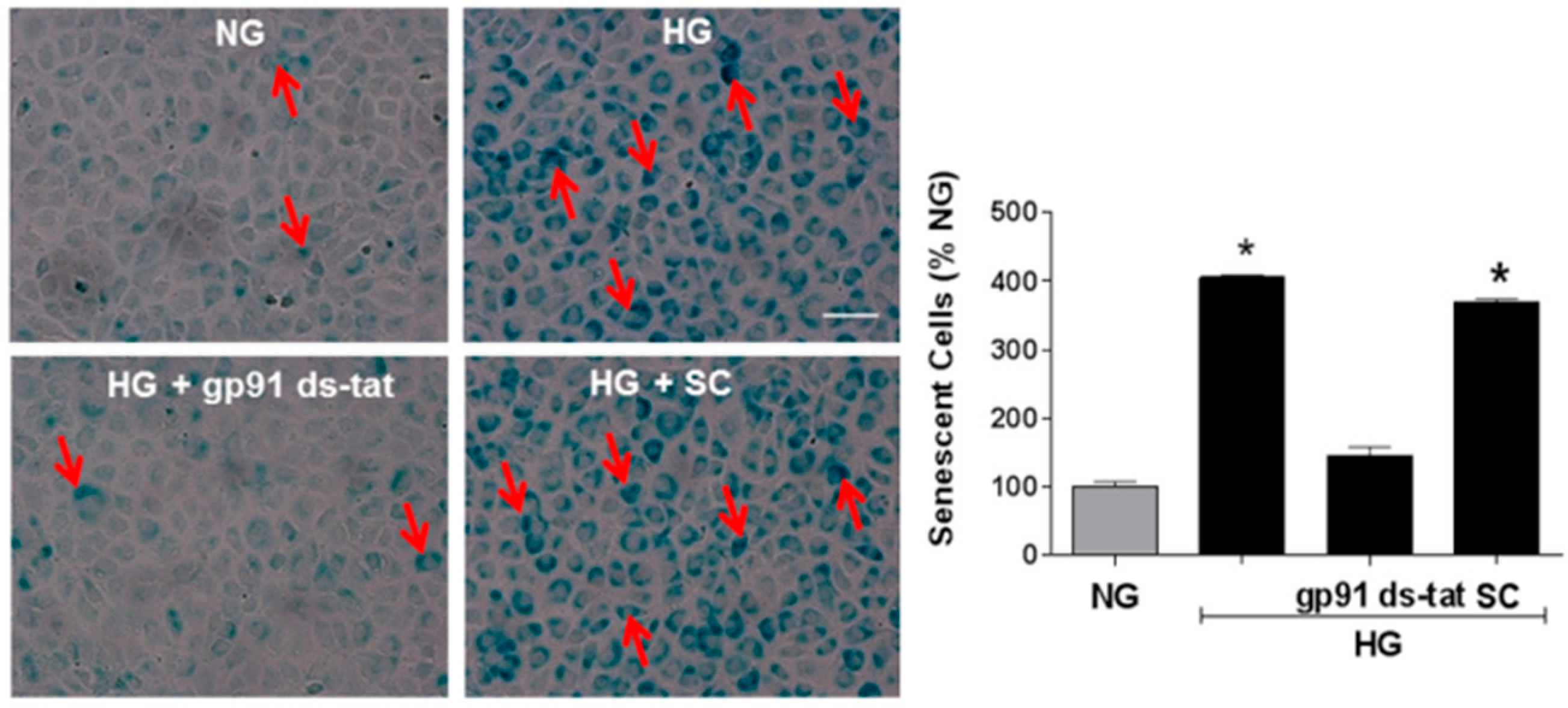
3.2. Effects of NOX2 Deletion/Blockade on Diabetes- or High Glucose-Induced Increases in SA-β-Gal Activity
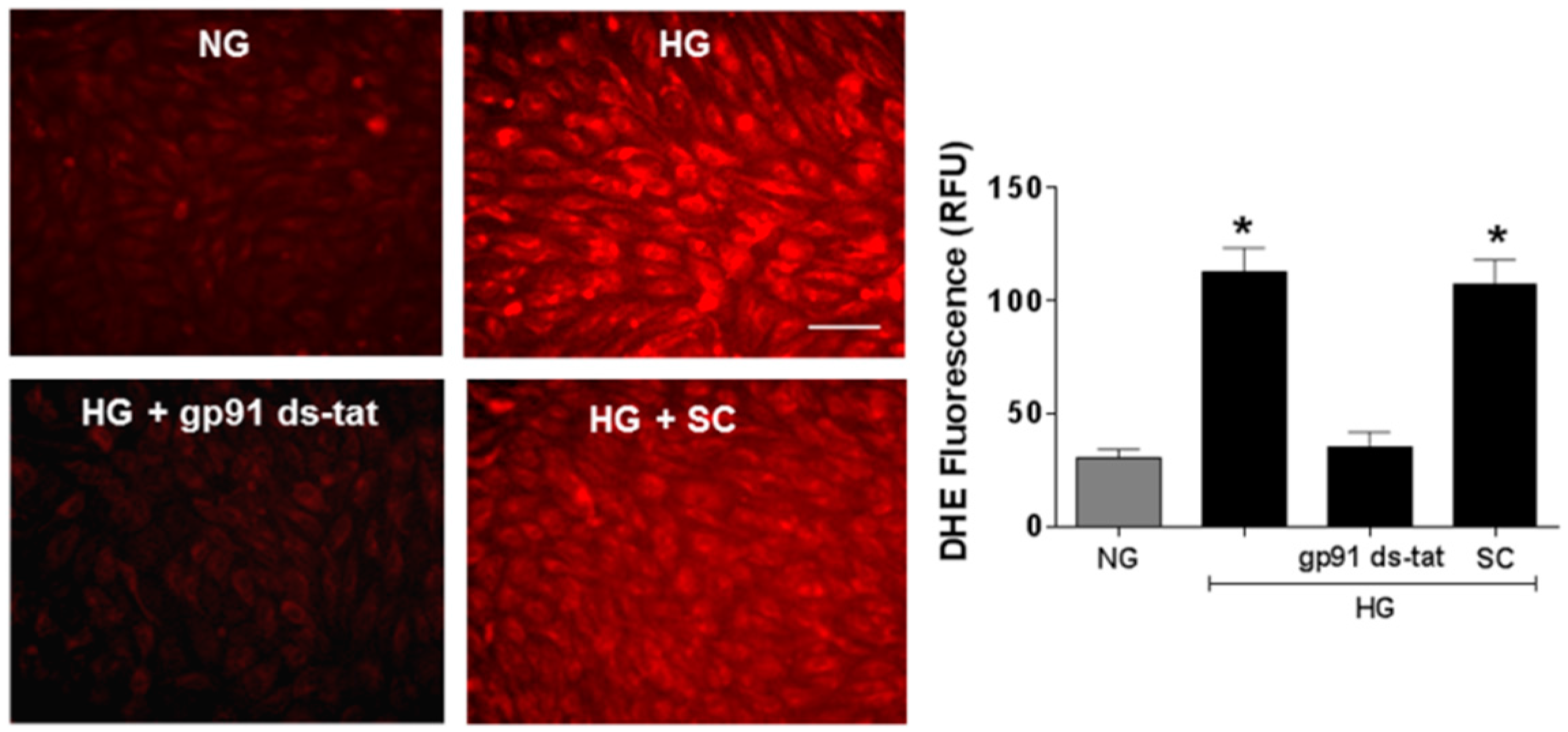
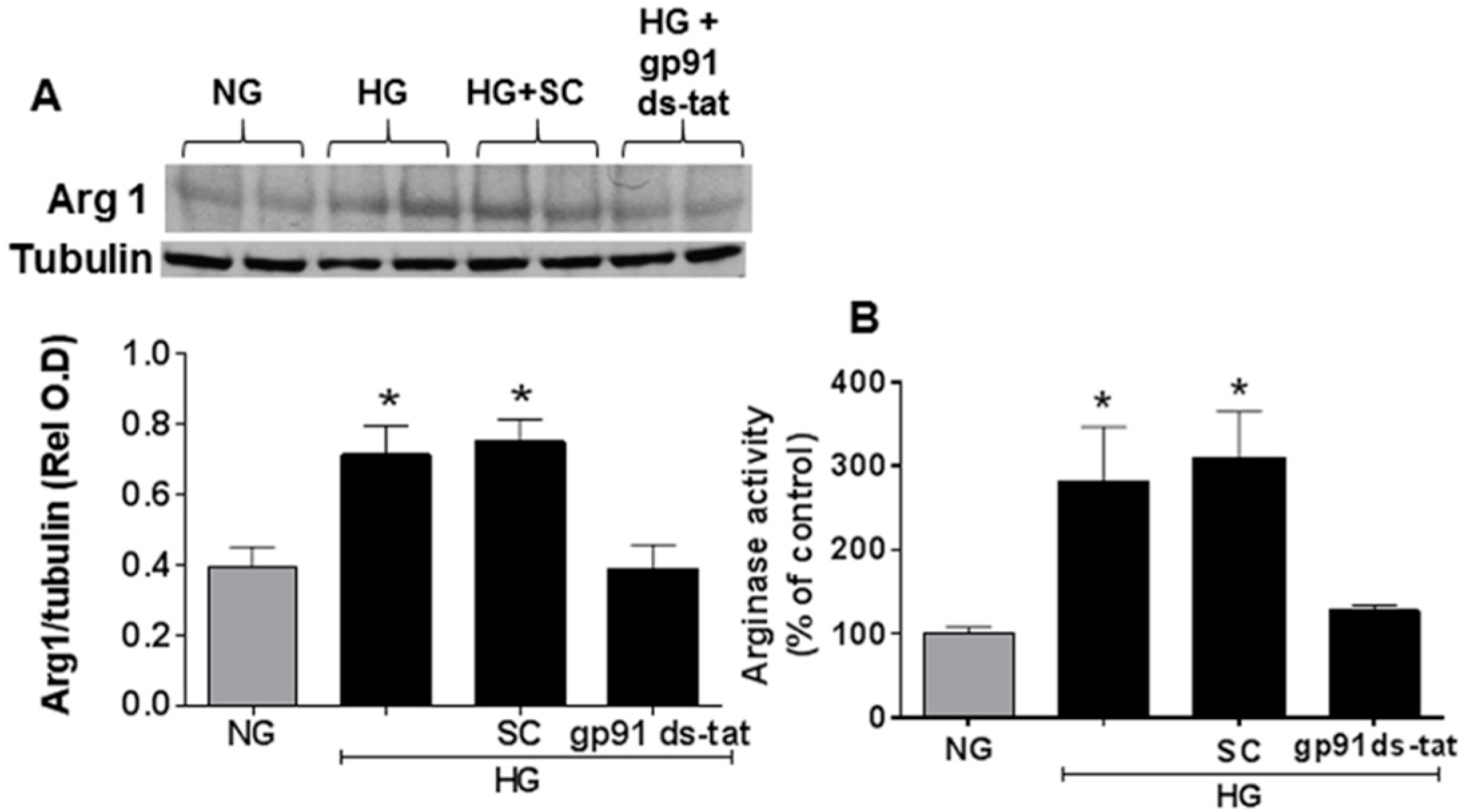
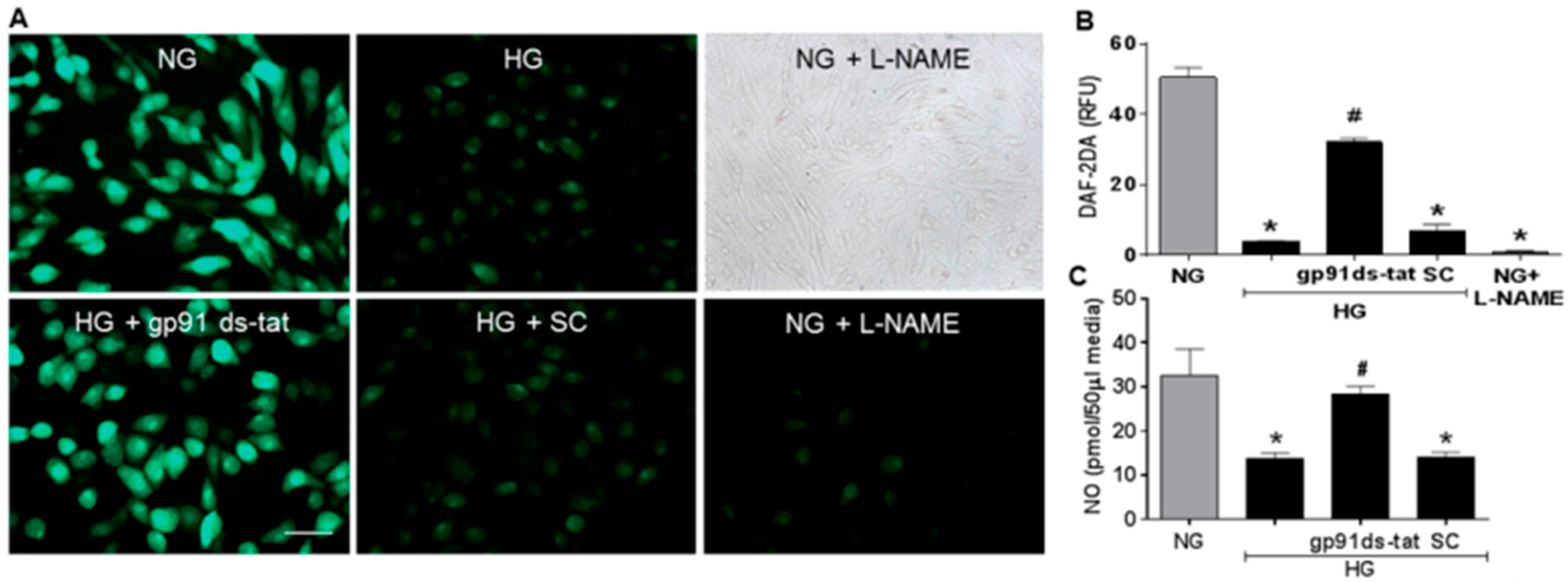
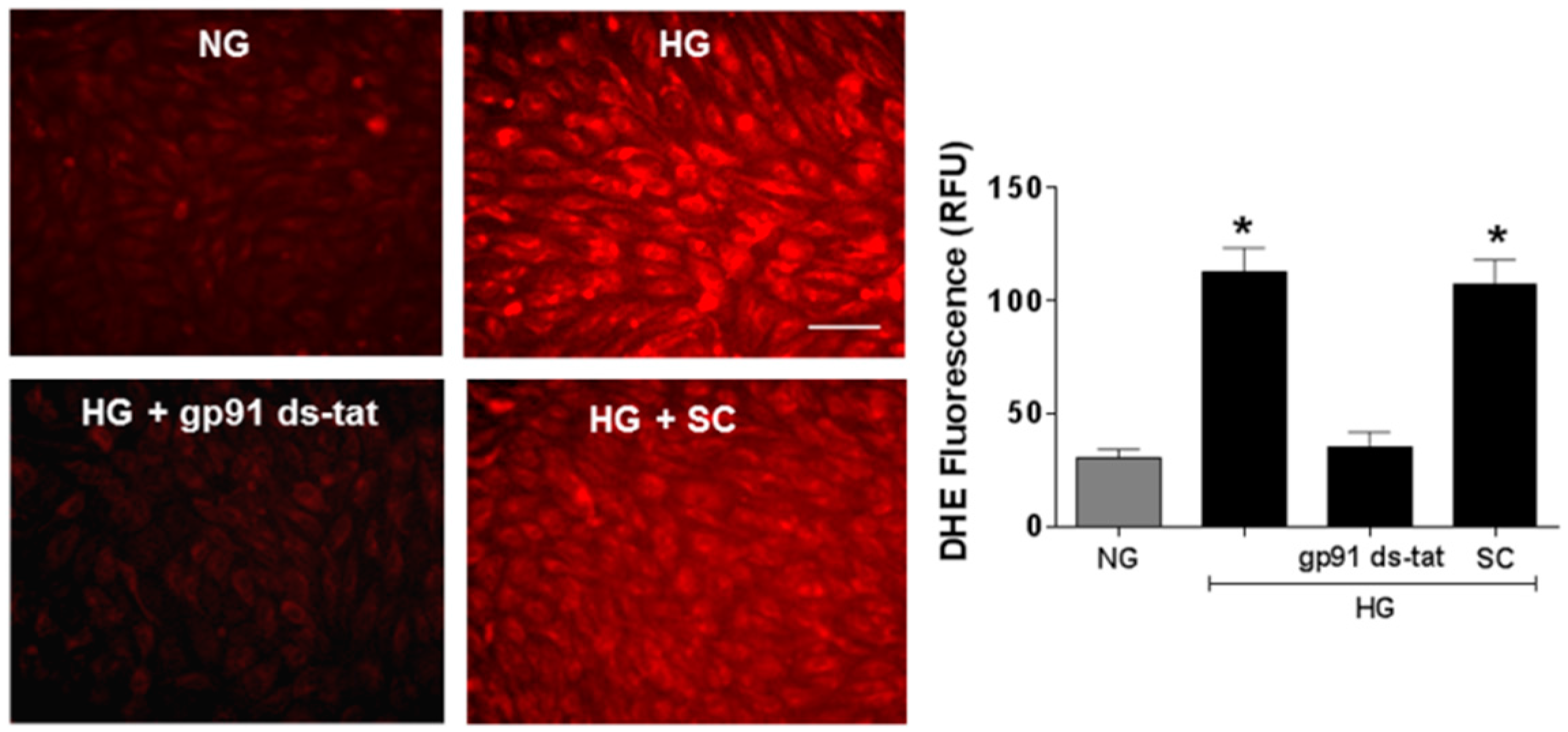
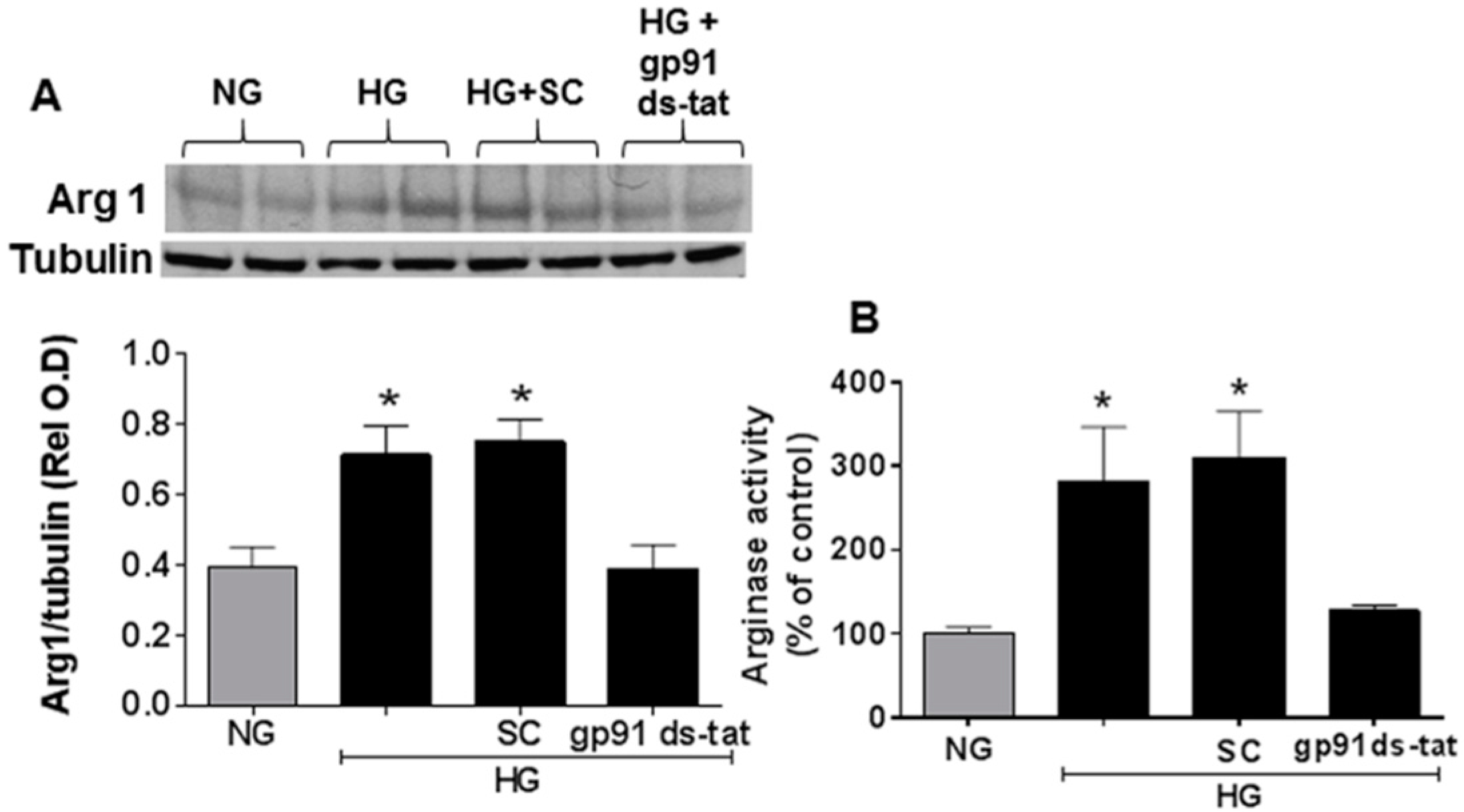
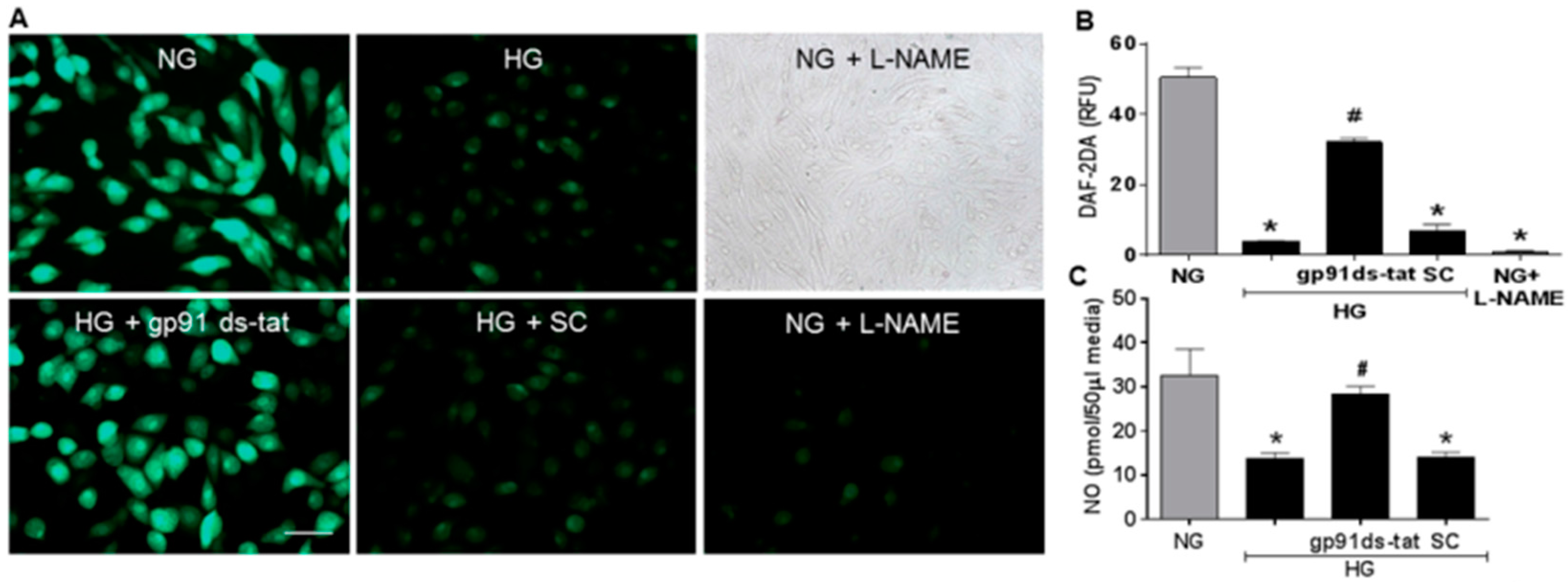
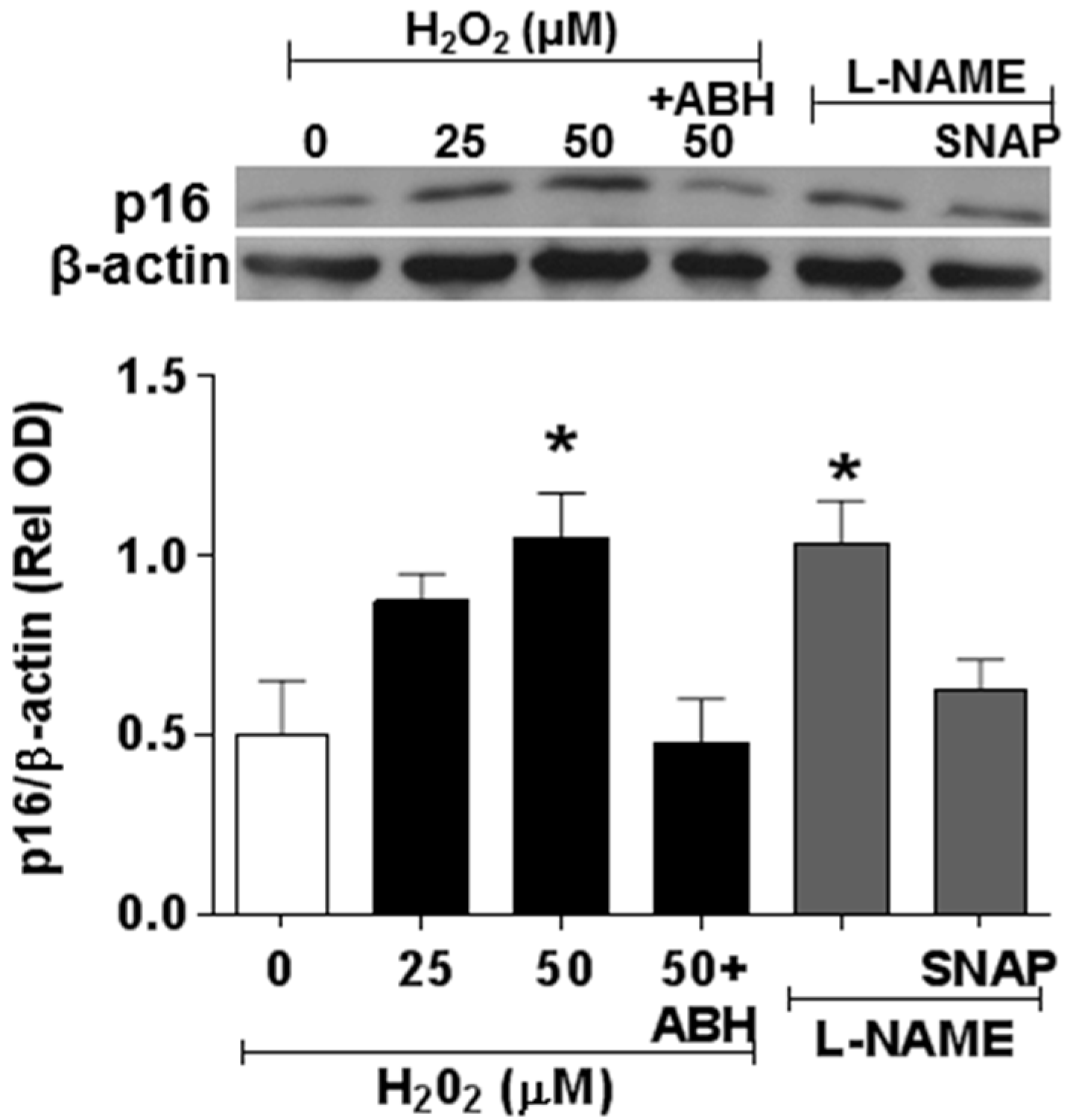
3.3. Effects of NOX2 Blockade on High Glucose-Induced Arginase Expression and Activity and NO Formation
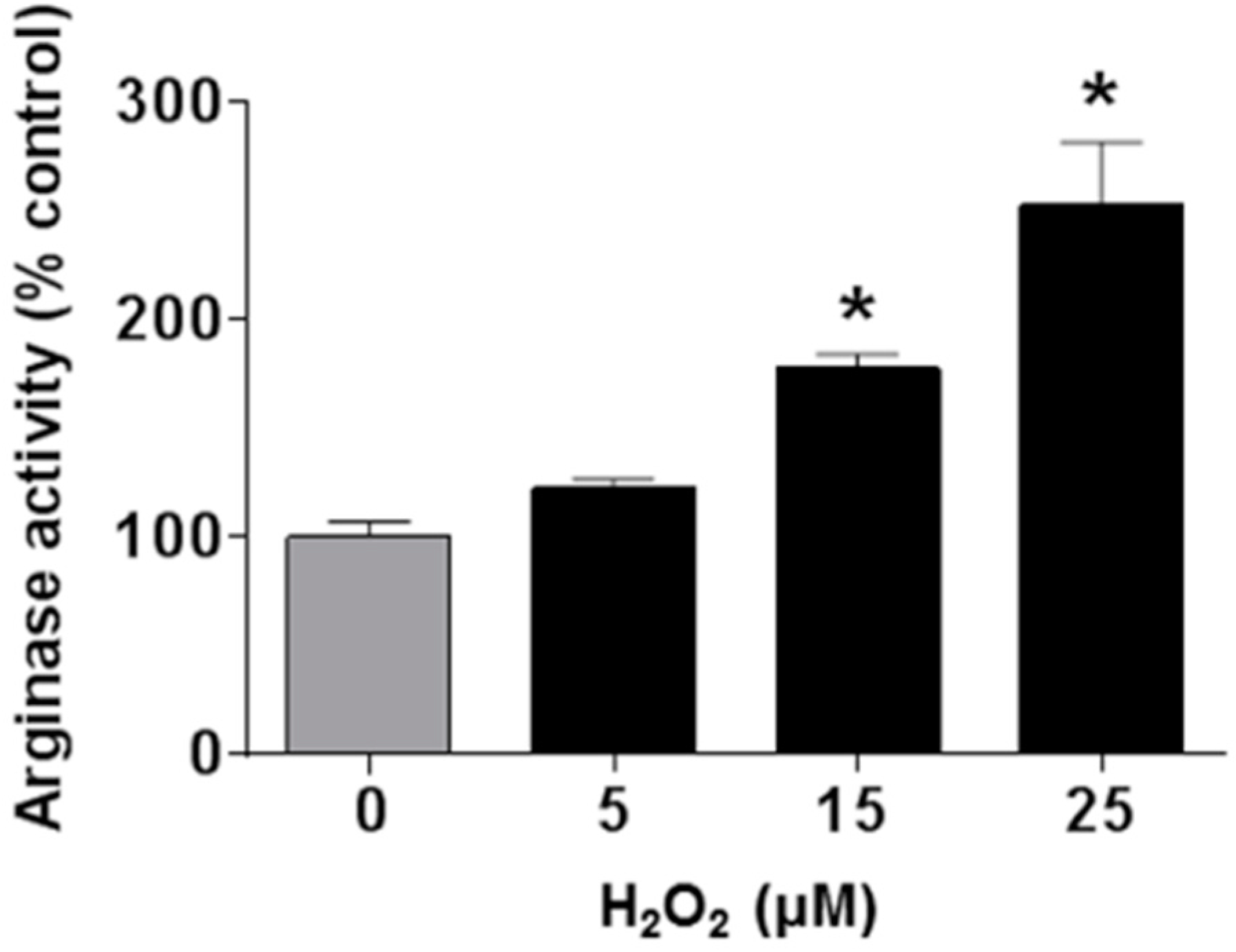
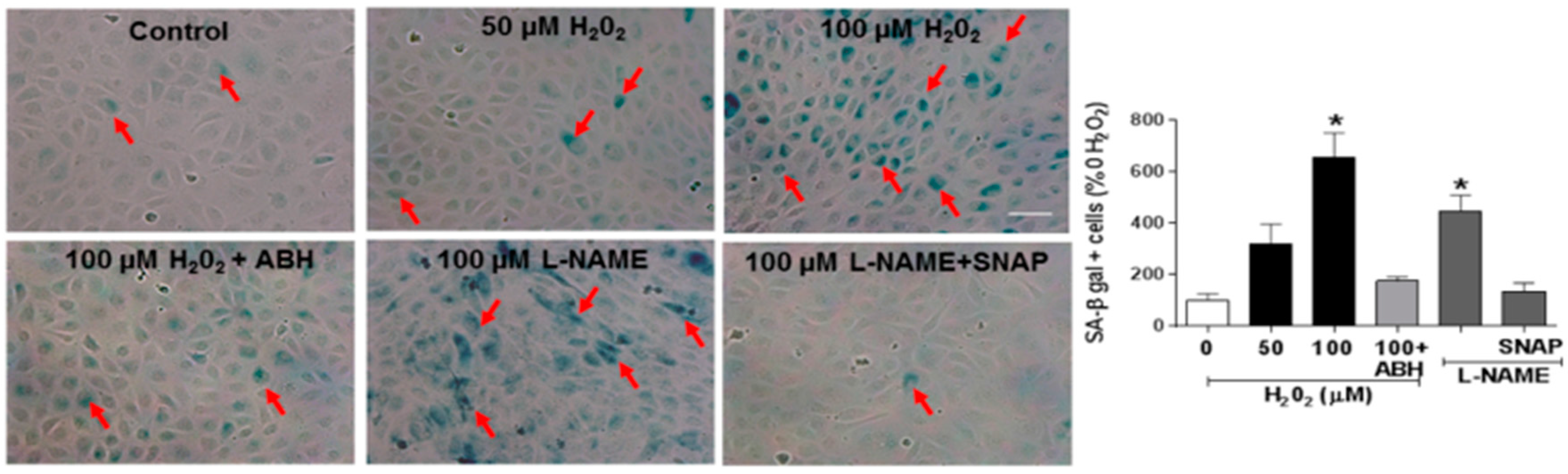
3.4. Involvement of Arginase and NOS Activity in ROS-Induced EC Senescence
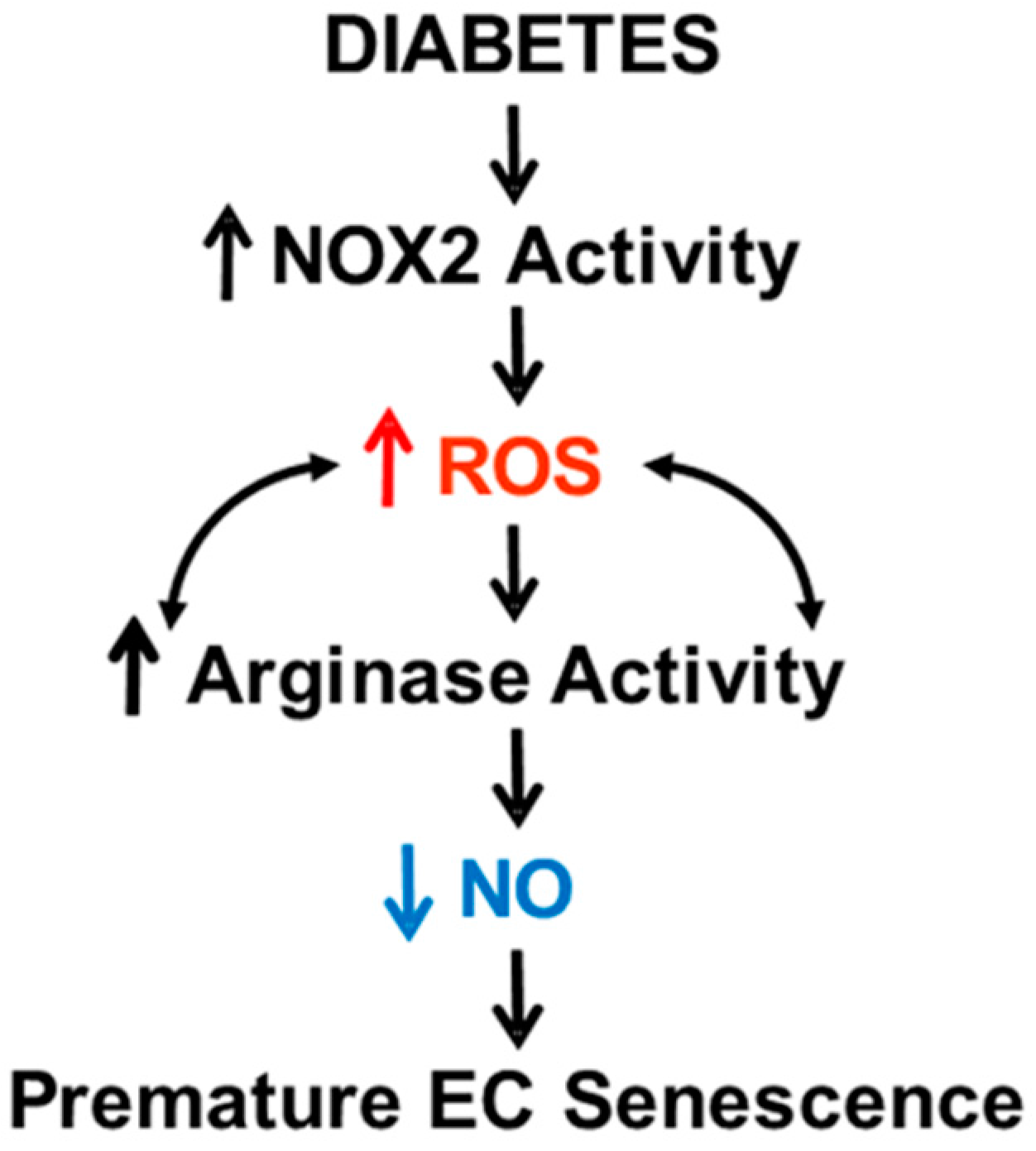
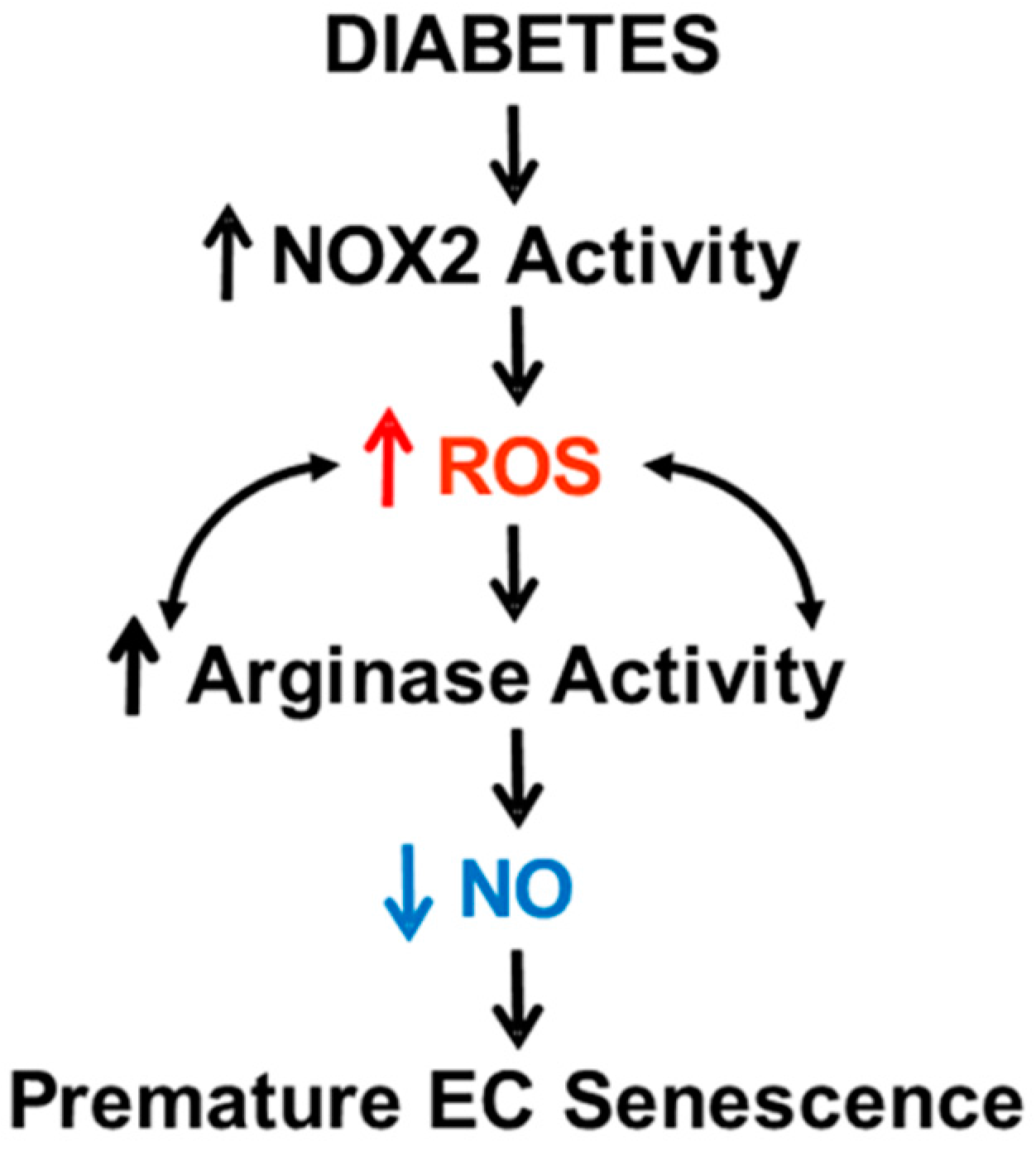
4. Discussion
Supplementary Materials
Acknowledgments
Funding
Author Contributions
Conflicts of Interest
Abbreviations
References
- Zhang, X.; Saaddine, J.B.; Chou, C.F.; Cotch, M.F.; Cheng, Y.J.; Geiss, L.S.; Gregg, E.W.; Albright, A.L.; Klein, B.E.; Klein, R. Prevalence of diabetic retinopathy in the United States, 2005–2008. JAMA 2010, 304, 649–656. [Google Scholar] [CrossRef] [PubMed]
- Hammes, H.P.; Feng, Y.; Pfister, F.; Brownlee, M. Diabetic retinopathy: Targeting vasoregression. Diabetes 2011, 60, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Stitt, A.W.; Curtis, T.M.; Chen, M.; Medina, R.J.; McKay, G.J.; Jenkins, A.; Gardiner, T.A.; Lyons, T.J.; Hammes, H.P.; Simo, R.; et al. The progress in understanding and treatment of diabetic retinopathy. Prog. Retin. Eye Res. 2016, 51, 156–186. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.R.; Gardner, T.W. Diabetic Retinopathy and Diabetic Macular Edema. Dev. Ophthalmol. 2016, 55, 137–146. [Google Scholar] [PubMed]
- Goldberg, M.F. Retinal detachment associated with proliferative retinopathies (sickle cell disease, retrolental fibroplasia and diabetes mellitus). Isr. J. Med. Sci. 1972, 8, 1447–1457. [Google Scholar] [PubMed]
- Kowluru, R.A.; Kowluru, A.; Mishra, M.; Kumar, B. Oxidative stress and epigenetic modifications in the pathogenesis of diabetic retinopathy. Prog. Retin. Eye Res. 2015, 48, 40–61. [Google Scholar] [CrossRef] [PubMed]
- Caldwell, R.B.; Bartoli, M.; Behzadian, M.A.; El-Remessy, A.E.; Al-Shabrawey, M.; Platt, D.H.; Liou, G.I.; Caldwell, R.W. Vascular endothelial growth factor and diabetic retinopathy: Role of oxidative stress. Curr. Drug Targets 2005, 6, 511–524. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Vanhoutte, P.M. Macro- and microvascular endothelial dysfunction in diabetes. J. Diabetes 2017, 9, 434–449. [Google Scholar] [CrossRef] [PubMed]
- Caldwell, R.B.; Toque, H.A.; Narayanan, S.P.; Caldwell, R.W. Arginase: An old enzyme with new tricks. Trends Pharmacol. Sci. 2015, 36, 395–405. [Google Scholar] [CrossRef] [PubMed]
- Durante, W.; Johnson, F.K.; Johnson, R.A. Arginase: A critical regulator of nitric oxide synthesis and vascular function. Clin. Exp. Pharmacol. Physiol. 2007, 34, 906–911. [Google Scholar] [CrossRef] [PubMed]
- Forstermann, U.; Nakane, M.; Tracey, W.R.; Pollock, J.S. Isoforms of nitric oxide synthase: Functions in the cardiovascular system. Eur. Heart J. 1993, 14 (Suppl. I), 10–15. [Google Scholar] [CrossRef]
- Kubes, P.; Suzuki, M.; Granger, D.N. Nitric oxide: An endogenous modulator of leukocyte adhesion. Proc. Natl. Acad. Sci. USA 1991, 88, 4651–4655. [Google Scholar] [CrossRef] [PubMed]
- Salama, R.; Sadaie, M.; Hoare, M.; Narita, M. Cellular senescence and its effector programs. Genes Dev. 2014, 28, 99–114. [Google Scholar] [CrossRef] [PubMed]
- Maeda, M.; Hayashi, T.; Mizuno, N.; Hattori, Y.; Kuzuya, M. Intermittent high glucose implements stress-induced senescence in human vascular endothelial cells: Role of superoxide production by NADPH oxidase. PLoS ONE 2015, 10, e0123169. [Google Scholar] [CrossRef] [PubMed]
- Yepuri, G.; Velagapudi, S.; Xiong, Y.; Rajapakse, A.G.; Montani, J.P.; Ming, X.F.; Yang, Z. Positive crosstalk between arginase-II and S6K1 in vascular endothelial inflammation and aging. Aging Cell 2012, 11, 1005–1016. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Yu, Y.; Montani, J.P.; Ming, X.F.; Yang, Z. Arginase-I enhances vascular endothelial inflammation and senescence through eNOS-uncoupling. BMC Res. Notes 2017, 10, 82. [Google Scholar] [CrossRef] [PubMed]
- Lamoke, F.; Shaw, S.; Yuan, J.; Ananth, S.; Duncan, M.; Martin, P.; Bartoli, M. Increased Oxidative and Nitrative Stress Accelerates Aging of the Retinal Vasculature in the Diabetic Retina. PLoS ONE 2015, 10, e0139664. [Google Scholar] [CrossRef] [PubMed]
- Oubaha, M.; Miloudi, K.; Dejda, A.; Guber, V.; Mawambo, G.; Germain, M.A.; Bourdel, G.; Popovic, N.; Rezende, F.A.; Kaufman, R.J.; et al. Senescence-associated secretory phenotype contributes to pathological angiogenesis in retinopathy. Sci. Transl. Med. 2016, 8, 362ra144. [Google Scholar] [CrossRef] [PubMed]
- Al-Shabrawey, M.; Bartoli, M.; El-Remessy, A.B.; Ma, G.; Matragoon, S.; Lemtalsi, T.; Caldwell, R.W.; Caldwell, R.B. Role of NADPH oxidase and Stat3 in statin-mediated protection against diabetic retinopathy. Investig. Ophthalmol. Vis. Sci. 2008, 49, 3231–3238. [Google Scholar] [CrossRef] [PubMed]
- Al-Shabrawey, M.; Rojas, M.; Sanders, T.; Behzadian, A.; El-Remessy, A.; Bartoli, M.; Parpia, A.K.; Liou, G.; Caldwell, R.B. Role of NADPH oxidase in retinal vascular inflammation. Investig. Ophthalmol. Vis. Sci. 2008, 49, 3239–3244. [Google Scholar] [CrossRef] [PubMed]
- Rojas, M.; Zhang, W.; Xu, Z.; Lemtalsi, T.; Chandler, P.; Toque, H.A.; Caldwell, R.W.; Caldwell, R.B. Requirement of NOX2 expression in both retina and bone marrow for diabetes-induced retinal vascular injury. PLoS ONE 2013, 8, e84357. [Google Scholar] [CrossRef] [PubMed]
- Patel, C.; Rojas, M.; Narayanan, S.P.; Zhang, W.; Xu, Z.; Lemtalsi, T.; Jittiporn, K.; Caldwell, R.W.; Caldwell, R.B. Arginase as a mediator of diabetic retinopathy. Front. Immunol. 2013, 4, 173. [Google Scholar] [CrossRef] [PubMed]
- Brodsky, S.V.; Gealekman, O.; Chen, J.; Zhang, F.; Togashi, N.; Crabtree, M.; Gross, S.S.; Nasjletti, A.; Goligorsky, M.S. Prevention and reversal of premature endothelial cell senescence and vasculopathy in obesity-induced diabetes by ebselen. Circ. Res. 2004, 94, 377–384. [Google Scholar] [CrossRef] [PubMed]
- Chandra, S.; Romero, M.J.; Shatanawi, A.; Alkilany, A.M.; Caldwell, R.B.; Caldwell, R.W. Oxidative species increase arginase activity in endothelial cells through the RhoA/Rho kinase pathway. Br. J. Pharmacol. 2012, 165, 506–519. [Google Scholar] [CrossRef] [PubMed]
- Shatanawi, A.; Lemtalsi, T.; Yao, L.; Patel, C.; Caldwell, R.B.; Caldwell, R.W. Angiotensin II limits NO production by upregulating arginase through a p38 MAPK-ATF-2 pathway. Eur. J. Pharmacol. 2015, 746, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Lai, A.K.; Lo, A.C. Animal models of diabetic retinopathy: Summary and comparison. J. Diabetes Res. 2013, 2013, 106594. [Google Scholar] [CrossRef] [PubMed]
- Behzadian, M.A.; Wang, X.L.; Jiang, B.; Caldwell, R.B. Angiostatic role of astrocytes: Suppression of vascular endothelial cell growth by TGF-beta and other inhibitory factor(s). Glia 1995, 15, 480–490. [Google Scholar] [CrossRef] [PubMed]
- Csanyi, G.; Cifuentes-Pagano, E.; Al Ghouleh, I.; Ranayhossaini, D.J.; Egana, L.; Lopes, L.R.; Jackson, H.M.; Kelley, E.E.; Pagano, P.J. Nox2 B-loop peptide, Nox2ds, specifically inhibits the NADPH oxidase Nox2. Free Radic. Biol. Med. 2011, 51, 1116–1125. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.D.; Kim, N.N.; Traish, A.M.; Christianson, D.W. Arginase-boronic acid complex highlights a physiological role in erectile function. Nat. Struct. Biol. 1999, 6, 1043–1047. [Google Scholar] [PubMed]
- Pudlo, M.; Demougeot, C.; Girard-Thernier, C. Arginase Inhibitors: A Rational Approach Over One Century. Med. Res. Rev. 2017, 37, 475–513. [Google Scholar] [CrossRef] [PubMed]
- Toque, H.A.; Nunes, K.P.; Rojas, M.; Bhatta, A.; Yao, L.; Xu, Z.; Romero, M.J.; Webb, R.C.; Caldwell, R.B.; Caldwell, R.W. Arginase 1 mediates increased blood pressure and contributes to vascular endothelial dysfunction in deoxycorticosterone acetate-salt hypertension. Front. Immunol. 2013, 4, 219. [Google Scholar] [CrossRef] [PubMed]
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O.; et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9363–9367. [Google Scholar] [CrossRef] [PubMed]
- Giacco, F.; Brownlee, M. Oxidative stress and diabetic complications. Circ. Res. 2010, 107, 1058–1070. [Google Scholar] [CrossRef] [PubMed]
- Goligorsky, M.S. Vascular endothelium in diabetes. Am. J. Physiol. Renal. Physiol. 2017, 312, F266–F275. [Google Scholar] [CrossRef] [PubMed]
- Paravicini, T.M.; Touyz, R.M. NADPH oxidases, reactive oxygen species, and hypertension: Clinical implications and therapeutic possibilities. Diabetes Care 2008, 31 (Suppl. 2), S170–S180. [Google Scholar] [CrossRef] [PubMed]
- Sankaralingam, S.; Xu, H.; Davidge, S.T. Arginase contributes to endothelial cell oxidative stress in response to plasma from women with preeclampsia. Cardiovasc. Res. 2010, 85, 194–203. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Huang, X.; Halicka, D.; Brodsky, S.; Avram, A.; Eskander, J.; Bloomgarden, N.A.; Darzynkiewicz, Z.; Goligorsky, M.S. Contribution of p16INK4a and p21CIP1 pathways to induction of premature senescence of human endothelial cells: Permissive role of p53. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H1575–H1586. [Google Scholar] [CrossRef] [PubMed]
- Krishnamurthy, J.; Torrice, C.; Ramsey, M.R.; Kovalev, G.I.; Al-Regaiey, K.; Su, L.; Sharpless, N.E. Ink4a/Arf expression is a biomarker of aging. J. Clin. Investig. 2004, 114, 1299–1307. [Google Scholar] [CrossRef] [PubMed]
- Selemidis, S.; Dusting, G.J.; Peshavariya, H.; Kemp-Harper, B.K.; Drummond, G.R. Nitric oxide suppresses NADPH oxidase-dependent superoxide production by S-nitrosylation in human endothelial cells. Cardiovasc. Res. 2007, 75, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Qian, J.; Chen, F.; Kovalenkov, Y.; Pandey, D.; Moseley, M.A.; Foster, M.W.; Black, S.M.; Venema, R.C.; Stepp, D.W.; Fulton, D.J. Nitric oxide reduces NADPH oxidase 5 (Nox5) activity by reversible S-nitrosylation. Free Radic. Biol. Med. 2012, 52, 1806–1819. [Google Scholar] [CrossRef] [PubMed]
- Bode-Boger, S.M.; Martens-Lobenhoffer, J.; Tager, M.; Schroder, H.; Scalera, F. Aspirin reduces endothelial cell senescence. Biochem. Biophys. Res. Commun. 2005, 334, 1226–1232. [Google Scholar] [CrossRef] [PubMed]
- Archer, S.L.; Huang, J.M.; Hampl, V.; Nelson, D.P.; Shultz, P.J.; Weir, E.K. Nitric oxide and cGMP cause vasorelaxation by activation of a charybdotoxin-sensitive K channel by cGMP-dependent protein kinase. Proc. Natl. Acad. Sci. USA 1994, 91, 7583–7587. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.L.; Strassmaier, T.; Brady, J.D.; Karpen, J.W. The pharmacology of cyclic nucleotide-gated channels: Emerging from the darkness. Curr. Pharm. Des. 2006, 12, 3597–3613. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rojas, M.; Lemtalsi, T.; Toque, H.A.; Xu, Z.; Fulton, D.; Caldwell, R.W.; Caldwell, R.B. NOX2-Induced Activation of Arginase and Diabetes-Induced Retinal Endothelial Cell Senescence. Antioxidants 2017, 6, 43. https://doi.org/10.3390/antiox6020043
Rojas M, Lemtalsi T, Toque HA, Xu Z, Fulton D, Caldwell RW, Caldwell RB. NOX2-Induced Activation of Arginase and Diabetes-Induced Retinal Endothelial Cell Senescence. Antioxidants. 2017; 6(2):43. https://doi.org/10.3390/antiox6020043
Chicago/Turabian StyleRojas, Modesto, Tahira Lemtalsi, Haroldo A. Toque, Zhimin Xu, David Fulton, Robert William Caldwell, and Ruth B. Caldwell. 2017. "NOX2-Induced Activation of Arginase and Diabetes-Induced Retinal Endothelial Cell Senescence" Antioxidants 6, no. 2: 43. https://doi.org/10.3390/antiox6020043
APA StyleRojas, M., Lemtalsi, T., Toque, H. A., Xu, Z., Fulton, D., Caldwell, R. W., & Caldwell, R. B. (2017). NOX2-Induced Activation of Arginase and Diabetes-Induced Retinal Endothelial Cell Senescence. Antioxidants, 6(2), 43. https://doi.org/10.3390/antiox6020043