Role of Presenilin in Mitochondrial Oxidative Stress and Neurodegeneration in Caenorhabditis elegans


{kind=link}
{kind=link}
Abstract
1. Introduction
2. Mitochondria: The Story of Energy and ROS Generation
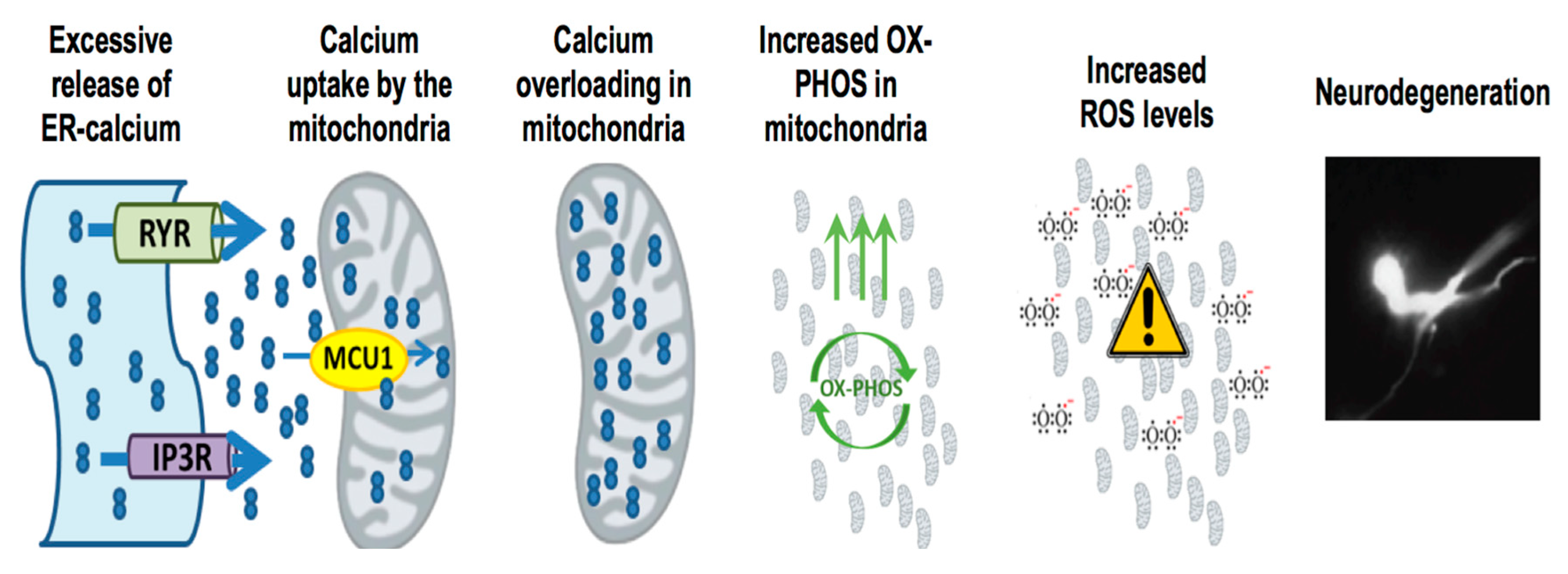
3. Mitochondria, Calcium and ROS: The Holy Trinity
4. Alzheimer’s Disease
5. Presenilin and the Calcium Hypothesis of Alzheimer’s Disease
6. C. elegans as a Model for Alzheimer’s Disease
7. Calcium Homeostasis and Mitochondrial Function Is Disrupted in C. elegans Presenilin Mutants
8. Oxidative Stress Mediated Neurodegeneration in sel-12 Mutants
9. Presenilin and γ-Secretase Function
10. Studying Aβ Toxicity in C. elegans
11. Future Directions and Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Dementia: A Public Health Priority. Available online: www.whoint/mental_health/publications/dementia_report_2012/en/ (accessed on 16 February 2017).
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxid. Med. Cell Longev. 2017, 2017, 8416763. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.J. Calcium regulation of neural rhythms, memory and Alzheimer’s disease. J. Physiol. 2014, 592, 281–293. [Google Scholar] [CrossRef] [PubMed]
- Verkhratsky, A.; Mattson, M.P.; Toescu, E.C. Aging in the mind. Trends Neurosci. 2004, 27, 577–578. [Google Scholar] [CrossRef] [PubMed]
- Jeyaraju, D.V.; Cisbani, G.; Pellegrini, L. Calcium regulation of mitochondria motility and morphology. Biochim. Biophys. Acta 2009, 1787, 1363–1373. [Google Scholar] [CrossRef] [PubMed]
- Das, A.M.; Harris, D.A. Control of mitochondrial ATP synthase in heart cells: Inactive to active transitions caused by beating or positive inotropic agents. Cardiovasc. Res. 1990, 24, 411–417. [Google Scholar] [CrossRef] [PubMed]
- Glancy, B.; Balaban, R.S. Role of mitochondrial Ca2+ in the regulation of cellular energetics. Biochemistry 2012, 51, 2959–2973. [Google Scholar] [CrossRef] [PubMed]
- Hansford, R.G.; Zorov, D. Role of mitochondrial calcium transport in the control of substrate oxidation. Mol. Cell Biochem. 1998, 184, 359–369. [Google Scholar] [CrossRef] [PubMed]
- McCormack, J.G.; Denton, R.M. The role of intramitochondrial Ca2+ in the regulation of oxidative phosphorylation in mammalian tissues. Biochem. Soc. Trans. 1993, 21, 793–799. [Google Scholar] [CrossRef] [PubMed]
- Mildaziene, V.; Baniene, R.; Nauciene, Z.; Bakker, B.M.; Brown, G.C.; Westerhoff, H.V.; Kholodenko, B.N. Calcium indirectly increases the control exerted by the adenine nucleotide translocator over 2-oxoglutarate oxidation in rat heart mitochondria. Arch. Biochem. Biophys. 1995, 324, 130–134. [Google Scholar] [CrossRef] [PubMed]
- Wernette, M.E.; Ochs, R.S.; Lardy, H.A. Ca2+ stimulation of rat liver mitochondrial glycerophosphate dehydrogenase. J. Biol. Chem. 1981, 256, 12767–12771. [Google Scholar] [PubMed]
- Shoshan-Barmatz, V.; Ben-Hail, D. VDAC, a multi-functional mitochondrial protein as a pharmacological target. Mitochondrion 2012, 2, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Báthori, G.; Csordás, G.; Garcia-Perez, C.; Davies, E.; Hajnóczky, G. Ca2+-dependent control of the permeability properties of the mitochondrial outer membrane and voltage-dependent anion-selective channel (VDAC). J. Biol. Chem. 2006, 281, 17347–17358. [Google Scholar] [CrossRef] [PubMed]
- Perocchi, F.; Gohil, V.M.; Girgis, H.S.; Bao, X.R.; McCombs, J.E.; Palmer, A.E.; Mootha, V.K. MICU1 encodes a mitochondrial EF hand protein required for Ca2+ uptake. Nature 2010, 467, 291–296. [Google Scholar] [CrossRef] [PubMed]
- De Stefani, D.; Raffaello, A.; Teardo, E.; Szabo, I.; Rizzuto, R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 2011, 476, 336–340. [Google Scholar] [CrossRef] [PubMed]
- Baughman, J.M.; Perocchi, F.; Girgis, H.S.; Plovanich, M.; Belcher-Timme, C.A.; Sancak, Y.; Bao, X.R.; Strittmatter, L.; Goldberger, O.; Bogorad, R.L.; et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 2011, 476, 341–345. [Google Scholar] [CrossRef] [PubMed]
- Csordas, G.; Golenar, T.; Seifert, E.L.; Kamer, K.J.; Sancak, Y.; Perocchi, F.; Moffat, C.; Weaver, D.; de la Fuente Perez, S.; Bogorad, R.; et al. MICU1 controls both the threshold and cooperative activation of the mitochondrial Ca(2)(+) uniporter. Cell Metab. 2013, 17, 976–987. [Google Scholar] [CrossRef] [PubMed]
- Mallilankaraman, K.; Cardenas, C.; Doonan, P.J.; Chandramoorthy, H.C.; Irrinki, K.M.; Golenar, T.; Csordas, G.; Madireddi, P.; Yang, J.; Muller, M.; et al. MCUR1 is an essential component of mitochondrial Ca2+ uptake that regulates cellular metabolism. Nat. Cell Biol. 2012, 14, 1336–1343. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Liu, J.; Nguyen, T.; Liu, C.; Sun, J.; Teng, Y.; Fergusson, M.M.; Rovira, I.I.; Allen, M.; Springer, D.A.; et al. The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nat. Cell Biol. 2013, 15, 1464–1472. [Google Scholar] [CrossRef] [PubMed]
- Herzig, S.; Maundrell, K.; Martinou, J.C. Life without the mitochondrial calcium uniporter. Nat. Cell Biol. 2013, 15, 1398–1400. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Chisholm, A.D. C. C. elegans epidermal wounding induces a mitochondrial ROS burst that promotes wound repair. Dev. Cell 2014, 31, 48–60. [Google Scholar] [CrossRef] [PubMed]
- Dedkova, E.N.; Blatter, L.A. Calcium signaling in cardiac mitochondria. J. Mol. Cell Cardiol. 2013, 58, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Li, H.; Tai, Y.; Huang, J.; Su, Y.; Abramowitz, J.; Zhu, M.X.; Birnbaumer, L.; Wang, Y. Canonical transient receptor potential 3 channels regulate mitochondrial calcium uptake. Proc. Natl. Acad. Sci. USA 2013, 110, 11011–11016. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Lytton, J. Molecular cloning of a sixth member of the K+-dependent Na+/Ca2+ exchanger gene family, NCKX6. J. Biol. Chem. 2004, 2004. 279, 5867–5876. [Google Scholar] [CrossRef]
- Palty, R.; Ohana, E.; Hershfinkel, M.; Volokita, M.; Elgazar, V.; Beharier, O.; Silverman, W.F.; Argaman, M.; Sekler, I. Lithium-calcium exchange is mediated by a distinct potassium-independent sodium-calcium exchanger. J. Biol. Chem. 2004, 279, 25234–25240. [Google Scholar] [CrossRef]
- Bonora, M.; Morganti, C.; Morciano, G.; Pedriali, G.; Lebiedzinska-Arciszewska, M.; Aquila, G.; Giorgi, C.; Rizzo, P.; Campo, G.; Ferrari, R.; et al. Mitochondrial permeability transition involves dissociation of F1FO ATP synthase dimers and C-ring conformation. EMBO Rep. 2017, 18, 1077–1089. [Google Scholar] [CrossRef] [PubMed]
- Kamboh, M.I.; Aston, C.E.; Perez-Tur, J.; Kokmen, E.; Ferrell, R.E.; Hardy, J.; DeKosky, S.T. A novel mutation in the apolipoprotein E gene (APOE*4 Pittsburgh) is associated with the risk of late-onset Alzheimer’s disease. Neurosci. Lett. 1999, 263, 129–132. [Google Scholar] [CrossRef]
- Loeffler, M.; Kroemer, G. The mitochondrion in cell death control: Certainties and incognita. Exp. Cell Res. 2000, 256, 19–26. [Google Scholar] [CrossRef] [PubMed]
- McMenemey, W.H.; Worster-Drought, C.; Flind, J.; Williams, H.G. Familial Presenile Dementia: Report of Case with Clinical and Pathological Features of Alzheimer’s Disease. J. Neurol. Psychiatry 1939, 2, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Schellenberg, G.D.; Bird, T.D.; Wijsman, E.M.; Orr, H.T.; Anderson, L.; Nemens, E.; White, J.A.; Bonnycastle, L.; Weber, J.L.; Alonso, M.E.; et al. Genetic linkage evidence for a familial Alzheimer’s disease locus on chromosome 14. Science 1992, 258, 668–671. [Google Scholar] [CrossRef] [PubMed]
- St George-Hyslop, P.; Haines, J.; Rogaev, E.; Mortilla, M.; Vaula, G.; Pericak-Vance, M.; Foncin, J.F.; Montesi, M.; Bruni, A.; Sorbi, S.; et al. Genetic evidence for a novel familial Alzheimer’s disease locus on chromosome 14. Nat. Genet. 1992, 2, 330–334. [Google Scholar] [CrossRef] [PubMed]
- Van Broeckhoven, C.; Backhovens, H.; Cruts, M.; De Winter, G.; Bruyland, M.; Cras, P.; Martin, J.J. Mapping of a gene predisposing to early-onset Alzheimer’s disease to chromosome 14q24.3. Nat. Genet. 1992, 2, 335–339. [Google Scholar] [CrossRef] [PubMed]
- Sherrington, R.; Rogaev, E.I.; Liang, Y.; Rogaeva, E.A.; Levesque, G.; Ikeda, M.; Chi, H.; Lin, C.; Li, G.; Holman, K.; et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature 1995, 375, 754–760. [Google Scholar] [CrossRef] [PubMed]
- Levy-Lahad, E.; Wasco, W.; Poorkaj, P.; Romano, D.M.; Oshima, J.; Pettingell, W.H.; Yu, C.E.; Jondro, P.D.; Schmidt, S.D.; Wang, K.; et al. Candidate gene for the chromosome 1 familial Alzheimer’s disease locus. Science 1995, 269, 973–977. [Google Scholar] [CrossRef] [PubMed]
- Haass, C. Take five-BACE and the gamma-secretase quartet conduct Alzheimer’s amyloid beta-peptide generation. EMBO J. 2004, 23, 483–488. [Google Scholar] [CrossRef] [PubMed]
- Seubert, P.; Vigo-Pelfrey, C.; Esch, F.; Lee, M.; Dovey, H.; Davis, D.; Sinha, S.; Schlossmacher, M.; Whaley, J.; Swindlehurst, C.; et al. Isolation and quantification of soluble Alzheimer’s beta-peptide from biological fluids. Nature 1992, 359, 325–327. [Google Scholar] [CrossRef] [PubMed]
- Schenk, D.; Basi, G.S.; Pangalos, M.N. Treatment strategies targeting amyloid beta-protein. Cold Spring Harb. Perspect. Med. 2012, 2, a006387. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. Alzheimers disease: Genes, proteins, and therapy. Physiol. Rev. 2001, 81, 741–766. [Google Scholar] [CrossRef] [PubMed]
- Chartier-Harlin, M.C.; Crawford, F.; Houlden, H.; Warren, A.; Hughes, D.; Fidani, L.; Goate, A.; Rossor, M.; Roques, P.; Hardy, J.; et al. Early-onset Alzheimer’s disease caused by mutations at codon 717 of the beta-amyloid precursor protein gene. Nature 1991, 353, 844–846. [Google Scholar] [CrossRef] [PubMed]
- Mullan, M. Familial Alzheimer’s disease: Second gene locus located. BMJ: Br. Med. J. 1992, 305, 1108–1109. [Google Scholar] [CrossRef]
- Citron, M.O.T.; Haass, C.; McConlogue, L.; Hung, A.Y.; Seubert, P.; Vigo-Pelfrey, C.; Lieberburg, I.; Selkoe, D.J. Mutation of the beta-amyloid precursor protein in familial Alzheimer’s disease increases beta-protein production. Nature 1992, 360, 672–674. [Google Scholar] [CrossRef] [PubMed]
- Shoji, M.; Golde, T.E.; Ghiso, J.; Cheung, T.T.; Estus, S.; Shaffer, L.M.; Cai, X.D.; McKay, D.M.; Tintner, R.; Frangione, B.; et al. Production of the Alzheimer amyloid beta protein by normal proteolytic processing. Science 1992, 258, 126–129. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, N.; Cheung, T.T.; Cai, X.D.; Odaka, A.; Otvos, L.J.; Eckman, C.; Golde, T.E.; Younkin, S.G. An increased percentage of long amyloid beta protein secreted by familial amyloid beta protein precursor (beta APP717) mutants. Science 1994, 264, 1336–1340. [Google Scholar] [CrossRef] [PubMed]
- Nilsberth, C.; Westlind-Danielsson, A.; Eckman, C.B.; Condron, M.M.; Axelman, K.; Forsell, C.; Stenh, C.; Luthman, J.; Teplow, D.B.; Younkin, S.G.; et al. The ‘Arctic’ APP mutation (E693G) causes Alzheimer’s disease by enhanced Abeta protofibril formation. Nat. Neurosci. 2001, 4, 887–893. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Yin, Y.I.; Li, Y.M.; Sisodia, S.S. Evidence that assembly of an active gamma-secretase complex occurs in the early compartments of the secretory pathway. J. Biol. Chem. 2004, 279, 48615–48619. [Google Scholar] [CrossRef] [PubMed]
- Takami, M.; Funamoto, S. gamma-Secretase-Dependent Proteolysis of Transmembrane Domain of Amyloid Precursor Protein: Successive Tri- and Tetrapeptide Release in Amyloid beta-Protein Production. Int. J. Alzheimers Dis. 2012, 2012, 591392. [Google Scholar]
- De Strooper, B.; Annaert, W.; Cupers, P.; Saftig, P.; Craessaerts, K.; Mumm, J.S.; Schroeter, E.H.; Schrijvers, V.; Wolfe, M.S.; Ray, W.J.; et al. A presenilin-1-dependent gamma-secretase-like protease mediates release of Notch intracellular domain. Nature 1999, 398, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Mutations. Available online: www.alzforumorg/mutations (accessed on 23 November 2016).
- Gandy, S.; Heppner, F.L. Microglia as dynamic and essential components of the amyloid hypothesis. Neuron 2013, 78, 575–577. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Doody, R.S.; Raman, R.; Farlow, M.; Iwatsubo, T.; Vellas, B.; Joffe, S.; Kieburtz, K.; He, F.; Sun, X.; Thomas, R.G.; et al. A phase 3 trial of semagacestat for treatment of Alzheimer's disease. N. Engl. J. Med. 2013, 369, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Le Couteur, D.G.; Hunter, S.; Brayne, C. Solanezumab and the amyloid hypothesis for Alzheimer’s disease. BMJ 2016, 355, i6771. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.P.; Clark, I.A.; Vissel, B. Inconsistencies and controversies surrounding the amyloid hypothesis of Alzheimer’s disease. Acta Neuropathol. Commun. 2014, 2, 135. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Zhou, R.; Yang, G.; Shi, Y. Analysis of 138 pathogenic mutations in presenilin-1 on the in vitro production of Abeta42 and Abeta40 peptides by gamma-secretase. Proc. Natl. Acad. Sci. USA 2017, 114, E476–E485. [Google Scholar] [CrossRef] [PubMed]
- Khachaturian, Z.S. Calcium, membranes, aging, and Alzheimer’s disease. Introduction and overview. Ann. N. Y. Acad. Sci. 1989, 568, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Ito, E.; Oka, K.; Etcheberrigaray, R.; Nelson, T.J.; McPhie, D.L.; Tofel-Grehl, B.; Gibson, G.E.; Alkon, D.L. Internal Ca2+ mobilization is altered in fibroblasts from patients with Alzheimer disease. Proc. Natl. Acad. Sci. USA 1994, 91, 534–538. [Google Scholar] [CrossRef] [PubMed]
- Etcheberrigaray, R.; Hirashima, N.; Nee, L.; Prince, J.; Govoni, S.; Racchi, M.; Tanzi, R.E.; Alkon, D.L. Calcium responses in fibroblasts from asymptomatic members of Alzheimer’s disease families. Neurobiol. Dis. 1998, 5, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Leissring, M.A.; Paul, B.A.; Parker, I.; Cotman, C.W.; LaFerla, F.M. Alzheimer’s presenilin-1 mutation potentiates inositol 1,4,5-trisphosphate-mediated calcium signaling in Xenopus oocytes. J. Neurochem. 1999, 72, 1061–1068. [Google Scholar] [CrossRef] [PubMed]
- Stutzmann, G.E.; Caccamo, A.; LaFerla, F.M.; Parker, I. Dysregulated IP3 signaling in cortical neurons of knock-in mice expressing an Alzheimer’s-linked mutation in presenilin1 results in exaggerated Ca2+ signals and altered membrane excitability. J. Neurosci. 2004, 24, 508–513. [Google Scholar] [CrossRef] [PubMed]
- Leissring, M.A.; Yamasaki, T.R.; Wasco, W.; Buxbaum, J.D.; Parker, I.; LaFerla, F.M. Calsenilin reverses presenilin-mediated enhancement of calcium signaling. Proc. Natl. Acad. Sci. USA 2000, 97, 8590–8593. [Google Scholar] [CrossRef] [PubMed]
- Chakroborty, S.; Goussakov, I.; Miller, M.B.; Stutzmann, G.E. Deviant ryanodine receptor-mediated calcium release resets synaptic homeostasis in presymptomatic 3xTg-AD mice. J. Neurosci. 2009, 29, 9458–9470. [Google Scholar] [CrossRef] [PubMed]
- Stutzmann, G.E.; Smith, I.; Caccamo, A.; Oddo, S.; Laferla, F.M.; Parker, I. Enhanced ryanodine receptor recruitment contributes to Ca2+ disruptions in young, adult, and aged Alzheimer’s disease mice. J. Neurosci. 2006, 26, 5180–5189. [Google Scholar] [CrossRef] [PubMed]
- Green, K.N.; Demuro, A.; Akbari, Y.; Hitt, B.D.; Smith, I.F.; Parker, I.; LaFerla, F.M. SERCA pump activity is physiologically regulated by presenilin and regulates amyloid beta production. J. Cell Biol. 2008, 181, 1107–1116. [Google Scholar] [CrossRef] [PubMed]
- Bandara, S.; Malmersjo, S.; Meyer, T. Regulators of calcium homeostasis identified by inference of kinetic model parameters from live single cells perturbed by siRNA. Sci. Signal. 2013, 6, ra56. [Google Scholar] [CrossRef] [PubMed]
- Tu, H.; Nelson, O.; Bezprozvanny, A.; Wang, Z.; Lee, S.F.; Hao, Y.H.; Serneels, L.; De Strooper, B.; Yu, G.; Bezprozvanny, I. Presenilins form ER Ca2+ leak channels, a function disrupted by familial Alzheimer’s disease-linked mutations. Cell 2006, 126, 981–993. [Google Scholar] [CrossRef] [PubMed]
- Nelson, O.; Tu, H.; Lei, T.; Bentahir, M.; de Strooper, B.; Bezprozvanny, I. Familial Alzheimer disease-linked mutations specifically disrupt Ca2+ leak function of presenilin 1. J. Clin. Investig. 2007, 117, 1230–1239. [Google Scholar] [CrossRef] [PubMed]
- De Brito, O.M.; Scorrano, L. An intimate liaison: Spatial organization of the endoplasmic reticulum-mitochondria relationship. EMBO J. 2010, 29, 2715–2723. [Google Scholar] [CrossRef] [PubMed]
- Rowland, A.A.; Voeltz, G.K. Endoplasmic reticulum-mitochondria contacts: Function of the junction. Nat. Rev. Mol. Cell Biol. 2012, 13, 607–625. [Google Scholar] [CrossRef] [PubMed]
- Area-Gomez, E.; de Groof, A.J.; Boldogh, I.; Bird, T.D.; Gibson, G.E.; Koehler, C.M.; Yu, W.H.; Duff, K.E.; Yaffe, M.P.; Pon, L.A.; et al. Presenilins are enriched in endoplasmic reticulum membranes associated with mitochondria. Am. J. Pathol. 2009, 175, 1810–1816. [Google Scholar] [CrossRef] [PubMed]
- Zampese, E.; Fasolato, C.; Kipanyula, M.J.; Bortolozzi, M.; Pozzan, T.; Pizzo, P. Presenilin 2 modulates endoplasmic reticulum (ER)-mitochondria interactions and Ca2+ cross-talk. Proc. Natl. Acad. Sci. USA 2011, 108, 2777–2782. [Google Scholar] [CrossRef] [PubMed]
- Area-Gomez, E.; Castillo, M.C.L.; Tambini, M.D.; Guardia-Laguarta, C.; de Groof, A.J.; Madra, M.; Ikenouchi, J.; Umeda, M.; Bird, T.D.; Sturley, S.L.; et al. Upregulated function of mitochondria-associated ER membranes in Alzheimer disease. EMBO J. 2012, 31, 4106–4123. [Google Scholar] [CrossRef] [PubMed]
- Kipanyula, M.J.; Contreras, L.; Zampese, E.; Lazzari, C.; Wong, A.K.; Pizzo, P.; Fasolato, C.; Pozzan, T. Ca2+ dysregulation in neurons from transgenic mice expressing mutant presenilin 2. Aging Cell 2012, 11, 885–893. [Google Scholar] [CrossRef] [PubMed]
- Smolarkiewicz, M.; Skrzypczak, T.; Wojtaszek, P. The very many faces of presenilins and the gamma-secretase complex. Protoplasma 2013, 250, 997–1011. [Google Scholar] [CrossRef] [PubMed]
- Khandelwal, A.; Chandu, D.; Roe, C.M.; Kopan, R.; Quatrano, R.S. Moonlighting activity of presenilin in plants is independent of gamma-secretase and evolutionarily conserved. Proc. Natl. Acad. Sci. USA 2007, 104, 13337–13342. [Google Scholar] [CrossRef] [PubMed]
- McMains, V.C.; Myre, M.; Kreppel, L.; Kimmel, A.R. Dictyostelium possesses highly diverged presenilin/gamma-secretase that regulates growth and cell-fate specification and can accurately process human APP: A system for functional studies of the presenilin/gamma-secretase complex. Dis. Model. Mech. 2010, 3, 581–594. [Google Scholar] [CrossRef] [PubMed]
- Ludtmann, M.H.; Otto, G.P.; Schilde, C.; Chen, Z.H.; Allan, C.Y.; Brace, S.; Beesley, P.W.; Kimmel, A.R.; Fisher, P.; Killick, R.; et al. An ancestral non-proteolytic role for presenilin proteins in multicellular development of the social amoeba Dictyostelium discoideum. J. Cell Sci. 2014, 127, 1576–1584. [Google Scholar] [CrossRef] [PubMed]
- Brenner, S. The genetics of Caenorhabditis elegans. Genetics 1974, 77, 71–94. [Google Scholar] [PubMed]
- Lewis, J.A.; Fleming, J.T. Basic culture methods. Methods Cell Biol. 1995, 48, 3–29. [Google Scholar] [PubMed]
- Sulston, J.E.; Horvitz, H.R. Post-embryonic cell lineages of the nematode, Caenorhabditis elegans. Dev. biol. 1977, 56, 110–156. [Google Scholar] [CrossRef]
- White, J.G.; Southgate, E.; Thomson, J.N.; Brenner, S. The structure of the nervous system of the nematode Caenorhabditis elegans. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1986, 314, 1–340. [Google Scholar] [CrossRef] [PubMed]
- Daigle, I.; Li, C. apl-1, a Caenorhabditis elegans gene encoding a protein related to the human beta-amyloid protein precursor. Proc. Natl. Acad. Sci. USA 1993, 90, 12045–12049. [Google Scholar] [CrossRef] [PubMed]
- McColl, G.; Roberts, B.R.; Pukala, T.L.; Kenche, V.B.; Roberts, C.M.; Link, C.D.; Ryan, T.M.; Masters, C.L.; Barnham, K.J.; Bush, A.I.; et al. Utility of an improved model of amyloid-beta (Abeta(1)(-)(4)(2)) toxicity in Caenorhabditis elegans for drug screening for Alzheimer’s disease. Mol. Neurodegener. 2012, 7, 57. [Google Scholar] [CrossRef] [PubMed]
- Arduengo, P.M.; Appleberry, O.K.; Chuang, P.; L’Hernault, S.W. The presenilin protein family member SPE-4 localizes to an ER/Golgi derived organelle and is required for proper cytoplasmic partitioning during Caenorhabditis elegans spermatogenesis. J. Cell Sci. 1998, 111, 3645–3654. [Google Scholar] [PubMed]
- Levitan, D.; Greenwald, I. Facilitation of lin-12-mediated signalling by sel-12, a Caenorhabditis elegans S182 Alzheimer’s disease gene. Nature 1995, 377, 351–354. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Greenwald, I. HOP-1, a Caenorhabditis elegans presenilin, appears to be functionally redundant with SEL-12 presenilin and to facilitate LIN-12 and GLP-1 signaling. Proc. Natl. Acad. Sci. USA 1997, 94, 12204–12209. [Google Scholar] [CrossRef] [PubMed]
- Levitan, D.; Greenwald, I. Effects of SEL-12 presenilin on LIN-12 localization and function in Caenorhabditis elegans. Development 1998, 125, 3599–3606. [Google Scholar] [PubMed]
- Sarasija, S.; Norman, K.R. A gamma-Secretase Independent Role for Presenilin in Calcium Homeostasis Impacts Mitochondrial Function and Morphology in Caenorhabditis elegans. Genetics 2015, 201, 1453–1466. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.L.; Mayne, M.; Holden, C.P.; Geiger, J.D.; Mattson, M.P. Presenilin-1 mutations increase levels of ryanodine receptors and calcium release in PC12 cells and cortical neurons. J. Biol. Chem. 2000, 275, 18195–18200. [Google Scholar] [CrossRef] [PubMed]
- Leissring, M.A.; Akbari, Y.; Fanger, C.M.; Cahalan, M.D.; Mattson, M.P.; LaFerla, F.M. Capacitative calcium entry deficits and elevated luminal calcium content in mutant presenilin-1 knockin mice. J. Cell Biol. 2000, 149, 793–798. [Google Scholar] [CrossRef] [PubMed]
- Smith, I.F.; Hitt, B.; Green, K.N.; Oddo, S.; LaFerla, F.M. Enhanced caffeine-induced Ca2+ release in the 3xTg-AD mouse model of Alzheimer’s disease. J. Neurochem. 2005, 94, 1711–1718. [Google Scholar] [CrossRef] [PubMed]
- Sarasija, S.; Laboy, J.T.; Ashkavand, Z.; Bonner, J.; Tang, Y.; Norman, K.R. Presenilin mutations deregulate mitochondrial Ca2+ homeostasis and metabolic activity causing neurodegeneration in Caenorhabditis elegans. eLife 2018, 7, e33052. [Google Scholar] [CrossRef] [PubMed]
- Van der Bliek, A.M.; Shen, Q.; Kawajiri, S. Mechanisms of mitochondrial fission and fusion. Cold Spring Harb. Perspect. Biol. 2013, 5, a011072. [Google Scholar] [CrossRef] [PubMed]
- Cereghetti, G.M.; Stangherlin, A.; Martins de Brito, O.; Chang, C.R.; Blackstone, C.; Bernardi, P.; Scorrano, L. Dephosphorylation by calcineurin regulates translocation of Drp1 to mitochondria. Proc. Natl. Acad. Sci. USA 2008, 105, 15803–15808. [Google Scholar] [CrossRef] [PubMed]
- Cribbs, J.T.; Strack, S. Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO Rep. 2007, 8, 939–944. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Pi, H.; Chen, Y.; Zhang, N.; Guo, P.; Lu, Y.; He, M.; Xie, J.; Zhong, M.; Zhang, Y.; et al. Cadmium induced Drp1-dependent mitochondrial fragmentation by disturbing calcium homeostasis in its hepatotoxicity. Cell Death Dis. 2013, 4, e540. [Google Scholar] [CrossRef] [PubMed]
- Hirai, K.; Aliev, G.; Nunomura, A.; Fujioka, H.; Russell, R.L.; Atwood, C.S.; Johnson, A.B.; Kress, Y.; Vinters, H.V.; Tabaton, M.; et al. Mitochondrial abnormalities in Alzheimer’s disease. J. Neurosci. 2001, 21, 3017–3023. [Google Scholar] [CrossRef] [PubMed]
- Oksanen, M.; Petersen, A.J.; Naumenko, N.; Puttonen, K.; Lehtonen, S.; Gubert Olive, M.; Shakirzyanova, A.; Leskela, S.; Sarajarvi, T.; Viitanen, M.; et al. PSEN1 Mutant iPSC-Derived Model Reveals Severe Astrocyte Pathology in Alzheimer’s Disease. Stem Cell Rep. 2017, 9, 1885–1897. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.H. Increased mitochondrial fission and neuronal dysfunction in Huntington’s disease: Implications for molecular inhibitors of excessive mitochondrial fission. Drug Discov. Today 2014, 19, 951–955. [Google Scholar] [CrossRef] [PubMed]
- Harman, D. Aging: A theory based on free radical and radiation chemistry. J. Gerontol. 1956, 11, 298–300. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.C.; Hsu, J.M.; Yen, C.P.; Chao, C.C.; Chen, R.H.; Pan, C.L. Neural activity and CaMKII protect mitochondria from fragmentation in aging Caenorhabditis elegans neurons. Proc. Natl. Acad. Sci. USA 2015, 112, 8768–8773. [Google Scholar] [CrossRef] [PubMed]
- Toth, M.L.; Melentijevic, I.; Shah, L.; Bhatia, A.; Lu, K.; Talwar, A.; Naji, H.; Ibanez-Ventoso, C.; Ghose, P.; Jevince, A.; et al. Neurite sprouting and synapse deterioration in the aging Caenorhabditis elegans nervous system. J. Neurosci. 2012, 32, 8778–8790. [Google Scholar] [CrossRef] [PubMed]
- Tank, E.M.; Rodgers, K.E.; Kenyon, C. Spontaneous age-related neurite branching in Caenorhabditis elegans. J. Neurosci. 2011, 31, 9279–9288. [Google Scholar] [CrossRef] [PubMed]
- Pan, C.L.; Peng, C.Y.; Chen, C.H.; McIntire, S. Genetic analysis of age-dependent defects of the Caenorhabditis elegans touch receptor neurons. Proc. Natl. Acad. Sci. USA. 2011, 108, 9274–9279. [Google Scholar] [CrossRef] [PubMed]
- De Strooper, B. Loss-of-function presenilin mutations in Alzheimer disease. Talking Point on the role of presenilin mutations in Alzheimer disease. EMBO Rep. 2007, 8, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Xia, D.; Watanabe, H.; Wu, B.; Lee, S.H.; Li, Y.; Tsvetkov, E.; Bolshakov, V.Y.; Shen, J.; Kelleher, R.J. Presenilin-1 knockin mice reveal loss-of-function mechanism for familial Alzheimer’s disease. Neuron 2015, 85, 967–981. [Google Scholar] [CrossRef] [PubMed]
- Greenwald, I.; Kovall, R. Notch signaling: Genetics and structure. WormBook 2013, 17, 1–28. [Google Scholar] [CrossRef] [PubMed]
- Cohen, E.; Bieschke, J.; Perciavalle, R.M.; Kelly, J.W.; Dillin, A. Opposing activities protect against age-onset proteotoxicity. Science 2006, 313, 1604–1610. [Google Scholar] [CrossRef] [PubMed]
- McColl, G.; Roberts, B.R.; Gunn, A.P.; Perez, K.A.; Tew, D.J.; Masters, C.L.; Barnham, K.J.; Cherny, R.A.; Bush, A.I. The Caenorhabditis elegans A beta 1-42 model of Alzheimer disease predominantly expresses A beta 3-42. J. Biol. Chem. 2009, 284, 22697–22702. [Google Scholar] [CrossRef] [PubMed]
- Treusch, S.; Hamamichi, S.; Goodman, J.L.; Matlack, K.E.; Chung, C.Y.; Baru, V.; Shulman, J.M.; Parrado, A.; Bevis, B.J.; Valastyan, J.S.; et al. Functional links between Abeta toxicity, endocytic trafficking, and Alzheimer’s disease risk factors in yeast. Science 2011, 334, 1241–1245. [Google Scholar] [CrossRef] [PubMed]
- Fong, S.; Teo, E.; Ng, L.F.; Chen, C.B.; Lakshmanan, L.N.; Tsoi, S.Y.; Moore, P.K.; Inoue, T.; Halliwell, B.; Gruber, J. Energy crisis precedes global metabolic failure in a novel Caenorhabditis elegans Alzheimer Disease model. Sci. Rep. 2016, 6, 33781. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, V.; Romani, M.; Mouchiroud, L.; Beck, J.S.; Zhang, H.; D’Amico, D.; Moullan, N.; Potenza, F.; Schmid, A.W.; Rietsch, S.; et al. Enhancing mitochondrial proteostasis reduces amyloid-beta proteotoxicity. Nature 2017, 552, 187–193. [Google Scholar] [PubMed]
- Butterfield, D.A.; Bader Lange, M.L.; Sultana, R. Involvements of the lipid peroxidation product, HNE, in the pathogenesis and progression of Alzheimer’s disease. Biochim. Biophys. Acta 2010, 1801, 924–929. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, W.; Li, L.; Perry, G.; Lee, H.G.; Zhu, X. Oxidative stress and mitochondrial dysfunction in Alzheimer’s disease. Biochim. Biophys. Acta 2014, 1842, 1240–1247. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sarasija, S.; Norman, K.R. Role of Presenilin in Mitochondrial Oxidative Stress and Neurodegeneration in Caenorhabditis elegans. Antioxidants 2018, 7, 111. https://doi.org/10.3390/antiox7090111
Sarasija S, Norman KR. Role of Presenilin in Mitochondrial Oxidative Stress and Neurodegeneration in Caenorhabditis elegans. Antioxidants. 2018; 7(9):111. https://doi.org/10.3390/antiox7090111
Chicago/Turabian StyleSarasija, Shaarika, and Kenneth R. Norman. 2018. "Role of Presenilin in Mitochondrial Oxidative Stress and Neurodegeneration in Caenorhabditis elegans" Antioxidants 7, no. 9: 111. https://doi.org/10.3390/antiox7090111
APA StyleSarasija, S., & Norman, K. R. (2018). Role of Presenilin in Mitochondrial Oxidative Stress and Neurodegeneration in Caenorhabditis elegans. Antioxidants, 7(9), 111. https://doi.org/10.3390/antiox7090111