Hydrogen Sulfide and Persulfides Oxidation by Biologically Relevant Oxidizing Species

1
Laboratorio de Enzimología, Instituto de Química Biológica, Facultad de Ciencias, Universidad de la República, Montevideo 11400, Uruguay
2
Center for Free Radical and Biomedical Research, Universidad de la República, Montevideo 11800, Uruguay
3
Unidad de Bioquímica Analítica, Centro de Investigaciones Nucleares, Facultad de Ciencias, Universidad de la República, Montevideo 11400, Uruguay
4
Laboratorio de Fisicoquímica Biológica, Instituto de Química Biológica, Facultad de Ciencias, Universidad de la República, Montevideo 11400, Uruguay
*
Author to whom correspondence should be addressed.
Antioxidants 2019, 8(2), 48; https://doi.org/10.3390/antiox8020048
Submission received: 22 January 2019
/
Revised: 15 February 2019
/
Accepted: 19 February 2019
/
Published: 22 February 2019
(This article belongs to the Special Issue H2S in Redox Signaling)
{kind=link}
{kind=link}
Abstract
:Hydrogen sulfide (H2S/HS–) can be formed in mammalian tissues and exert physiological effects. It can react with metal centers and oxidized thiol products such as disulfides (RSSR) and sulfenic acids (RSOH). Reactions with oxidized thiol products form persulfides (RSSH/RSS–). Persulfides have been proposed to transduce the signaling effects of H2S through the modification of critical cysteines. They are more nucleophilic and acidic than thiols and, contrary to thiols, also possess electrophilic character. In this review, we summarize the biochemistry of hydrogen sulfide and persulfides, focusing on redox aspects. We describe biologically relevant one- and two-electron oxidants and their reactions with H2S and persulfides, as well as the fates of the oxidation products. The biological implications are discussed.
1. Hydrogen Sulfide: An Ancient Metabolite with Novel Regulatory Roles
Hydrogen sulfide (H2S) is the simplest molecule containing reduced sulfur among the wide diversity of sulfur compounds. It was first synthesized as a gas, and its composition was identified in the eighteenth century [1]. It is naturally occurring in geothermal sources [2] and has been related to the emergence and evolution of organisms in Earth [3]. It is a crucial molecule in the metabolism of several organisms, acting both as an electron donor in energetic pathways (anoxygenic photosynthesis and oxidative phosphorylation in lithotrophs [4]) and as a sulfur donor in synthetic pathways (cysteine and methionine synthesis, among others [5]). Furthermore, sulfur- and sulfate-reducing organisms are able to produce H2S in assimilative or dissimilative processes [6].
H2S was identified as a toxic compound for humans centuries ago. High levels of H2S can induce inhibition of cellular respiration [7]. Exposure of mammals to H2S from sewage, industrial sources and domestic accidents have produced numerous cases of loss of consciousness and death [8]. However, some animals are able to tolerate elevated levels of H2S and have efficient systems for its detoxification [9]. Indeed, in the human intestine, mitochondria are able to deal with the H2S produced by the enteric flora [10], which diffuses easily through membranes [11,12,13] and represents a hazard for neighboring tissues. The enterocytes are able to fuel the electronic transport chain by oxidizing H2S to sulfate (SO42–) and thiosulfate (S2O32–) [14]. The discovery of endogenous biogenesis of H2S in mammals [15,16] and the evidence of modulatory effects when administered at low concentrations led to the hypothesis of a regulatory role [17,18,19]. Numerous physiological responses to H2S have been reported since, including interactions with several cation channels in the nervous system, the attenuation of myocardial ischemia reperfusion injury, promotion of angiogenesis, and the induction of pro-/anti-inflammatory responses [20]. Several reports assigned an antioxidant role for H2S, which could be related to cytoprotective effects [21,22,23,24,25,26]; nevertheless, consideration of the kinetics of the reactions suggests that the protective role is not due to direct reactions of H2S with oxidants, as will be described below [27].
2. Persulfides as Potential Transducers of H2S Signaling
The observed physiopathological effects focused the research in the field on the identification of cellular targets and on the underlying signaling mechanisms in order to develop plausible routes for pharmacological intervention. H2S shows nucleophilicity and is analogous to thiols, albeit weaker [27,28]. Among some possible targets in cells, H2S can react with metallic centers (iron, copper, zinc, molybdenum) producing coordination complexes and, in some circumstances, donating electrons. H2S is also able to react with oxidized forms of protein thiols such as disulfides (RSSR) or sulfenic acids (RSOH), thus leading to the formation of persulfides (RSSH/RSS–) [28,29].
The persulfides, which are also intermediates in the synthesis of particular sulfur biomolecules [30,31], are nowadays being considered candidate species for the transduction of the signals triggered by H2S. Not only are these species suggested to be more nucleophilic and acidic than the original thiols, but they also show an electrophilic character that is lacking in the original thiols [32]. In addition, enzymatic pathways for the H2S–independent formation of persulfides have been reported [33,34,35,36,37,38,39,40,41,42]. The generation of persulfides on critical protein cysteines constitutes a posttranslational modification which could affect relevant activities in cells [43,44] and thus allow for signal transduction and the modulation of processes. Furthermore, persulfides are able to react with oxidants. Direct and indirect protective effects of persulfides have been hypothesized and possible antioxidant roles are being surveyed [45,46,47,48].
The biochemistry of H2S and persulfides is now under intense scrutiny. In this work we describe the characteristics of these species from a redox biochemistry perspective, focusing on the reactions of H2S and persulfides with reactive species of biological interest.
3. Chemistry and Biology of H2S
H2S is recognized by its odor of rotten eggs. Humans can detect as low as 0.0005–0.3 ppm, whereas concentrations higher than 10 ppm start to cause discomfort and higher than 500 ppm cause rapid loss of consciousness and death [49].
The recommended names by IUPAC are dihydrogen sulfide and sulfane for H2S, and sulfanide or hydrogen(sulfide)(1–) for HS– [20]. In this review, “H2S” will be used for the gas and for the mixture of H2S and HS– in aqueous solution.
H2S may be considered at first sight to be similar to water, but there are several important differences that lead to dissimilar physical and chemical properties. Sulfur is larger than oxygen (van der Waals radii of 1.80 and 1.42 Å, respectively) [50], has a lower electronegativity (2.58 and 3.44 in the Pauling scale, respectively), and is more polarizable [51]. Hence, the dipole moment of H2S is lower than that of water, and hydrogen bonds in H2S are not formed at room temperature [51]; this explains why H2S is a gas at ambient temperature and pressure. H2S can dissolve in water with a relatively high solubility (101.3 mM/atm at 25 °C) [52], and the solvation is dominated by dispersive forces with no hydrogen bonds with water [51]. The slightly hydrophobic character of H2S is further supported by its twice higher solubility in organic solvents such as octanol and hexane than in water [12].
H2S is a weak diprotic acid with pKa1 = 6.98 at 25 °C and 6.76 at 37 °C (Equation 1) [53]. The second pKa2 has been more difficult to determine exactly but lies between 17 and 19 (Equation 2) in aqueous solution at 25 °C [20,53]. Therefore, S2– concentrations will be very low in biological settings and will not contribute significantly to H2S reactivity. At pH 7.4 and 25 °C, total sulfide exists 28% as H2S, 72% as HS–, and as insignificant amounts of S2–.
H2S ⇌ HS− + H+
HS− ⇌ S2− + H+
The hydrophobic character of H2S results in a higher solubility in membrane lipids than in water that results in a rapid diffusion through cell membranes without assistance by protein channels [11,12,13]. Permeability coefficients are estimated to be larger than 0.5 cm/s and probably larger than 10 cm/s [11,12,13], which indicates that membranes could slow down H2S diffusion 10–100 times compared to an equally thick layer of water. This is a low barrier that does not impede free diffusion across cellular membranes and only becomes significant when considering diffusion across a large number of membranes such as in a tissue [12].
The oxidation state of sulfur in H2S and in HS– is –2. Thus, H2S can only be oxidized; it cannot act as an oxidant. Therefore, the statements in the literature regarding H2S oxidation of protein cysteines to persulfides are not correct; either the thiol or H2S need to have undergone previous oxidation for cysteine persulfide formation to occur. Oxidation of H2S can lead to various products in which the sulfur can have oxidation numbers up to +6. The oxidation products include sulfate (SO42−), sulfite (SO32−), thiosulfate (S2O32−), persulfides (RSS−), organic (RSSnSR) and inorganic (HSSnS−) polysulfides, and elemental sulfur (Sn).
3.1. Biological Sources of H2S
Mammals obtain reduced sulfur from cysteine and methionine in the diet. The latter is an essential amino acid, but cysteine can be synthesized from methionine through the transsulfuration pathway. Two proteins involved in this metabolic pathway, cystathionine β-synthase (CBS) and cystathionine γ-lyase (also called γ-cystathionase, CSE), can produce H2S. Another enzyme from sulfur metabolism, 3-mercaptopyruvate sulfurtransferase (MST), also produces H2S [20,32].
The canonical reaction of CBS is the β-replacement of L-serine by L-homocysteine to generate L-cystathionine and H2O, but CBS also catalyzes other reactions using alternative substrates such as cysteine instead of serine that leads to cystathionine and H2S [54]. Mutations in this protein are the most common cause of hereditary homocystinuria [55]. CSE catalyzes the γ-elimination of cystathionine to give cysteine, α-ketobutyrate, and ammonia, but, like CBS, can also catalyze other reactions that lead to formation of H2S, such as the β-elimination of cysteine that is estimated to be the major reaction involved in H2S synthesis by CSE [54]. MST catalyzes the transfer of the sulfur atom from 3-mercaptopyruvate to a nucleophilic acceptor such as thioredoxin or glutathione (GSH), which subsequently releases H2S [56].
There are some differences in tissue distribution and subcellular localization. CSE is considered a cytosolic protein that predominates in cardiovascular tissue and liver; CBS is also cytosolic and is considered an important source of H2S in the nervous system; MST has been found both in the cytosol and the mitochondria and is the other important source of H2S in the nervous system [20].
3.2. Biological Sinks of H2S
There are several mechanisms that decompose H2S. The main pathway for H2S detoxification is located in the mitochondria and involves oxidation of sulfide to sulfate and thiosulfate [20]. The enzyme sulfide quinone oxidoreductase (SQR) oxidizes H2S and reduces coenzyme Q. An intermediate SQR-persulfide is formed and transferred either to glutathione to make glutathione persulfide (GSSH) or to sulfite to form thiosulfate. GSSH is subsequently metabolized by persulfide dioxygenase (PDO, also called ethylmalonic encephalopathy protein 1, ETHE1) to glutathione and sulfite (SO32–), and the latter is oxidized to sulfate (SO42–) by sulfite oxidase. Rhodanese could use GSSH and sulfite to make thiosulfate. Alternatively, thiosulfate could be utilized by rhodanese to produce GSSH [57,58,59].
H2S also reacts with ferric hemoglobin and myoglobin (methemoglobin and metmyoglobin, respectively) through a complex mechanism that results in the formation of thiosulfate and iron-bound polysulfides [60,61]. Though the proportion of methemoglobin is usually below 1% [62], the concentration of total hemoglobin subunits in red blood cells is 20 mM, and thus up to 200 µM methemoglobin may be available to react with H2S. Besides, several medications and diseases are known to increase methemoglobin [62], so this may be an important sink for H2S in circulation. Furthermore, in the presence of oxygen or hydrogen peroxide, H2S can modify hemes covalently in these globins and some other hemeproteins, thus forming green sulfheme in which the sulfur is covalently bound to one pyrrole ring. These processes will be further analyzed in Section 4.2.6.
3.3. Physiology and Pharmacology of H2S
Although early studies indicated that physiological concentrations of H2S in different tissues were in the 30–100 µM range, improved detection methods suggest that actual concentrations are in the 4–55 nM range for most tissues [63,64,65,66].
Externally added H2S at higher concentrations has been observed to have different effects. In the cardiovascular system, H2S has multiple nitric oxide-dependent and independent effects that include the relaxation of smooth muscle cells, regulation of blood pressure, promotion of angiogenesis, and attenuation of myocardial ischemia reperfusion injury [19,67,68,69,70,71,72]. In the nervous system, H2S is considered to be a neuromodulator. For example, it affects hippocampal long-term potentiation [18]. It has also been shown to inhibit HOCl- and peroxynitrite-mediated toxicity in neuroblastoma cells [21,23]. Moreover, H2S protects neurons from oxidative stress by increasing γ-glutamylcysteine synthetase activity and cystine transport, which induces the production of reduced glutathione [22]. In the respiratory system, H2S has potential therapeutic uses against fibrosis and chronic obstructive pulmonary disease [73,74]. In the liver, a hepatoprotective effect of H2S has been observed against ethanol-induced liver injury in mice [75].
H2S has shown both pro- and anti-inflammatory effects. In sepsis and acute pancreatitis, a pro-inflammatory effect of H2S has been observed [76,77,78], whereas an anti-inflammatory effect has been reported for intestinal ischemic damage and ethanol-induced gastritis [79,80,81].
The antioxidant effects cannot be attributed to direct reactions with oxidants since, from a kinetic perspective, H2S may not be able to trap oxidative species, as rationalized below. The indirect mechanisms underlying the observed antioxidant effects may be various. For example, H2S was proposed to inhibit oxidative stress through the persulfidation of Keap1, which activates nuclear factor (erythroid-derived 2)-like 2 (Nrf2) signaling and promotes its translocation to the nucleus and the expression of antioxidant pathways [75,82,83,84,85].
4. Oxidizing Species in Biology and Their Reactions with H2S
The formation of partial oxygen reduction products can occur in biological systems. These free radical and non-free radical species are superoxide radical (O2•–), hydrogen peroxide (H2O2), and hydroxyl radical (HO•). Together with secondary species such as peroxynitrite (ONOO–), hypochlorous acid (HOCl), or organic peroxyl radicals (ROO•), they are in general called reactive oxygen species (ROS); however, this term should be used with caution because it gives the erroneous idea that there is only one ill-defined species that oxidizes all biomolecules. In contrast, the reactivity of the oxidants varies, and their targets in biological settings are determined by kinetic aspects of rate constants multiplied by the concentration of the potential target as well as compartmentalization and membrane permeability aspects. The different species can be divided into two-electron or one-electron oxidants. The latter are often free radicals that start oxygen-dependent chain reactions.
One important site of reactive species formation is the mitochondrial electron transport chain. They can also be formed by ionizing and UV radiation, through the redox cycling of certain xenobiotics, and enzymatically through the action of NADPH oxidases, xanthine oxidase, hemeperoxidases, and cytochromes P450. Nitric oxide synthase generates nitric oxide, which, by itself, is not highly oxidizing, but it can react with superoxide radical generating peroxynitrite and secondary oxidants derived from it.
The high reactivity of some of these species with biomolecules can lead to cell damage; this aspect is exploited by immune cells in their warfare against microorganisms. Oxidant action can be counteracted, in some cases, by antioxidants; for example, in the free radical process of lipid peroxidation, peroxyl radical intermediates can be scavenged by the antioxidant vitamins E and C. In addition to their cell damaging action, oxidants can have signaling roles, as becomes clear from the evidence for the actions of hydrogen peroxide as a second messenger [86]. Thus, the classical concept of oxidative stress as a disbalance between oxidant formation and antioxidant action has been extended to include the concept of a disbalance in regulatory pathways.
In the following paragraphs, we briefly describe the oxidants potentially involved in the two- and one-electron oxidation of H2S, we provide the rate constants of the reactions, and we discuss the fate of some of the products formed. The reactions are summarized in Scheme 1.
4.1. Two-Electron Oxidation
H2S is a strong reductant according to the two-electron reduction potential E°’(HSS–, H+/2HS–) = –0.23 V [87,88]. The disulfide HSSH/HSS– is not formed directly but in at least two steps. Rather, sulfenic acid (HSOH) is formed, an unstable intermediate that reacts fast with a second H2S to form HSSH/HSS–. The latter is also unstable and can disproportionate yielding polysulfides of different chain lengths, elemental sulfur and H2S. Polysulfides can also react with reductants or be oxidized to oxyacids. Several two-electron oxidants can react with H2S.
4.1.1. Hydrogen Peroxide
This two-electron oxidant (E°’(H2O2, 2H+/2H2O) = +1.35 V) [89] is formed mainly by the dismutation of superoxide radical, which can be spontaneous or catalyzed by superoxide dismutases. It can also be formed directly by some oxidases. Though it is a strong oxidant, hydrogen peroxide reacts relatively slowly with most biomolecules, with the exception of peroxidases containing selenocysteine, cysteine or heme. Accordingly, the reaction between H2S and hydrogen peroxide has a rate constant of 0.73 M–1 s–1 at pH 7.4 and 37 °C [27]. The primary product is HSOH. The final products depend on the initial ratios of hydrogen peroxide to H2S and consist mainly of polysulfides, elemental sulfur and, in the presence of excess oxidant, sulfate. Curiously, the system can behave like a chemical oscillator [90,91].
4.1.2. Hypochlorous Acid and Chloramines
Hypochlorous acid (HOCl) is formed from the oxidation of chloride ions by the neutrophil enzyme myeloperoxidase. HOCl is a highly oxidizing species (E°’(HClO, H+/Cl–, H2O) = +1.28 V) [92] and reacts fast with HS– with a second order rate constant of 0.8–20 × 108 M–1 s–1 at pH 7.4 [27,93]. Hypochlorous acid usually reacts by transferring a Cl+ species to a nucleophile. Thus, the reaction with HS– is likely to proceed through the formation of HSCl, which quickly hydrolyzes to HSOH. The hypochlorite ion is in equilibrium with hypochlorous acid (pKa 7.47). Hypochlorite can also react with HS–, albeit several orders of magnitude more slowly [93]. At an excess of H2S with respect to hypochlorous acid, the main product formed is elemental sulfur, with the intermediacy of polysulfides [93].
The reaction of hypochlorous acid with amines leads to chloramines (RHNCl and R2NCl). These are less reactive and more selective oxidants than hypochlorous acid. Taurine chloramine reacts with H2S with a rate constant of 3 × 102 M–1 s–1 at pH 7.4 and 37 °C [27].
4.1.3. Peroxynitrite
This anion (ONOO–) and its conjugate acid, peroxynitrous acid (ONOOH, pKa 6.8), are formed from the reaction of superoxide radical with nitric oxide, which occurs at nearly diffusion controlled rates. Peroxynitrous acid is unstable and can decay in buffer to nitric acid, hydroxyl radical, and nitrogen dioxide. In vivo, peroxynitrite can either react directly with targets such as thiols or metal centers, or it can react with carbon dioxide, leading to the formation of the secondary radicals, nitrogen dioxide and carbonate radical [94]. Peroxynitrous acid reacts directly with HS– with a second order rate constant of 6.7 × 103 M–1 s–1 (pH 7.4, 37 °C) [95] and, after a short lag, yellow products are formed [96]. According to computational simulations, the process starts with the nucleophilic substitution of HS– on ONOOH to give HSOH and NO2– as initial products. In the presence of excess H2S, HSOH reacts with a second HS– to yield HSS–/HSSH. It was proposed that the yellow products originate from the secondary reaction of HSS–/HSSH with peroxynitrite, and that at least one of the yellow products is HSNO2 or its isomer HSONO [95].
In addition to the direct reaction of peroxynitrous acid with HS–, the free radicals derived from peroxyinitrite decay can also react with H2S and trigger oxygen dependent chain reactions that can be evidenced, for example, by the consumption of oxygen [27].
4.2. One-Electron Oxidation
The one-electron reduction potential of H2S is (E°’(HS•, H+/H2S) = +0.91–0.94 V) [87,97]. This value means that, in principle, only relatively strong one-electron oxidants such as hydroxyl radical (HO•), carbonate radical (CO3•–), nitrogen dioxide (NO2•), and peroxidase oxoferryl compounds I and II can oxidize H2S to HS•/S•–. However, further reactions of HS•/S•– can drive the oxidation processes, extending the list of possible initial oxidants to, for example, metal centers with lower one-electron reduction potentials than that of the HS•/H2S couple.
The initial product of the one-electron oxidation of H2S is the sulfiyl radical (HS•/S•–). This is an oxidizing radical and can react with several electron or hydrogen atom donors (AH, Equation 3). In addition, it can react with a second HS•/S•– radical and form HSSH/HSS– (Equation 4). Alternatively, HS•/S•– can react with HS– to form HSS•2–, a reducing radical that can react with oxygen forming superoxide radical (Equations 5,6). HS•/S•– can also react with oxygen and form SO2•–, which can again react with oxygen to form superoxide radical (Equations 7,8). The latter dismutates to hydrogen peroxide and oxygen, which helps to drive the process.
| HS•/S•− + AH → H2S/HS− + A• | (3) | |
| HS•/S•− + HS•/S•− → HSSH/HSS– | k = 6.5 × 109 M–1 s–1 [98] | (4) |
| HS•/S•− + HS− ⇌ HSS•2− | kf = 5.4 × 109 M–1 s–1; kr = 5.3 × 105 s–1 [98] | (5) |
| HSS•2– + O2 → HSS– + O2•− | k = 4 × 108 M–1 s–1 [98] | (6) |
| HS•/S•− + O2 → SO2•− | k = 7.5 × 109 M–1 s–1 [98] | (7) |
| SO2•− + O2 → SO2 + O2•− | k = 1 × 108 M–1 s–1 [99] | (8) |
In addition to these relatively well characterized reactions, several other reactions can occur, which lead to a wide variety of possible radical and non-radical products and unstable intermediates. It is clear that the one-electron oxidation of H2S can trigger oxygen-dependent free radical chain reactions that can lead to the amplification of initial oxidation events. Several one-electron oxidants can initiate these events.
4.2.1. Oxygen
The one-electron oxidation of H2S by oxygen (O2) is thermodynamically uphill, since the E°’(O2/O2•–) = −0.35 V (–0.18 V for a 1 M O2 standard state) [89]. It is also very slow because of the spin barrier imposed by the diradical character of O2. Nevertheless, H2S autoxidation occurs and is a major concern in practical work with H2S. The process is likely driven by the reactivity of the initial products, HS• and O2•–, as well as that of secondary intermediates. Autoxidation is very slow and dependent on pH. It is critically affected by the presence of trace metal ions that can act as catalysts. It is also mechanistically complex due to the involvement of radical and non-radical intermediates. The products formed are usually elemental sulfur (Sn), sulfate (SO42–), sulfite (SO32–), and thiosulfate (S2O32–) [100,101].
4.2.2. Superoxide Radical
This species is the primary free radical formed from the partial reduction of oxygen. Superoxide radical (O2•–) can dismutate to oxygen and hydrogen peroxide, spontaneously or enzymatically. It can also react with nitric oxide yielding peroxynitrite. This radical can be both oxidizing and reducing, with E°’(O2•–, 2H+/H2O2) = +0.91 V and E°’(O2/O2•–) = –0.18 V (1 M O2 standard state) [89]. It can react with H2S with a rate constant of ~208 M–1 s–1 in dimethyl sulfoxide [102]. However, rather than being scavenged by H2S, superoxide radical can be formed in H2S oxidation processes due to free radical chain reactions, as described in Section 4.2.
4.2.3. Hydroxyl Radical
The highly oxidizing hydroxyl radical (HO•) is formed in biological systems mainly through the Fenton reaction, i.e., through the reduction of hydrogen peroxide to HO• and H2O by reduced metal ions (Fe2+ or Cu+). With a reduction potential of +2.31 V at pH 7 [89], hydroxyl radical is so oxidizing that it can react with most biomolecules, and the rate constants are close to the diffusion limit. It can react very fast with both H2S and HS–, according to Equations 9,10 [98,103].
| HO• + H2S → H2O + HS• | k = 1.1 × 1010 M–1 s–1 [103] | (9) |
| HO• + HS− → HSOH•− → S•−/HS• + H2O/OH− | k = 5.4 × 109 M–1 s–1 [103] | (10) |
4.2.4. Nitrogen Dioxide
This free radical (NO2•) can be formed from the autoxidation of nitric oxide, from the decay of peroxynitrite in the presence of carbon dioxide and from the oxidation of nitrite. It is an oxidizing species with E°’(NO2•/NO2–) = +1.04 V [88]. The reaction between H2S and nitrogen dioxide was studied by pulse radiolysis. The rate constant is 3 × 106 M–1 s–1 at pH 6 (25 °C) and increases at more alkaline pH, suggesting that HS– is the reacting species [27].
4.2.5. Carbonate Radical
The formation of carbonate radical (CO3•–) depends mainly on the decay of peroxynitrite in the presence of carbon dioxide. It also depends on the oxidation of carbon dioxide/bicarbonate by the peroxidase activity of CuZn superoxide dismutase or by xanthine oxidase [94]. Carbonate radical is a strong one-electron oxidant (E°’(CO3•–, H+/HCO3–) = +1.77 V) [88]. It reacts with H2S with a rate constant of 2.0 × 108 M–1 s–1 (pH 7, 20 °C) [97].
4.2.6. Metal Centers
Ferric hemeproteins with an open sixth coordination position, such as methemoglobin, metmyoglobin, myeloperoxidase, and catalase, can oxidize H2S [60,61,104,105,106].
Depending on the protein, either protonated H2S or HS– can bind to ferric heme [107]. The binding is stable enough in some hemeproteins to suggest a function in H2S storage and transport, as in hemoglobin I from Lucina pectinata [108]. In the case of methemoglobin and metmyoglobin, H2S binds quickly and reversibly to ferric heme. Deprotonation leads to a ferric-HS– complex that can slowly evolve to ferrous-bound HS•. The latter species can react with a second H2S or with oxygen, which leads to the formation of polysulfides and thiosulfate as products [60,61,107,109,110].
Remarkably, the hexacoordinated protein ferric cytochrome c can also oxidize H2S. The process is mediated by reducing species derived from the HS•/S•– radical [111].
The oxoferryl compounds I and II of myeloperoxidase are formed in the peroxidase cycle of the enzyme and have reduction potentials of +1.35 and +0.97 V [92]. They can react with H2S with rate constants of 1 × 106 M–1 s–1 and 2.0 × 105 M–1 s–1, respectively (pH 7.4 and 25 °C). Ferric myeloperoxidase can also oxidize H2S and is reduced to the ferrous state [105]. Under aerobic conditions, compound III (ferric-superoxide or ferrous-dioxygen complexes in resonance) is formed. This compound slowly evolves to the ferric enzyme, allowing the enzyme to turnover. Myeloperoxidase-catalyzed oxidation of H2S leads to the formation of polysulfides and protein persulfides [106].
In many cases, the interaction between a hemeprotein and H2S leads to the formation of sulfheme, where one of the pyrroles in the porphyrin ring is covalently modified with a sulfur atom. Such modification has been reported for some globins, lactoperoxidase, and catalase [112]. Many details need to be filled in regarding the mechanism of sulfheme formation. An adequately positioned histidine in the distal heme site has been proposed to be critical [112]. It has also been proposed that, in the case of myeloperoxidase, the presence of a sulfonium ion linkage between a methionine and the heme ring determines that, in contrast to lactoperoxidase, the sulfheme cannot be formed [106]. Computational simulations suggest that sulfheme formation occurs through a concerted interaction between the distal histidine, H2S, and a ferric-bound hydroperoxide to form oxoferryl compound II, H2O and HS•; the latter subsequently adds to a specific pyrrole site [113].
4.3. Other Oxidants
Along with the species described above, other oxidants of biological relevance are likely to react with H2S, by analogy to their reactions with thiols. Among these other oxidants, organic hydroperoxides, other hypohalous acids such as HOBr and HOSCN, and free radicals such as peroxyl and phenoxyl radicals are to be considered.
4.4. Nitric Oxide
4.5. Biological Implications of H2S Oxidation by Reactive Species
At the physiological pH of 7.4, the rate constants of the reactions of H2S with oxidants have similar values as those of typical biological thiols such as cysteine and glutathione [27]. The pH-independent rate constants corresponding to the HS– species are lower than those of the thiolates species (RS–), probably due to the lack of the inductive effect of the methylene. However, the higher acidity of H2S with respect to thiols (pKa of 6.98 for H2S compared to 8.29 for cysteine and 8.94 for glutathione [53,122]), determines a higher availability of the ionized species. The net effect is that the apparent rate constants at physiological pH are comparable.
To analyze whether H2S can be a scavenger of oxidant species, the rates instead of the rate constants need to be taken into account, i.e., the products of rate constants times concentration. While the rate constants are similar to those of thiols, the concentration of H2S in tissues is considered to be submicromolar and several orders of magnitude lower than that of thiols, which is millimolar, so H2S cannot compete with thiols for oxidants. Similar considerations apply for the comparison between H2S and other oxidant scavengers such as ascorbic acid. Hence, H2S cannot be considered a typical antioxidant scavenger except under exceptional situations that lead to high concentrations such as exogenous administration together with enhanced oxidant production. Other mechanisms are probably underlying the observed antioxidant effects of H2S; for example, interference with signaling pathways. Furthermore, the one-electron oxidation of H2S can trigger free radical chain reactions, opening the possibility for pro-oxidant effects of H2S.
5. Chemistry and Biology of Persulfides
Persulfides (RSSH/RSS–) are sulfane sulfur compounds, meaning that they have a sulfur atom bound to two sulfurs or to a sulfur and an ionizable hydrogen [123]; the outer sulfur is the sulfane sulfur according to this criterion. RSSH can also be referred to with the terms hydropersulfide or hydrodisulfide. In this text, “persulfide” is used to designate mixtures of RSSH and RSS−. Their chemical properties differ from those of thiols; hydropersulfides are more acidic and can either act as nucleophiles when deprotonated (RSS–) or electrophiles when protonated (RSSH). They are formed in biological systems through H2S-dependent and independent pathways. It is worth noting that the reaction between H2S and thiols does not occur in the absence of an oxidant.
5.1. Reactivity of Persulfides
5.1.1. Persulfide Acidity
Protonated persulfides ionize in water and form the corresponding anionic persulfides (Equation 11). Their acidity is predicted to be higher than that of the analogous thiols. Thus, at physiological pH, the anionic species would predominate. For instance, the pKa of 2-[(3-aminopropyl)amino]ethane persulfide was estimated to be 6.2 ± 0.1, while the pKa of the analogous thiol is 7.6 ± 0.1 [124].
RSSH ⇌ RSS− + H+
5.1.2. Persulfides as Nucleophiles
Anionic persulfides can act as nucleophiles (Equation 12). This is evidenced by their reactions with thiol alkylating agents such as monobromobimane and iodoacetamide, which yield disulfides. This is also evidenced by their reactions with disulfides such as 5,5-dithiobis(2-nitrobenzoic acid) and 4,4-dithiodipyridine to produce trisulfides [20,28,33,125,126,127,128,129]. Moreover, persulfides react with two- and one-electron oxidants as discussed in Section 6.
RSS− + E+ → RSSE
Persulfides have been proposed to have an increased nucleophilicity compared to analogous thiolates [126,130]. This can be explained by the alpha effect, namely, the increased reactivity of an atom due to the presence of unshared pairs of electrons in the adjacent atom [131,132]. This effect has been observed in the nucleophilicity of HOO– compared to HO– and NH2NH2 and NH2OH compared to NH3 [131]. The first determination of the alpha effect in persulfides was observed in human serum albumin, in which persulfide has an increased reactivity towards the disulfide 4-4’-dithiodipyridine compared to the thiolate derivative [28]. The alpha effect, together with the predominance of RSS– versus RSSH at physiological pH due to the relatively high acidity, make persulfides better nucleophiles than thiols.
5.1.3. Persulfides as Electrophiles
Protonated persulfides are weak electrophiles. They can react with nucleophiles to give thiols when the outer sulfur is attacked (Equation 13) or H2S when the inner sulfur is attacked (Equation 14). Whether the attack is on one sulfur or the other appears to be determined by steric hindrance [133,134] and by the acidity of the leaving group, with the better leaving group being the one with lower pKa. Hence, as low molecular weight (LMW) thiols have higher pKas than H2S, i.e., the pKa of cysteine is 8.29 [122] and the pKa of H2S is 6.98 [53], the attack on the inner sulfur would predominate. In protein persulfides formed in acidic cysteines, the attack on the outer sulfur would predominate and restore the reduced protein.
RSSH + Nu− → RS− + NuSH
RSSH + Nu− → RSNu + HS−
The outer sulfur can react with cyanide, thiolates, sulfite [133,135], amines [136], hydroxide [137], and tertiary phosphines [138,139]. Persulfides and cyanide react to form the corresponding thiol and thiocyanate, which, in the presence of ferric ions, forms a colored complex that can be quantified spectrophotometrically. This is the basis of the method of cold cyanolysis used to quantify persulfides [123,140].
In the reaction of a persulfide with a thiol, the attack on the outer sulfur constitutes a transpersulfidation reaction. In addition to forming thiol, the sulfane sulfur is transferred and a new persulfide at the attacking thiol is formed (Equation 15). The reactions between persulfides and thiols are plausible to occur in vivo. Intermediate persulfides formed in the proteins MST, SQR, and rhodanese can be transferred to several acceptors including cyanide, sulfite, and thiols [34,57,58,141,142,143,144]. Cysteine desulfurases, such as those that participate in the synthesis of iron sulfur clusters, are another example of enzymes transferring the sulfane sulfur to a thiol in specific compounds [145]. The reduction of protein persulfides by thiols could constitute a protective mechanism to avoid irreversible cysteine modifications under oxidative stress [45,111,146]. A depersulfidase role for thioredoxin has been suggested. One of the proposed mechanisms involves the transfer of the sulfane sulfur from either LMW or protein persulfides to the nucleophilic cysteine of thioredoxin, which regenerates the original thiol and forms thioredoxin persulfide. Subsequently, H2S and oxidized thioredoxin are formed [29,147,148].
On the other hand, when the inner sulfur of a persulfide is attacked by a thiol, H2S is released and a mixed disulfide is formed (Equation 16) [125,139,149]. This mechanism has been suggested for the activation of Nrf2 by Keap1. Keap1 persulfide/Nrf2 was proposed to react with a protein thiolate to form a mixed disulfide and release H2S and Nrf2, inducing its activation to protect neuroblastoma cells from methylglyoxal-induced toxicity [150].
RSSH + R’S− ⇌ RS− + R’SSH
RSSH + R’S− ⇌ RSSR’ + HS−
5.1.4. Disproportionation of Persulfides
Persulfides are highly unstable in aqueous solution, making their study difficult. This is related to their nucleophilic and electrophilic character that facilitate the reactions between them causing spontaneous decay through a disproportionation process. Depending on which sulfur is attacked, different products can be formed. The attack on the outer sulfur generates thiol and hydropolysulfides that evolve to elemental sulfur (Equations 17,18), while the attack on the inner sulfur yields polysulfides and H2S (Equation 19) [137]. The decay products are also likely to depend on steric effects and on the acidity of the leaving groups. For example, cysteine persulfide predominantly decays through the release of H2S instead of cysteine, which has a higher pKa [35].
RSSH + RSS− ⇌ RSSS− + RSH
RSSSH → RSH + Sn
RSSH + RSS− ⇌ RSSSR + HS−
5.2. Biological Sources of Persulfides
Several processes can give rise to persulfides in biological context; some are dependent on H2S and some are not. These processes are described below. In addition, it was recently proposed that cysteine persulfide can be generated from cysteine via cysteinyl-tRNA synthetase [41].
5.2.1. Radical Processes
Sulfiyl (HS•) and thiyl radicals (RS•) could be formed by the reaction of strong one-electron oxidants and H2S or RSH, respectively. These radicals could rapidly react yielding a persulfide (Equation 20). Nevertheless, this reaction is not expected to be relevant in vivo due to the low concentration of radical species. A more likely radical pathway to form persulfides is the formation of the reducing radical RSSH•– via the reaction of either HS• and RS– or RS• and HS– and the subsequent reaction with oxygen (Equations 21–23) [20,151].
HS• + RS• → RSSH
HS• + RS− → RSSH•−
RS• + HS− → RSSH•−
RSSH•− + O2 → RSSH + O2•−
5.2.2. H2S and Oxidized Thiols
The reactions of H2S with oxidized thiols constitute biologically significant pathways for the formation of persulfides. H2S can react with disulfides releasing thiol (Equation 24) or with sulfenic acid eliminating water (Equation 25).
HS− + RSSR’ ⇌ RSSH + R’S−
HS− + RSOH → RSSH + OH−
Protein persulfides are formed endogenously through these pathways. For example, a persulfide intermediate of SQR is formed by the reaction of H2S with the active site disulfide of the enzyme. This reaction is ~106 fold accelerated compared to the reaction with cystine [57,58,152]. In the endoplasmic reticulum, an environment with high concentration of disulfides, the reaction of H2S with disulfides could be more relevant than in compartments with scarce disulfides like the cytosol [151]. In cultured cells, an increase in persulfide concentration was observed after treatment with H2O2 that led to an increase in sulfenic acid and disulfide. However, this was not observed upon treatment with diamide that only increased disulfide concentration, which suggests that persulfide was formed by H2S and sulfenic acid [28,29]. The reaction of H2S with sulfenic acids could be a mechanism of protection against its overoxidation to sulfinic acid (RSO2H) and sulfonic acid (RSO3H), which are irreversible modifications. On the contrary, the oxidation of persulfidated residues leads to RSSO2H and RSSO3H species that can be reduced to thiols restoring the protein [20,29,45,46,153].
The reactions of H2S with disulfides and sulfenic acids are common strategies to prepare persulfides in the laboratory. Mixtures of LMW disulfides and H2S have been used as sources of persulfides [28,66,126,154,155]. However, the facts that reaction (24) is reversible and that the persulfides formed can react with disulfides to form trisulfides and thiols, need to be taken into account [128,129]. The kinetics of the reactions between H2S and LMW disulfides such as cystine, glutathione disulfide, homocystine, and 5,5’-dithiobis-(2-nitrobenzoic acid) to form LMW persulfides are slower than the reactions of thiols with disulfides [28]. The incubation of H2S and mixed disulfides of protein cysteines and acidic LMW thiols has been used to produce persulfides in papain [126], glutathione peroxidase Gpx3 [125], and albumin [28]. The reaction of protein sulfenic acid with H2S has also been used to prepare persulfides in albumin [28,151].
5.2.3. Thiolates and Oxidized Sulfur Derivatives
Thiolates and oxidized sulfur derivatives of H2S react to form persulfides. As described previously, the attack of thiolates on the outer sulfur of persulfides produces a new persulfide at the attacking thiolate (Equation 15). For example, the persulfide intermediate formed in SQR has been proposed to react with different molecules. When it reacts with a thiol, a new persulfide is generated [42,58]. In addition, organic and inorganic polysulfides (RSSnSR, RSSnS−, HSSnS−) can be sources of persulfides. A few polysulfide-containing proteins have been detected, such as SOD1, IL-6, and IgG2 [156]. Recently, the reaction between dibenzyl trisulfide and tetrasulfide with cysteine and glutathione was shown to release H2S with a brief lag-time that was attributed to the formation of persulfides as intermediates [157]. Cysteine trisulfide treatment of cultured cells led to an increase in intracellular cysteine and glutathione persulfides [129].
5.2.4. Elimination of Disulfide
The disulfide cystine in the presence of the pyridoxal phosphate-dependent enzymes, CBS and CSE, forms cysteine persulfide, ammonia, and pyruvate. In addition, CSE also catalyzes the formation of homocysteine persulfide from homocystine. Nevertheless, these reactions are limited in vivo [33,35,37,38,39,158]. Disulfides can also suffer a β-elimination process under alkaline conditions yielding persulfide, a dehydroalanine (2-aminoacrylate) derivative, and water [159,160,161].
6. Reactivity of Persulfides with Oxidants
6.1. Two-Electron Oxidation
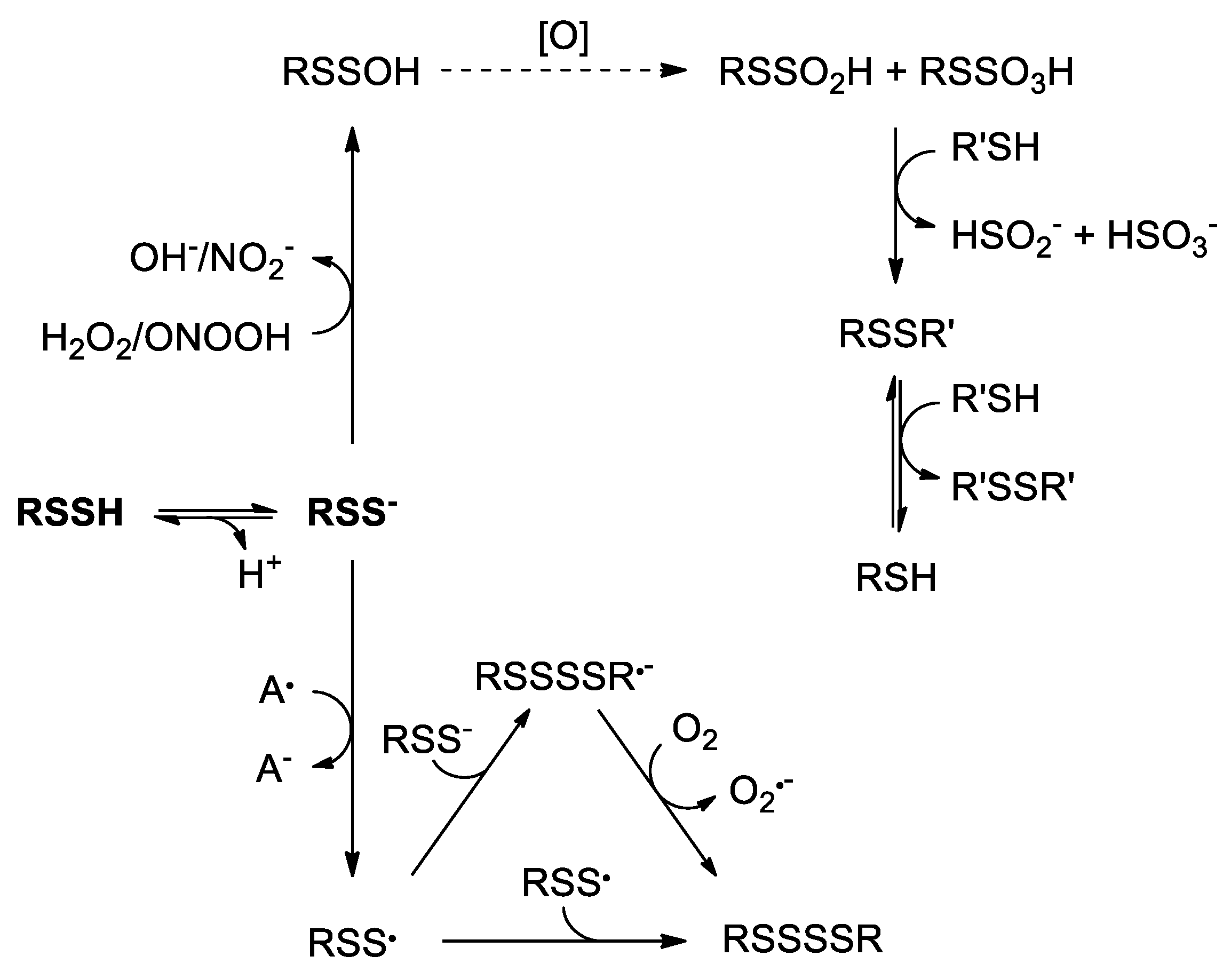
The enhanced nucleophilicity of persulfides versus thiols is also evidenced by the reactions with two-electron oxidizing electrophiles such as H2O2 and peroxynitrite. Though the reducing abilities of persulfides have not been fully studied, they seem to be better reductants than the corresponding thiols [162]. The reaction of peroxynitrite with albumin persulfide is one order of magnitude faster ((1.2 ± 0.4) × 104 M–1 s–1 at 20 °C) than the reduced protein at pH 7.4 ((2.7 ± 0.3) × 103 M–1 s–1) [28]. Furthermore, it was observed that, under certain conditions, H2O2 did not react with glutathione but rapidly reacted with glutathione persulfide [33]. Analogously to the reaction of H2O2 with thiols that forms sulfenic acid (RSOH), the reaction with persulfides is expected to produce perthiosulfenic acid (RSSOH). In fact, density functional theory (DFT) calculations predicted that RSSH can be readily oxidized to RSSOH. The reaction of H2O2 with the persulfide model ethyldisulfane (Et-SSH) to form Et-SSOH was ~6.2 kcal/mol more favorable compared to the oxidation of the corresponding thiol, ethanethiol (Et-SH) to Et-SOH. These calculations also predicted that RSOH and RSSOH are reactive towards dimedone to form stable products. Moreover, the oxidation of cysteine persulfide residues to perthiosulfenic acid was observed in tyrosine kinases such as epidemial growth factor receptor (EGFR) in response to NADPH oxidase activation [163]. In the presence of excess oxidant, the perthiosulfenic acid could be oxidized to perthiosulfinic and perthiosulfonic acids (RSSO2H and RSSO3H) [20,163]. These species have been detected in papain, albumin and glutathione peroxidase [28,125,151]. It is noteworthy that while sulfinic and sulfonic acids (RSO2H and RSO3H) are generally considered irreversible modification products, RSSO2H and RSSO3H can be reduced to thiols by common reductants such as dithiothreitol and enzymes such as thioredoxin, and they can therefore allow the restoration of the reduced protein. Thus, as it was previously mentioned, persulfides could protect protein thiols [29,45,46,153]. The reactions of persulfides with two-electron oxidants are summarized in Scheme 2.
6.2. One-Electron Oxidation
Persulfides have enhanced one-electron reducing capability compared to thiols and H2S [164]. Firstly, persulfides have a lower S-H bond dissociation energy (70 kcal/mol) than thiols and H2S (92 kcal/mol) [165]. Secondly, the one-electron reduction potential of persulfides (E°’(RSS•/RSS–) = +0.68 V) is lower than those of thiols (E°’(RS•, H+/RSH) = +0.96 V) and H2S (E°’(S•–, H+/HS–) = +0.91 V) [87]. Finally, the resonance stabilization energy of RSS• is higher than that of RS•, as the unpaired electron in RSS• is delocalized between the two sulfur atoms [124,164]. Hence, compared to thiols and H2S, persulfides can be oxidized by weaker oxidants; the one-electron oxidation products RSS• are less oxidizing than RS• and HS•, react more slowly, and possess an inherent stability. In summary, persulfides constitute better reductants than thiols and H2S.
The reaction with one-electron oxidants gives rise to RSS• and can occur through hydrogen atom or electron transfer mechanisms [164]. Persulfides can be oxidized by ferric ions and cannot oxidize ferrous ions [166]. Ferricyanide [47], ferric cytochrome c [126,149,167], and metmyoglobin [47] can oxidize persulfides. RSS• is also formed in the reactions with carbon centered radicals [124], peroxyl radicals [164,168], and the nitroxide TEMPOL [47]. These oxidants react more slowly or not at all with the corresponding thiols, exemplifying the better reductant character of persulfides. For instance, TEMPOL and metmyoglobin can be reduced by N-methoxycarbonyl penicillamine persulfide but not by an analogous thiol, N-acetyl penicillamine [47]. Furthermore, glutathione persulfide was reported to efficiently reduce ferric cytochrome c, while glutathione and H2S only slowly reduced it [126]. A theoretical study of the reactions of HO• towards a set of persulfides proposed that they could occur through disulfide bond cleavage. In this scenario, RSS• would not be formed [169].
Perthiyl radicals rapidly decay forming tetrasulfide species (RSSSSR) with rate constants of 1–6 × 109 M–1 s–1 [47,168,170]. In fact, dialkyl tetrasulfide was photolytically cleaved to produce the corresponding RSS•, but dimerization rapidly occurred [168]. The efficiency of this reaction could explain why some thermodynamically unfavorable RSS• formation reactions do occur, such as those of TEMPOL and CH3SSH [47], N-methoxycarbonyl penicillamine persulfide and ferric cytochrome c [87,149], or RSS– and O2 [171,172]. Moreover, the resulting RSSSSR can decompose to form RSSR and Sn [20].
Perthiyl radicals can react with RSS– to give the intermediate RSSSSR•– that reacts with O2 to yield O2•– and RSSSSR. However, RSSSSR•– have not been experimentally evidenced [173]. Analogously, RSS• and RS– react producing RSSSR•– [164]. The reactions are summarized in Scheme 2.
Perthiyl radicals, generated via radiolytic reduction of dialkyl trisulfide, were initially reported to react with O2 with a rate constant of 5.1 × 106 M–1 s–1. Such reaction was proposed to form RSSOO• species with subsequent formation of inorganic sulfate [170], which is analogous to thiyl radicals that initially form RSOO• in reversible processes [174,175,176,177]. However, more recent studies suggest that RSS• and O2 do not react. Photolytically produced RSS• exhibited the same decay in absence than in presence of O2 [168]; besides, oxygen consumption was not altered by addition of perthiyl radicals [47]. Furthermore, computational analysis showed that the reaction between RSS• and O2 is endergonic by 16 kcal/mol [47].
The reaction of RS• and NO• to form S-nitrosothiols (RSNO) has been deeply studied [178]. The expected product from the reaction of RSS• with NO• would be S-nitrosopersulfide (RSSNO); however, these radicals do not appear to react. Experimental approaches to synthesize RSSNO failed, with formation of NO• and tetrasulfide. In addition, a solution generating RSS• did not consume NO• [47]. Considering the weakness of the S-N bond determined by computational analyses (3–4 kcal/mol) and the high stability of the radicals, RSSNO is unlikely to exist at room temperature [47,179].
To conclude, persulfides are excellent one-electron reductants. In these reactions, relatively stable perthiyl radicals are formed and can undergo different reactions that ultimately yield RSSSSR.
7. Conclusions
In the previous sections, we have summarized the properties of hydrogen sulfide and persulfides, with a focus on redox aspects. A rationalization of the interactions of these species with biologically-relevant reactive oxidants requires an understanding of the kinetics of the processes as well as the elucidation of the fates of the oxidation products formed. While it is becoming clear that direct antioxidant scavenging roles cannot be attributed to H2S except under exceptionally high concentrations, many aspects are yet to be understood about persulfides and their rich biochemistry, including their reactions with oxidants.
Author Contributions
All authors wrote and edited the manuscript.
Funding
This work was supported in part by grants from the Comisión Sectorial de Investigación Científica (CSIC) from Universidad de la República (GRUPOS_I+D_2014_C632-348 and I+D_2016_541), from the Agencia Nacional de Investigación e Innovación (ANII) (to Matías N. Möller, FCE_1_2017_1_136043) and from Fondo Vaz Ferreira, Ministerio de Educación y Cultura (to Ernesto Cuevasanta, I/FVF2017/069). Dayana Benchoam and Ernesto Cuevasanta were partially supported by Comisión Académica de Posgrado (CAP) from Universidad de la República and by PEDECIBA (Uruguay).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Beauchamp, R.O.; Bus, J.S.; Popp, J.A.; Boreiko, C.J.; Andjelkovich, D.A. A critical review of the literature on hydrogen sulfide toxicity. Crit. Rev. Toxicol. 1984, 13, 25–97. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.S.; Beehler, C.L.; Sakamoto-Arnold, C.M.; Childress, J.J. In situ measurements of chemical distributions in a deep-sea hydrothermal vent field. Science 1986, 231, 1139–1141. [Google Scholar] [CrossRef] [PubMed]
- Olson, K.R.; Straub, K.D. The role of hydrogen sulfide in evolution and the evolution of hydrogen sulfide in metabolism and signaling. Physiology (Bethesda) 2016, 31, 60–72. [Google Scholar] [CrossRef] [PubMed]
- Griesbeck, C.; Hauska, G.; Schütz, M. Biological Sulfide Oxidation: Sulfide-Quinone Reductase (SQR), the Primary Reaction. Recent Res. Dev. Microbiol. 2000, 4, 179–203. [Google Scholar]
- Mozzarelli, A.; Bettati, S.; Campanini, B.; Salsi, E.; Raboni, S.; Singh, R.; Spyrakis, F.; Kumar, V.P.; Cook, P.F. The multifaceted pyridoxal 5′-phosphate-dependent O-acetylserine sulfhydrylase. Biochim. Biophys. Acta 2011, 1814, 1497–1510. [Google Scholar] [CrossRef] [PubMed]
- Barton, L.L.; Fardeau, M.-L.; Fauque, G.D. Hydrogen sulfide: A toxic gas produced by dissimilatory sulfate and sulfur reduction and consumed by microbial oxidation. Met. Ions Life Sci. 2014, 14, 237–277. [Google Scholar] [PubMed]
- Cooper, C.E.; Brown, G.C. The inhibition of mitochondrial cytochrome oxidase by the gases carbon monoxide, nitric oxide, hydrogen cyanide and hydrogen sulfide: Chemical mechanism and physiological significance. J. Bioenerg. Biomembr. 2008, 40, 533–539. [Google Scholar] [CrossRef] [PubMed]
- Reiffenstein, R.J.; Hulbert, W.C.; Roth, S.H. Toxicology of hydrogen sulfide. Annu. Rev. Pharmacol. Toxicol. 1992, 32, 109–134. [Google Scholar] [CrossRef] [PubMed]
- Grieshaber, M.K.; Völkel, S. Animal adaptations for tolerance and exploitation of poisonous sulfide. Annu. Rev. Physiol. 1998, 60, 33–53. [Google Scholar] [CrossRef]
- Leschelle, X.; Goubern, M.; Andriamihaja, M.; Blottière, H.M.; Couplan, E.; Gonzalez-Barroso, M.-D.-M.; Petit, C.; Pagniez, A.; Chaumontet, C.; Mignotte, B.; et al. Adaptative metabolic response of human colonic epithelial cells to the adverse effects of the luminal compound sulfide. Biochim. Biophys. Acta 2005, 1725, 201–212. [Google Scholar] [CrossRef]
- Mathai, J.C.; Missner, A.; Kügler, P.; Saparov, S.M.; Zeidel, M.L.; Lee, J.K.; Pohl, P. No facilitator required for membrane transport of hydrogen sulfide. Proc. Natl. Acad. Sci. USA 2009, 106, 16633–16638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuevasanta, E.; Denicola, A.; Alvarez, B.; Möller, M.N. Solubility and permeation of hydrogen sulfide in lipid membranes. PLoS ONE 2012, 7, e34562. [Google Scholar] [CrossRef] [PubMed]
- Riahi, S.; Rowley, C.N. Why can hydrogen sulfide permeate cell membranes? J. Am. Chem. Soc. 2014, 136, 15111–15113. [Google Scholar] [CrossRef] [PubMed]
- Goubern, M.; Andriamihaja, M.; Nübel, T.; Blachier, F.; Bouillaud, F. Sulfide, the first inorganic substrate for human cells. FASEB J. 2007, 21, 1699–1706. [Google Scholar] [CrossRef] [Green Version]
- Kabil, O.; Vitvitsky, V.; Xie, P.; Banerjee, R. The quantitative significance of the transsulfuration enzymes for H2S production in murine tissues. Antioxid. Redox Signal. 2011, 15, 363–372. [Google Scholar] [CrossRef]
- Fromageot, C. Oxidation of Organic Sulfur in Animals. In Advances in Enzymology and Related Areas of Molecular Biology; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 1947; pp. 369–407. ISBN 978-0-470-12252-5. [Google Scholar]
- Kruszyna, H.; Kruszyna, R.; Smith, R.P. Cyanide and sulfide interact with nitrogenous compounds to influence the relaxation of various smooth muscles. Proc. Soc. Exp. Biol. Med. 1985, 179, 44–49. [Google Scholar] [CrossRef]
- Abe, K.; Kimura, H. The possible role of hydrogen sulfide as an endogenous neuromodulator. J. Neurosci. 1996, 16, 1066–1071. [Google Scholar] [CrossRef]
- Hosoki, R.; Matsuki, N.; Kimura, H. The possible role of hydrogen sulfide as an endogenous smooth muscle relaxant in synergy with nitric oxide. Biochem. Biophys. Res. Commun. 1997, 237, 527–531. [Google Scholar] [CrossRef]
- Filipovic, M.R.; Zivanovic, J.; Alvarez, B.; Banerjee, R. Chemical Biology of H2S Signaling through Persulfidation. Chem. Rev. 2018, 118, 1253–1337. [Google Scholar] [CrossRef]
- Whiteman, M.; Armstrong, J.S.; Chu, S.H.; Jia-Ling, S.; Wong, B.-S.; Cheung, N.S.; Halliwell, B.; Moore, P.K. The novel neuromodulator hydrogen sulfide: An endogenous peroxynitrite “scavenger”? J. Neurochem. 2004, 90, 765–768. [Google Scholar] [CrossRef]
- Kimura, Y.; Kimura, H. Hydrogen sulfide protects neurons from oxidative stress. FASEB J. 2004, 18, 1165–1167. [Google Scholar] [CrossRef] [PubMed]
- Whiteman, M.; Cheung, N.S.; Zhu, Y.-Z.; Chu, S.H.; Siau, J.L.; Wong, B.S.; Armstrong, J.S.; Moore, P.K. Hydrogen sulphide: A novel inhibitor of hypochlorous acid-mediated oxidative damage in the brain? Biochem. Biophys. Res. Commun. 2005, 326, 794–798. [Google Scholar] [CrossRef] [PubMed]
- Yonezawa, D.; Sekiguchi, F.; Miyamoto, M.; Taniguchi, E.; Honjo, M.; Masuko, T.; Nishikawa, H.; Kawabata, A. A protective role of hydrogen sulfide against oxidative stress in rat gastric mucosal epithelium. Toxicology 2007, 241, 11–18. [Google Scholar] [CrossRef]
- Laggner, H.; Muellner, M.K.; Schreier, S.; Sturm, B.; Hermann, M.; Exner, M.; Gmeiner, B.M.K.; Kapiotis, S. Hydrogen sulphide: A novel physiological inhibitor of LDL atherogenic modification by HOCl. Free Radic. Res. 2007, 41, 741–747. [Google Scholar] [CrossRef] [PubMed]
- Jha, S.; Calvert, J.W.; Duranski, M.R.; Ramachandran, A.; Lefer, D.J. Hydrogen sulfide attenuates hepatic ischemia-reperfusion injury: Role of antioxidant and antiapoptotic signaling. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H801–H806. [Google Scholar] [CrossRef]
- Carballal, S.; Trujillo, M.; Cuevasanta, E.; Bartesaghi, S.; Möller, M.N.; Folkes, L.K.; García-Bereguiaín, M.A.; Gutiérrez-Merino, C.; Wardman, P.; Denicola, A.; et al. Reactivity of hydrogen sulfide with peroxynitrite and other oxidants of biological interest. Free Radic. Biol. Med. 2011, 50, 196–205. [Google Scholar] [CrossRef] [PubMed]
- Cuevasanta, E.; Lange, M.; Bonanata, J.; Coitiño, E.L.; Ferrer-Sueta, G.; Filipovic, M.R.; Alvarez, B. Reaction of hydrogen sulfide with disulfide and sulfenic acid to form the strongly nucleophilic persulfide. J. Biol. Chem. 2015, 290, 26866–26880. [Google Scholar] [CrossRef] [PubMed]
- Wedmann, R.; Onderka, C.; Wei, S.; Szijártó, I.A.; Miljkovic, J.L.; Mitrovic, A.; Lange, M.; Savitsky, S.; Yadav, P.K.; Torregrossa, R.; et al. Improved tag-switch method reveals that thioredoxin acts as depersulfidase and controls the intracellular levels of protein persulfidation. Chem. Sci. 2016, 7, 3414–3426. [Google Scholar] [CrossRef] [Green Version]
- Kessler, D. Enzymatic activation of sulfur for incorporation into biomolecules in prokaryotes. FEMS Microbiol. Rev. 2006, 30, 825–840. [Google Scholar] [CrossRef] [Green Version]
- Pandey, A.; Pain, J.; Ghosh, A.K.; Dancis, A.; Pain, D. Fe-S cluster biogenesis in isolated mammalian mitochondria: Coordinated use of persulfide sulfur and iron and requirements for GTP, NADH, and ATP. J. Biol. Chem. 2015, 290, 640–657. [Google Scholar] [CrossRef]
- Cuevasanta, E.; Möller, M.N.; Alvarez, B. Biological chemistry of hydrogen sulfide and persulfides. Arch. Biochem. Biophys. 2017, 617, 9–25. [Google Scholar] [CrossRef] [PubMed]
- Ida, T.; Sawa, T.; Ihara, H.; Tsuchiya, Y.; Watanabe, Y.; Kumagai, Y.; Suematsu, M.; Motohashi, H.; Fujii, S.; Matsunaga, T.; et al. Reactive cysteine persulfides and S-polythiolation regulate oxidative stress and redox signaling. Proc. Natl. Acad. Sci. USA 2014, 111, 7606–7611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hylin, J.W.; Wood, J.L. Enzymatic formation of polysulfides from mercaptopyruvate. J. Biol. Chem. 1959, 234, 2141–2144. [Google Scholar] [PubMed]
- Yadav, P.K.; Martinov, M.; Vitvitsky, V.; Seravalli, J.; Wedmann, R.; Filipovic, M.R.; Banerjee, R. Biosynthesis and reactivity of cysteine persulfides in signaling. J. Am. Chem. Soc. 2016, 138, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Cavallini, D.; De Marco, C.; Mondovi, B. Cleavage of cystine by a pyridoxal model. Arch. Biochem. Biophys. 1960, 87, 281–288. [Google Scholar] [CrossRef]
- Cavallini, D.; Mondovi, B.; De Marco, C.; Sciosciasantoro, A. Inhibitory effect of mercaptoethanol and hypotaurine on the desulfhydration of cysteine by cystathionase. Arch. Biochem. Biophys. 1962, 96, 456–457. [Google Scholar] [CrossRef]
- Flavin, M. Microbial transsulfuration: The mechanism of an enzymatic disulfide elimination reaction. J. Biol. Chem. 1962, 237, 768–777. [Google Scholar]
- Yamanishi, T.; Tuboi, S. The mechanism of the L-cystine cleavage reaction catalyzed by rat liver gamma-cystathionase. J. Biochem. 1981, 89, 1913–1921. [Google Scholar] [CrossRef]
- Szczepkowski, T.W.; Wood, J.L. The cystathionase-rhodanese system. Biochim. Biophys. Acta 1967, 139, 469–478. [Google Scholar] [CrossRef]
- Akaike, T.; Ida, T.; Wei, F.-Y.; Nishida, M.; Kumagai, Y.; Alam, M.M.; Ihara, H.; Sawa, T.; Matsunaga, T.; Kasamatsu, S.; et al. Cysteinyl-tRNA synthetase governs cysteine polysulfidation and mitochondrial bioenergetics. Nat. Commun. 2017, 8, 1177. [Google Scholar] [CrossRef]
- Mishanina, T.V.; Libiad, M.; Banerjee, R. Biogenesis of reactive sulfur species for signaling by hydrogen sulfide oxidation pathways. Nat. Chem. Biol. 2015, 11, 457–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valentine, W.N.; Toohey, J.I.; Paglia, D.E.; Nakatani, M.; Brockway, R.A. Modification of erythrocyte enzyme activities by persulfides and methanethiol: Possible regulatory role. Proc. Natl. Acad. Sci. USA 1987, 84, 1394–1398. [Google Scholar] [CrossRef] [PubMed]
- Toohey, J.I. Persulfide sulfur is a growth factor for cells defective in sulfur metabolism. Biochem. Cell Biol. 1986, 64, 758–765. [Google Scholar] [CrossRef] [PubMed]
- Millikin, R.; Bianco, C.L.; White, C.; Saund, S.S.; Henriquez, S.; Sosa, V.; Akaike, T.; Kumagai, Y.; Soeda, S.; Toscano, J.P.; et al. The chemical biology of protein hydropersulfides: Studies of a possible protective function of biological hydropersulfide generation. Free Radic. Biol. Med. 2016, 97, 136–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ono, K.; Akaike, T.; Sawa, T.; Kumagai, Y.; Wink, D.A.; Tantillo, D.J.; Hobbs, A.J.; Nagy, P.; Xian, M.; Lin, J.; et al. Redox chemistry and chemical biology of H2S, hydropersulfides, and derived species: Implications of their possible biological activity and utility. Free Radic. Biol. Med. 2014, 77, 82–94. [Google Scholar] [CrossRef] [PubMed]
- Bianco, C.L.; Chavez, T.A.; Sosa, V.; Saund, S.S.; Nguyen, Q.N.N.; Tantillo, D.J.; Ichimura, A.S.; Toscano, J.P.; Fukuto, J.M. The chemical biology of the persulfide (RSSH)/perthiyl (RSS·) redox couple and possible role in biological redox signaling. Free Radic. Biol. Med. 2016, 101, 20–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ezeriņa, D.; Takano, Y.; Hanaoka, K.; Urano, Y.; Dick, T.P. N-Acetyl Cysteine Functions as a Fast-Acting Antioxidant by Triggering Intracellular H2S and Sulfane Sulfur Production. Cell Chem. Biol. 2018, 25, 447–459.e4. [Google Scholar] [CrossRef]
- Agency for Toxic Substances and Disease Registry (ATSDR). Toxicological Profile for Hydrogen Sulfide/Carbonyl Sulfide; U.S. Department of Health and Human Services, Public Health Service: Atlanta, GA, USA, 2016.
- Bondi, A. van der Waals Volumes and Radii. J. Phys. Chem. 1964, 68, 441–451. [Google Scholar] [CrossRef]
- Riahi, S.; Rowley, C.N. Solvation of Hydrogen Sulfide in Liquid Water and at the Water–Vapor Interface Using a Polarizable Force Field. J. Phys. Chem. B 2014, 118, 1373–1380. [Google Scholar] [CrossRef]
- Fogg, P.G.T.; Young, C.L. Hydrogen Sulfide, Deuterium Sulfide and Hydrogen Selenide; Pergamon Press: Oxford, UK, 1988; Volume 32, ISBN 978-0-08-032481-4. [Google Scholar]
- Hughes, M.N.; Centelles, M.N.; Moore, K.P. Making and working with hydrogen sulfide: The chemistry and generation of hydrogen sulfide in vitro and its measurement in vivo: A review. Free Radic. Biol. Med. 2009, 47, 1346–1353. [Google Scholar] [CrossRef]
- Singh, S.; Padovani, D.; Leslie, R.A.; Chiku, T.; Banerjee, R. Relative contributions of cystathionine beta-synthase and gamma-cystathionase to H2S biogenesis via alternative trans-sulfuration reactions. J. Biol. Chem. 2009, 284, 22457–22466. [Google Scholar] [CrossRef] [PubMed]
- Mudd, S.H.; Skovby, F.; Levy, H.L.; Pettigrew, K.D.; Wilcken, B.; Pyeritz, R.E.; Andria, G.; Boers, G.H.; Bromberg, I.L.; Cerone, R. The natural history of homocystinuria due to cystathionine beta-synthase deficiency. Am. J. Hum. Genet. 1985, 37, 1–31. [Google Scholar] [PubMed]
- Shibuya, N.; Tanaka, M.; Yoshida, M.; Ogasawara, Y.; Togawa, T.; Ishii, K.; Kimura, H. 3-Mercaptopyruvate sulfurtransferase produces hydrogen sulfide and bound sulfane sulfur in the brain. Antioxid. Redox Signal. 2009, 11, 703–714. [Google Scholar] [CrossRef] [PubMed]
- Jackson, M.R.; Melideo, S.L.; Jorns, M.S. Human sulfide:quinone oxidoreductase catalyzes the first step in hydrogen sulfide metabolism and produces a sulfane sulfur metabolite. Biochemistry 2012, 51, 6804–6815. [Google Scholar] [CrossRef] [PubMed]
- Libiad, M.; Yadav, P.K.; Vitvitsky, V.; Martinov, M.; Banerjee, R. Organization of the human mitochondrial hydrogen sulfide oxidation pathway. J. Biol. Chem. 2014, 289, 30901–30910. [Google Scholar] [CrossRef] [PubMed]
- Landry, A.P.; Ballou, D.P.; Banerjee, R. H2S oxidation by nanodisc-embedded human sulfide quinone oxidoreductase. J. Biol. Chem. 2017, 292, 11641–11649. [Google Scholar] [CrossRef] [PubMed]
- Vitvitsky, V.; Yadav, P.K.; Kurthen, A.; Banerjee, R. Sulfide oxidation by a noncanonical pathway in red blood cells generates thiosulfate and polysulfides. J. Biol. Chem. 2015, 290, 8310–8320. [Google Scholar] [CrossRef] [PubMed]
- Bostelaar, T.; Vitvitsky, V.; Kumutima, J.; Lewis, B.E.; Yadav, P.K.; Brunold, T.C.; Filipovic, M.; Lehnert, N.; Stemmler, T.L.; Banerjee, R. Hydrogen Sulfide Oxidation by Myoglobin. J. Am. Chem. Soc. 2016, 138, 8476–8488. [Google Scholar] [CrossRef]
- Ash-Bernal, R.; Wise, R.; Wright, S.M. Acquired methemoglobinemia: A retrospective series of 138 cases at 2 teaching hospitals. Medicine (Baltimore) 2004, 83, 265–273. [Google Scholar] [CrossRef]
- Olson, K.R.; DeLeon, E.R.; Liu, F. Controversies and conundrums in hydrogen sulfide biology. Nitric Oxide 2014, 41, 11–26. [Google Scholar] [CrossRef]
- Furne, J.; Saeed, A.; Levitt, M.D. Whole tissue hydrogen sulfide concentrations are orders of magnitude lower than presently accepted values. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2008, 295, R1479–R1485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levitt, M.D.; Abdel-Rehim, M.S.; Furne, J. Free and acid-labile hydrogen sulfide concentrations in mouse tissues: Anomalously high free hydrogen sulfide in aortic tissue. Antioxid. Redox Signal. 2011, 15, 373–378. [Google Scholar] [CrossRef] [PubMed]
- Vitvitsky, V.; Kabil, O.; Banerjee, R. High Turnover Rates for Hydrogen Sulfide Allow for Rapid Regulation of Its Tissue Concentrations. Antioxid. Redox Signal. 2012, 17, 22–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, G.; Wu, L.; Jiang, B.; Yang, W.; Qi, J.; Cao, K.; Meng, Q.; Mustafa, A.K.; Mu, W.; Zhang, S.; et al. H2S as a physiologic vasorelaxant: Hypertension in mice with deletion of cystathionine gamma-lyase. Science 2008, 322, 587–590. [Google Scholar] [CrossRef] [PubMed]
- Predmore, B.L.; Lefer, D.J. Development of hydrogen sulfide-based therapeutics for cardiovascular disease. J. Cardiovasc. Transl. Res. 2010, 3, 487–498. [Google Scholar] [CrossRef] [PubMed]
- Elsey, D.J.; Fowkes, R.C.; Baxter, G.F. Regulation of cardiovascular cell function by hydrogen sulfide (H(2)S). Cell Biochem. Funct. 2010, 28, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Elrod, J.W.; Calvert, J.W.; Morrison, J.; Doeller, J.E.; Kraus, D.W.; Tao, L.; Jiao, X.; Scalia, R.; Kiss, L.; Szabo, C.; et al. Hydrogen sulfide attenuates myocardial ischemia-reperfusion injury by preservation of mitochondrial function. Proc. Natl. Acad. Sci. USA 2007, 104, 15560–15565. [Google Scholar] [CrossRef] [Green Version]
- Shen, Y.; Shen, Z.; Luo, S.; Guo, W.; Zhu, Y.Z. The Cardioprotective Effects of Hydrogen Sulfide in Heart Diseases: From Molecular Mechanisms to Therapeutic Potential. Oxid. Med. Cell. Longev. 2015, 2015, 925167. [Google Scholar] [CrossRef]
- Szabó, C.; Papapetropoulos, A. Hydrogen sulphide and angiogenesis: Mechanisms and applications. Br. J. Pharmacol. 2011, 164, 853–865. [Google Scholar] [CrossRef]
- Fang, L.; Li, H.; Tang, C.; Geng, B.; Qi, Y.; Liu, X. Hydrogen sulfide attenuates the pathogenesis of pulmonary fibrosis induced by bleomycin in rats. Can. J. Physiol. Pharmacol. 2009, 87, 531–538. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, R. The message in the air: Hydrogen sulfide metabolism in chronic respiratory diseases. Respir. Physiol. Neurobiol. 2012, 184, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Zeng, T.; Zhang, C.-L.; Zhu, Z.-P.; Yu, L.-H.; Zhao, X.-L.; Xie, K.-Q. Diallyl trisulfide (DATS) effectively attenuated oxidative stress-mediated liver injury and hepatic mitochondrial dysfunction in acute ethanol-exposed mice. Toxicology 2008, 252, 86–91. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, M. H2S and Inflammation: An Overview. Handb. Exp. Pharmacol. 2015, 230, 165–180. [Google Scholar] [PubMed]
- Ang, A.D.; Rivers-Auty, J.; Hegde, A.; Ishii, I.; Bhatia, M. The effect of CSE gene deletion in caerulein-induced acute pancreatitis in the mouse. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 305, G712–G721. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhi, L.; Moore, P.K.; Bhatia, M. Role of hydrogen sulfide in cecal ligation and puncture-induced sepsis in the mouse. Am. J. Physiol. Lung Cell Mol. Physiol. 2006, 290, L1193–L1201. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Kalogeris, T.; Wang, M.; Zuidema, M.Y.; Wang, Q.; Dai, H.; Davis, M.J.; Hill, M.A.; Korthuis, R.J. Hydrogen sulfide preconditioning or neutrophil depletion attenuates ischemia-reperfusion-induced mitochondrial dysfunction in rat small intestine. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 302, G44–G54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mard, S.A.; Neisi, N.; Solgi, G.; Hassanpour, M.; Darbor, M.; Maleki, M. Gastroprotective effect of NaHS against mucosal lesions induced by ischemia-reperfusion injury in rat. Dig. Dis. Sci. 2012, 57, 1496–1503. [Google Scholar] [CrossRef]
- Medeiros, J.V.R.; Bezerra, V.H.; Gomes, A.S.; Barbosa, A.L.R.; Lima-Júnior, R.C.P.; Soares, P.M.G.; Brito, G.A.C.; Ribeiro, R.A.; Cunha, F.Q.; Souza, M.H.L.P. Hydrogen Sulfide Prevents Ethanol-Induced Gastric Damage in Mice: Role of ATP-Sensitive Potassium Channels and Capsaicin-Sensitive Primary Afferent Neurons. J. Pharmacol. Exp. Ther. 2009, 330, 764–770. [Google Scholar] [CrossRef] [Green Version]
- Xie, L.; Gu, Y.; Wen, M.; Zhao, S.; Wang, W.; Ma, Y.; Meng, G.; Han, Y.; Wang, Y.; Liu, G.; et al. Hydrogen Sulfide Induces Keap1 S-sulfhydration and Suppresses Diabetes-Accelerated Atherosclerosis via Nrf2 Activation. Diabetes 2016, 65, 3171–3184. [Google Scholar] [CrossRef]
- Calvert, J.W.; Elston, M.; Nicholson, C.K.; Gundewar, S.; Jha, S.; Elrod, J.W.; Ramachandran, A.; Lefer, D.J. Genetic and pharmacologic hydrogen sulfide therapy attenuates ischemia-induced heart failure in mice. Circulation 2010, 122, 11–19. [Google Scholar] [CrossRef]
- Calvert, J.W.; Jha, S.; Gundewar, S.; Elrod, J.W.; Ramachandran, A.; Pattillo, C.B.; Kevil, C.G.; Lefer, D.J. Hydrogen sulfide mediates cardioprotection through Nrf2 signaling. Circ. Res. 2009, 105, 365–374. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; An, G.; Chen, J. Inhibitory effects of hydrogen sulphide on pulmonary fibrosis in smoking rats via attenuation of oxidative stress and inflammation. J. Cell. Mol. Med. 2014, 18, 1098–1103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sundaresan, M.; Yu, Z.X.; Ferrans, V.J.; Irani, K.; Finkel, T. Requirement for generation of H2O2 for platelet-derived growth factor signal transduction. Science 1995, 270, 296–299. [Google Scholar] [CrossRef] [PubMed]
- Koppenol, W.H.; Bounds, P.L. Signaling by sulfur-containing molecules. Quantitative aspects. Arch. Biochem. Biophys. 2017, 617, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, D.A.; Huie, R.E.; Koppenol, W.H.; Lymar, S.V.; Merenyi, G.; Neta, P.; Ruscic, B.; Stanbury, D.M.; Steenken, S.; Wardman, P. Standard electrode potentials involving radicals in aqueous solution: Inorganic radicals (IUPAC Technical Report). Pure Appl. Chem. 2015, 87, 1139–1150. [Google Scholar] [CrossRef]
- Koppenol, W.H.; Stanbury, D.M.; Bounds, P.L. Electrode potentials of partially reduced oxygen species, from dioxygen to water. Free Radic. Biol. Med. 2010, 49, 317–322. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.R. Kinetics and mechanism of oxidation of hydrogen sulfide by hydrogen peroxide in acidic solution. Environ. Sci. Technol. 1977, 11, 61–66. [Google Scholar] [CrossRef]
- Rabai, G.; Orban, M.; Epstein, I.R. Systematic design of chemical oscillators. 77. A model for the pH-regulated oscillatory reaction between hydrogen peroxide and sulfide ion. J. Phys. Chem. 1992, 96, 5414–5419. [Google Scholar] [CrossRef]
- Davies, M.J.; Hawkins, C.L.; Pattison, D.I.; Rees, M.D. Mammalian heme peroxidases: From molecular mechanisms to health implications. Antioxid. Redox Signal. 2008, 10, 1199–1234. [Google Scholar] [CrossRef]
- Nagy, P.; Winterbourn, C.C. Rapid reaction of hydrogen sulfide with the neutrophil oxidant hypochlorous acid to generate polysulfides. Chem. Res. Toxicol. 2010, 23, 1541–1543. [Google Scholar] [CrossRef]
- Ferrer-Sueta, G.; Campolo, N.; Trujillo, M.; Bartesaghi, S.; Carballal, S.; Romero, N.; Alvarez, B.; Radi, R. Biochemistry of Peroxynitrite and Protein Tyrosine Nitration. Chem. Rev. 2018, 118, 1338–1408. [Google Scholar] [CrossRef] [PubMed]
- Cuevasanta, E.; Zeida, A.; Carballal, S.; Wedmann, R.; Morzan, U.N.; Trujillo, M.; Radi, R.; Estrin, D.A.; Filipovic, M.R.; Alvarez, B. Insights into the mechanism of the reaction between hydrogen sulfide and peroxynitrite. Free Radic. Biol. Med. 2015, 80, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Filipovic, M.R.; Miljkovic, J.; Allgäuer, A.; Chaurio, R.; Shubina, T.; Herrmann, M.; Ivanovic-Burmazovic, I. Biochemical insight into physiological effects of H2S: Reaction with peroxynitrite and formation of a new nitric oxide donor, sulfinyl nitrite. Biochem. J. 2012, 441, 609–621. [Google Scholar] [CrossRef] [PubMed]
- Das, T.N.; Huie, R.E.; Neta, P.; Padmaja, S. Reduction potential of the sulfhydryl radical: pulse radiolysis and laser flash photolysis studies of the formation and reactions of ·SH and HSSH·—In aqueous solutions. J. Phys. Chem. A 1999, 103, 5221–5226. [Google Scholar] [CrossRef]
- Mills, G.; Schmidt, K.H.; Matheson, M.S.; Meisel, D. Thermal and photochemical reactions of sulfhydryl radicals. Implications for colloid photocorrosion. J. Phys. Chem. 1987, 91, 1590–1596. [Google Scholar] [CrossRef]
- Creutz, C.; Sutin, N. Kinetics of the reactions of sodium dithionite with dioxygen and hydrogen peroxide. Inorg. Chem. 1974, 13, 2041–2043. [Google Scholar] [CrossRef]
- Chen, K.Y.; Morris, J.C. Kinetics of oxidation of aqueous sulfide by O2. Environ. Sci. Technol. 1972, 6, 529–537. [Google Scholar] [CrossRef]
- O’Brien, D.J.; Birkner, F.B. Kinetics of oxygenation of reduced sulfur species in aqueous solution. Environ. Sci. Technol. 1977, 11, 1114–1120. [Google Scholar] [CrossRef]
- Wedmann, R.; Bertlein, S.; Macinkovic, I.; Böltz, S.; Miljkovic, J.L.; Muñoz, L.E.; Herrmann, M.; Filipovic, M.R. Working with “H2S”: Facts and apparent artifacts. Nitric Oxide 2014, 41, 85–96. [Google Scholar] [CrossRef]
- Karmann, W.; Meissner, G.; Henglein, A. Pulsradiolyse des Schwefelwasserstoffs in wäßriger Lösung. Zeitschrift für Naturforschung B 1967, 22, 273–282. [Google Scholar] [CrossRef]
- Olson, K.R.; Gao, Y.; DeLeon, E.R.; Arif, M.; Arif, F.; Arora, N.; Straub, K.D. Catalase as a sulfide-sulfur oxido-reductase: An ancient (and modern?) regulator of reactive sulfur species (RSS). Redox Biol. 2017, 12, 325–339. [Google Scholar] [CrossRef] [PubMed]
- Pálinkás, Z.; Furtmüller, P.G.; Nagy, A.; Jakopitsch, C.; Pirker, K.F.; Magierowski, M.; Jasnos, K.; Wallace, J.L.; Obinger, C.; Nagy, P. Interactions of hydrogen sulfide with myeloperoxidase. Br. J. Pharmacol. 2015, 172, 1516–1532. [Google Scholar] [CrossRef] [PubMed]
- Garai, D.; Ríos-González, B.B.; Furtmüller, P.G.; Fukuto, J.M.; Xian, M.; López-Garriga, J.; Obinger, C.; Nagy, P. Mechanisms of myeloperoxidase catalyzed oxidation of H2S by H2O2 or O2 to produce potent protein Cys-polysulfide-inducing species. Free Radic. Biol. Med. 2017, 113, 551–563. [Google Scholar] [CrossRef] [PubMed]
- Boubeta, F.M.; Bieza, S.A.; Bringas, M.; Estrin, D.A.; Boechi, L.; Bari, S.E. Mechanism of Sulfide Binding by Ferric Hemeproteins. Inorg. Chem. 2018, 57, 7591–7600. [Google Scholar] [CrossRef] [PubMed]
- Kraus, D.W.; Wittenberg, J.B. Hemoglobins of the Lucina pectinata/bacteria symbiosis. I. Molecular properties, kinetics and equilibria of reactions with ligands. J. Biol. Chem. 1990, 265, 16043–16053. [Google Scholar]
- Vitvitsky, V.; Yadav, P.K.; An, S.; Seravalli, J.; Cho, U.-S.; Banerjee, R. Structural and Mechanistic Insights into Hemoglobin-catalyzed Hydrogen Sulfide Oxidation and the Fate of Polysulfide Products. J. Biol. Chem. 2017, 292, 5584–5592. [Google Scholar] [CrossRef] [PubMed]
- Jensen, B.; Fago, A. Reactions of ferric hemoglobin and myoglobin with hydrogen sulfide under physiological conditions. J. Inorg. Biochem. 2018, 182, 133–140. [Google Scholar] [CrossRef]
- Vitvitsky, V.; Miljkovic, J.L.; Bostelaar, T.; Adhikari, B.; Yadav, P.K.; Steiger, A.K.; Torregrossa, R.; Pluth, M.D.; Whiteman, M.; Banerjee, R.; et al. Cytochrome c Reduction by H2S Potentiates Sulfide Signaling. ACS Chem. Biol. 2018, 13, 2300–2307. [Google Scholar] [CrossRef]
- Ríos-González, B.B.; Román-Morales, E.M.; Pietri, R.; López-Garriga, J. Hydrogen sulfide activation in hemeproteins: The sulfheme scenario. J. Inorg. Biochem. 2014, 133, 78–86. [Google Scholar] [CrossRef] [Green Version]
- Arbelo-Lopez, H.D.; Simakov, N.A.; Smith, J.C.; Lopez-Garriga, J.; Wymore, T. Homolytic Cleavage of Both Heme-Bound Hydrogen Peroxide and Hydrogen Sulfide Leads to the Formation of Sulfheme. J. Phys. Chem. B 2016, 120, 7319–7331. [Google Scholar] [CrossRef]
- Searcy, D.G.; Whitehead, J.P.; Maroney, M.J. Interaction of Cu, Zn superoxide dismutase with hydrogen sulfide. Arch. Biochem. Biophys. 1995, 318, 251–263. [Google Scholar] [CrossRef] [PubMed]
- Olson, K.R.; Gao, Y.; Arif, F.; Arora, K.; Patel, S.; DeLeon, E.R.; Sutton, T.R.; Feelisch, M.; Cortese-Krott, M.M.; Straub, K.D. Metabolism of hydrogen sulfide (H2S) and Production of Reactive Sulfur Species (RSS) by superoxide dismutase. Redox Biol. 2018, 15, 74–85. [Google Scholar] [CrossRef] [PubMed]
- Filipovic, M.R.; Miljkovic, J.L.; Nauser, T.; Royzen, M.; Klos, K.; Shubina, T.; Koppenol, W.H.; Lippard, S.J.; Ivanović-Burmazović, I. Chemical characterization of the smallest S-nitrosothiol, HSNO; cellular cross-talk of H2S and S-nitrosothiols. J. Am. Chem. Soc. 2012, 134, 12016–12027. [Google Scholar] [CrossRef]
- Cortese-Krott, M.M.; Kuhnle, G.G.C.; Dyson, A.; Fernandez, B.O.; Grman, M.; DuMond, J.F.; Barrow, M.P.; McLeod, G.; Nakagawa, H.; Ondrias, K.; et al. Key bioactive reaction products of the NO/H2S interaction are S/N-hybrid species, polysulfides, and nitroxyl. Proc. Natl. Acad. Sci. USA 2015, 112, E4651–E4660. [Google Scholar] [CrossRef] [PubMed]
- Wedmann, R.; Ivanovic-Burmazovic, I.; Filipovic, M.R. Nitrosopersulfide (SSNO−) decomposes in the presence of sulfide, cyanide or glutathione to give HSNO/SNO−: Consequences for the assumed role in cell signalling. Interface Focus 2017, 7, 20160139. [Google Scholar] [CrossRef]
- Wedmann, R.; Zahl, A.; Shubina, T.E.; Dürr, M.; Heinemann, F.W.; Bugenhagen, B.E.C.; Burger, P.; Ivanovic-Burmazovic, I.; Filipovic, M.R. Does Perthionitrite (SSNO−) Account for Sustained Bioactivity of NO? A (Bio)chemical Characterization. Inorg. Chem. 2015, 54, 9367–9380. [Google Scholar] [CrossRef]
- Cortese-Krott, M.M.; Butler, A.R.; Woollins, J.D.; Feelisch, M. Inorganic sulfur–nitrogen compounds: From gunpowder chemistry to the forefront of biological signaling. Dalton Trans. 2016, 45, 5908–5919. [Google Scholar] [CrossRef]
- Marcolongo, J.P.; Zeida, A.; Slep, L.D.; Olabe, J.A. Chapter Seven—Thionitrous Acid/Thionitrite and Perthionitrite Intermediates in the “Crosstalk” of NO and H2S. In Advances in Inorganic Chemistry; van Eldik, R., Hubbard, C.D., Eds.; Inorganic Reaction Mechanisms; Academic Press: Cambridge, MA, USA, 2017; Volume 70, pp. 277–309. [Google Scholar]
- Portillo-Ledesma, S.; Sardi, F.; Manta, B.; Tourn, M.V.; Clippe, A.; Knoops, B.; Alvarez, B.; Coitiño, E.L.; Ferrer-Sueta, G. Deconstructing the catalytic efficiency of peroxiredoxin-5 peroxidatic cysteine. Biochemistry 2014, 53, 6113–6125. [Google Scholar] [CrossRef]
- Wood, J.L. Sulfane sulfur. Meth. Enzymol. 1987, 143, 25–29. [Google Scholar]
- Everett, S.A.; Folkes, L.K.; Wardman, P.; Asmus, K.D. Free-radical repair by a novel perthiol: Reversible hydrogen transfer and perthiyl radical formation. Free Radic. Res. 1994, 20, 387–400. [Google Scholar] [CrossRef]
- Pan, J.; Carroll, K.S. Persulfide reactivity in the detection of protein s-sulfhydration. ACS Chem. Biol. 2013, 8, 1110–1116. [Google Scholar] [CrossRef] [PubMed]
- Francoleon, N.E.; Carrington, S.J.; Fukuto, J.M. The reaction of H(2)S with oxidized thiols: Generation of persulfides and implications to H(2)S biology. Arch. Biochem. Biophys. 2011, 516, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Park, C.-M.; Johnson, B.A.; Duan, J.; Park, J.-J.; Day, J.J.; Gang, D.; Qian, W.-J.; Xian, M. 9-Fluorenylmethyl (Fm) disulfides: Biomimetic precursors for persulfides. Org. Lett. 2016, 18, 904–907. [Google Scholar] [CrossRef] [PubMed]
- Bogdándi, V.; Ida, T.; Sutton, T.R.; Bianco, C.; Ditrói, T.; Koster, G.; Henthorn, H.A.; Minnion, M.; Toscano, J.P.; van der Vliet, A.; et al. Speciation of reactive sulfur species and their reactions with alkylating agents: Do we have any clue about what is present inside the cell? Br. J. Pharmacol. 2019, 176, 646–670. [Google Scholar] [CrossRef] [PubMed]
- Bianco, C.L.; Akaike, T.; Ida, T.; Nagy, P.; Bogdandi, V.; Toscano, J.P.; Kumagai, Y.; Henderson, C.F.; Goddu, R.N.; Lin, J.; et al. The reaction of hydrogen sulfide with disulfides: Formation of a stable trisulfide and implications for biological systems. Br. J. Pharmacol. 2019, 176, 671–683. [Google Scholar] [CrossRef] [PubMed]
- Fukuto, J.M.; Carrington, S.J.; Tantillo, D.J.; Harrison, J.G.; Ignarro, L.J.; Freeman, B.A.; Chen, A.; Wink, D.A. Small molecule signaling agents: The integrated chemistry and biochemistry of nitrogen oxides, oxides of carbon, dioxygen, hydrogen sulfide, and their derived species. Chem. Res. Toxicol. 2012, 25, 769–793. [Google Scholar] [CrossRef] [PubMed]
- Jencks, W.P.; Carriuolo, J. Reactivity of nucleophilic reagents toward esters. J. Am. Chem. Soc. 1960, 82, 1778–1786. [Google Scholar] [CrossRef]
- Edwards, J.O.; Pearson, R.G. The factors determining nucleophilic reactivities. J. Am. Chem. Soc. 1962, 84, 16–24. [Google Scholar] [CrossRef]
- Kawamura, S.; Nakabayashi, T.; Kitao, T.; Tsurugi, J. Aralkyl hydrodisulfides. VI. The Reaction of Benzhydryl Hydrosulfide with Several Neucleophiles. J. Org. Chem. 1966, 31, 1985–1987. [Google Scholar] [CrossRef]
- Bailey, T.S.; Zakharov, L.N.; Pluth, M.D. Understanding hydrogen sulfide storage: Probing conditions for sulfide release from hydrodisulfides. J. Am. Chem. Soc. 2014, 136, 10573–10576. [Google Scholar] [CrossRef]
- Kawamura, S.; Otsuji, Y.; Nakabayashi, T.; Kitao, T.; Tsurugi, J. Aralkyl hydrodisulfides. IV. The Reaction of Benzyl Hydrodisulfide with Several Nucleophiles. J. Org. Chem. 1965, 30, 2711–2714. [Google Scholar] [CrossRef]
- Tsurugi, J.; Abe, Y.; Nakabayashi, T.; Kawamura, S.; Kitao, T.; Niwa, M. Aralkyl hydrodisulfides. XI. Reaction with amines. J. Org. Chem. 1970, 35, 3263–3266. [Google Scholar] [CrossRef]
- Kawamura, S.; Kitao, T.; Nakabayashi, T.; Horii, T.; Tsurugi, J. Aralkyl hydrodisulfides. VIII. Alkaline decomposition and its competition with nucleophiles. J. Org. Chem. 1968, 33, 1179–1181. [Google Scholar] [CrossRef]
- Tsurugi, J.; Nakabayashi, T.; Ishihara, T. Aralkyl hydrodisulfides. III. The Reaction with Tertiary Phosphines. J. Org. Chem. 1965, 30, 2707–2710. [Google Scholar] [CrossRef]
- Bailey, T.S.; Pluth, M.D. Reactions of isolated persulfides provide insights into the interplay between H2S and persulfide reactivity. Free Radic. Biol. Med. 2015, 89, 662–667. [Google Scholar] [CrossRef] [PubMed]
- Hiskey, R.G.; Carroll, F.I. Chemistry of aliphatic disulfides. II. Cyanide cleavage of unsymmetrical disulfides. J. Am. Chem. Soc. 1961, 83, 4644–4647. [Google Scholar] [CrossRef]
- Yadav, P.K.; Yamada, K.; Chiku, T.; Koutmos, M.; Banerjee, R. Structure and kinetic analysis of H2S production by human mercaptopyruvate sulfurtransferase. J. Biol. Chem. 2013, 288, 20002–20013. [Google Scholar] [CrossRef]
- Sorbo, B. Enzymic transfer of sulfur from mercaptopyruvate to sulfate or sulfinates. Biochim. Biophys. Acta 1957, 24, 324–329. [Google Scholar] [CrossRef]
- Hildebrandt, T.M.; Grieshaber, M.K. Three enzymatic activities catalyze the oxidation of sulfide to thiosulfate in mammalian and invertebrate mitochondria. FEBS J. 2008, 275, 3352–3361. [Google Scholar] [CrossRef] [Green Version]
- Libiad, M.; Sriraman, A.; Banerjee, R. Polymorphic Variants of Human Rhodanese Exhibit Differences in Thermal Stability and Sulfur Transfer Kinetics. J. Biol. Chem. 2015, 290, 23579–23588. [Google Scholar] [CrossRef]
- Kim, S.; Park, S. Structural changes during cysteine desulfurase CsdA and sulfur acceptor CsdE interactions provide insight into the trans-persulfuration. J. Biol. Chem. 2013, 288, 27172–27180. [Google Scholar] [CrossRef] [PubMed]
- Aroca, A.; Gotor, C.; Romero, L.C. Hydrogen Sulfide Signaling in Plants: Emerging Roles of Protein Persulfidation. Front. Plant Sci. 2018, 9, 1369. [Google Scholar] [CrossRef] [PubMed]
- Dóka, É.; Pader, I.; Bíró, A.; Johansson, K.; Cheng, Q.; Ballagó, K.; Prigge, J.R.; Pastor-Flores, D.; Dick, T.P.; Schmidt, E.E.; et al. A novel persulfide detection method reveals protein persulfide- and polysulfide-reducing functions of thioredoxin and glutathione systems. Sci. Adv. 2016, 2, e1500968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ju, Y.; Wu, L.; Yang, G. Thioredoxin 1 regulation of protein S-desulfhydration. Biochem. Biophys. Rep. 2016, 5, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Artaud, I.; Galardon, E. A persulfide analogue of the nitrosothiol SNAP: Formation, characterization and reactivity. ChemBioChem 2014, 15, 2361–2364. [Google Scholar] [CrossRef] [PubMed]
- Koike, S.; Nishimoto, S.; Ogasawara, Y. Cysteine persulfides and polysulfides produced by exchange reactions with H2S protect SH-SY5Y cells from methylglyoxal-induced toxicity through Nrf2 activation. Redox Biol. 2017, 12, 530–539. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Macinkovic, I.; Devarie-Baez, N.O.; Pan, J.; Park, C.-M.; Carroll, K.S.; Filipovic, M.R.; Xian, M. Detection of protein S-sulfhydration by a tag-switch technique. Angew. Chem. Int. Ed. Engl. 2014, 53, 575–581. [Google Scholar] [CrossRef]
- Mishanina, T.V.; Yadav, P.K.; Ballou, D.P.; Banerjee, R. Transient Kinetic Analysis of Hydrogen Sulfide Oxidation Catalyzed by Human Sulfide Quinone Oxidoreductase. J. Biol. Chem. 2015, 290, 25072–25080. [Google Scholar] [CrossRef]
- Filipovic, M.R. Persulfidation (S-sulfhydration) and H2S. Handb. Exp. Pharmacol. 2015, 230, 29–59. [Google Scholar]
- Liu, D.K.; Chang, S.G. Kinetic study of the reaction between cystine and sulfide in alkaline solutions. Can. J. Chem. 1987, 65, 770–774. [Google Scholar] [CrossRef] [Green Version]
- Vasas, A.; Dóka, É.; Fábián, I.; Nagy, P. Kinetic and thermodynamic studies on the disulfide-bond reducing potential of hydrogen sulfide. Nitric Oxide 2015, 46, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, R.W.; Tachibana, C.; Hansen, N.E.; Winther, J.R. Trisulfides in Proteins. Antioxid. Redox Signal. 2010, 15, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Bolton, S.G.; Cerda, M.M.; Gilbert, A.K.; Pluth, M.D. Effects of sulfane sulfur content in benzyl polysulfides on thiol-triggered H2S release and cell proliferation. Free Radic. Biol. Med. 2019, 131, 393–398. [Google Scholar] [CrossRef] [PubMed]
- Toohey, J.I. Sulfur signaling: Is the agent sulfide or sulfane? Anal. Biochem. 2011, 413, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Florence, T.M. Degradation of protein disulphide bonds in dilute alkali. Biochem. J. 1980, 189, 507–520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Federici, G.; Duprè, S.; Matarese, R.M.; Solinas, S.P.; Cavallini, D. Is the alkaline cleavage of disulfide bonds in peptides an alpha-beta elimination reaction or a hydrolysis? Int. J. Pept. Protein Res. 1977, 10, 185–189. [Google Scholar] [CrossRef] [PubMed]
- Jones, A.J.; Helmerhorst, E.; Stokes, G.B. The formation of dehydroalanine residues in alkali-treated insulin and oxidized glutathione. A nuclear-magnetic-resonance study. Biochem. J. 1983, 211, 499–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Álvarez, L.; Bianco, C.L.; Toscano, J.P.; Lin, J.; Akaike, T.; Fukuto, J.M. Chemical Biology of Hydropersulfides and Related Species: Possible Roles in Cellular Protection and Redox Signaling. Antioxid. Redox Signal. 2017, 27, 622–633. [Google Scholar] [CrossRef] [PubMed]
- Heppner, D.E.; Hristova, M.; Ida, T.; Mijuskovic, A.; Dustin, C.M.; Bogdándi, V.; Fukuto, J.M.; Dick, T.P.; Nagy, P.; Li, J.; et al. Cysteine perthiosulfenic acid (Cys-SSOH): A novel intermediate in thiol-based redox signaling? Redox Biol. 2018, 14, 379–385. [Google Scholar] [CrossRef]
- Everett, S.A.; Wardman, P. Perthiols as antioxidants: Radical-scavenging and prooxidative mechanisms. Meth. Enzymol. 1995, 251, 55–69. [Google Scholar]
- Benson, S.W. Thermochemistry and kinetics of sulfur-containing molecules and radicals. Chem. Rev. 1978, 78, 23–35. [Google Scholar] [CrossRef]
- Kawamura, S.; Abe, Y.; Tsurugi, J. Aralkyl hydrodisulfides. X. Reactions with iron salts. J. Org. Chem. 1969, 34, 3633–3635. [Google Scholar] [CrossRef]
- Prütz, W.A. Catalytic reduction of Fe(III)-cytochrome-c involving stable radiolysis products derived from disulphides, proteins and thiols. Int. J. Radiat. Biol. 1992, 61, 593–602. [Google Scholar] [CrossRef] [PubMed]
- Chauvin, J.-P.R.; Haidasz, E.A.; Griesser, M.; Pratt, D.A. Polysulfide-1-oxides react with peroxyl radicals as quickly as hindered phenolic antioxidants and do so by a surprising concerted homolytic substitution. Chem. Sci. 2016, 7, 6347–6356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anglada, J.M.; Crehuet, R.; Adhikari, S.; Francisco, J.S.; Xia, Y. Reactivity of hydropersulfides toward the hydroxyl radical unraveled: Disulfide bond cleavage, hydrogen atom transfer, and proton-coupled electron transfer. Phys. Chem. Chem. Phys. 2018, 20, 4793–4804. [Google Scholar] [CrossRef] [PubMed]
- Everett, S.A.; Schoeneich, C.; Stewart, J.H.; Asmus, K.D. Perthiyl radicals, trisulfide radical ions, and sulfate formation: A combined photolysis and radiolysis study on redox processes with organic di- and trisulfides. J. Phys. Chem. 1992, 96, 306–314. [Google Scholar] [CrossRef]
- Chatterji, T.; Keerthi, K.; Gates, K.S. Generation of reactive oxygen species by a persulfide (BnSSH). Bioorg. Med. Chem. Lett. 2005, 15, 3921–3924. [Google Scholar] [CrossRef]
- Saund, S.S.; Sosa, V.; Henriquez, S.; Nguyen, Q.N.N.; Bianco, C.L.; Soeda, S.; Millikin, R.; White, C.; Le, H.; Ono, K.; et al. The chemical biology of hydropersulfides (RSSH): Chemical stability, reactivity and redox roles. Arch. Biochem. Biophys. 2015, 588, 15–24. [Google Scholar] [CrossRef] [Green Version]
- Kende, I.; Pickering, T.L.; Tobolsky, A.V. The Dissociation Energy of the Tetrasulfide Linkage. J. Am. Chem. Soc. 1965, 87, 5582–5586. [Google Scholar] [CrossRef]
- Quintiliani, M.; Badiello, R.; Tamba, M.; Esfandi, A.; Gorin, G. Radiolysis of glutathione in oxygen-containing solutions of pH7. Int. J. Radiat. Biol. Relat. Stud. Phys. Chem. Med. 1977, 32, 195–202. [Google Scholar] [CrossRef]
- Tamba, M.; Simone, G.; Quintiliani, M. Interactions of thiyl free radicals with oxygen: A pulse radiolysis study. Int. J. Radiat. Biol. Relat. Stud. Phys. Chem. Med. 1986, 50, 595–600. [Google Scholar] [CrossRef] [PubMed]
- Sevilla, M.D.; Becker, D.; Yan, M. The formation and structure of the sulfoxyl radicals RSO(.), RSOO(.), RSO2(.), and RSO2OO(.) from the reaction of cysteine, glutathione and penicillamine thiyl radicals with molecular oxygen. Int. J. Radiat. Biol. 1990, 57, 65–81. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, N.; Schuchmann, H.-P.; von Sonntag, C. Pulse Radiolysis of 2-Mercaptoethanol in Oxygenated Aqueous Solution. Generation and Reactions of the Thiylperoxyl Radical. J. Phys. Chem. 1994, 98, 6541–6547. [Google Scholar] [CrossRef]
- Madej, E.; Folkes, L.K.; Wardman, P.; Czapski, G.; Goldstein, S. Thiyl radicals react with nitric oxide to form S-nitrosothiols with rate constants near the diffusion-controlled limit. Free Radic. Biol. Med. 2008, 44, 2013–2018. [Google Scholar] [CrossRef] [PubMed]
- Bailey, T.S.; Henthorn, H.A.; Pluth, M.D. The Intersection of NO and H2S: Persulfides Generate NO from Nitrite through Polysulfide Formation. Inorg. Chem. 2016, 55, 12618–12625. [Google Scholar] [CrossRef] [PubMed]
Scheme 1.
Hydrogen sulfide oxidation by reactive species. The dashed lines represent processes that are accomplished in more than one step. Protons are sometimes omitted for simplicity.
Scheme 1.
Hydrogen sulfide oxidation by reactive species. The dashed lines represent processes that are accomplished in more than one step. Protons are sometimes omitted for simplicity.

Scheme 2.
Persulfide oxidation by reactive species. A• represents a one-electron oxidant. The dashed lines represent processes that are accomplished in more than one step. Protons are sometimes omitted for simplicity.
Scheme 2.
Persulfide oxidation by reactive species. A• represents a one-electron oxidant. The dashed lines represent processes that are accomplished in more than one step. Protons are sometimes omitted for simplicity.

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Benchoam, D.; Cuevasanta, E.; Möller, M.N.; Alvarez, B. Hydrogen Sulfide and Persulfides Oxidation by Biologically Relevant Oxidizing Species. Antioxidants 2019, 8, 48. https://doi.org/10.3390/antiox8020048
AMA Style
Benchoam D, Cuevasanta E, Möller MN, Alvarez B. Hydrogen Sulfide and Persulfides Oxidation by Biologically Relevant Oxidizing Species. Antioxidants. 2019; 8(2):48. https://doi.org/10.3390/antiox8020048
Chicago/Turabian StyleBenchoam, Dayana, Ernesto Cuevasanta, Matías N. Möller, and Beatriz Alvarez. 2019. "Hydrogen Sulfide and Persulfides Oxidation by Biologically Relevant Oxidizing Species" Antioxidants 8, no. 2: 48. https://doi.org/10.3390/antiox8020048
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.