Gelidium amansii Attenuates Hypoxia/Reoxygenation-Induced Oxidative Injury in Primary Hippocampal Neurons through Suppressing GluN2B Expression

 ,
,  ,
, 
 ,
, 
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
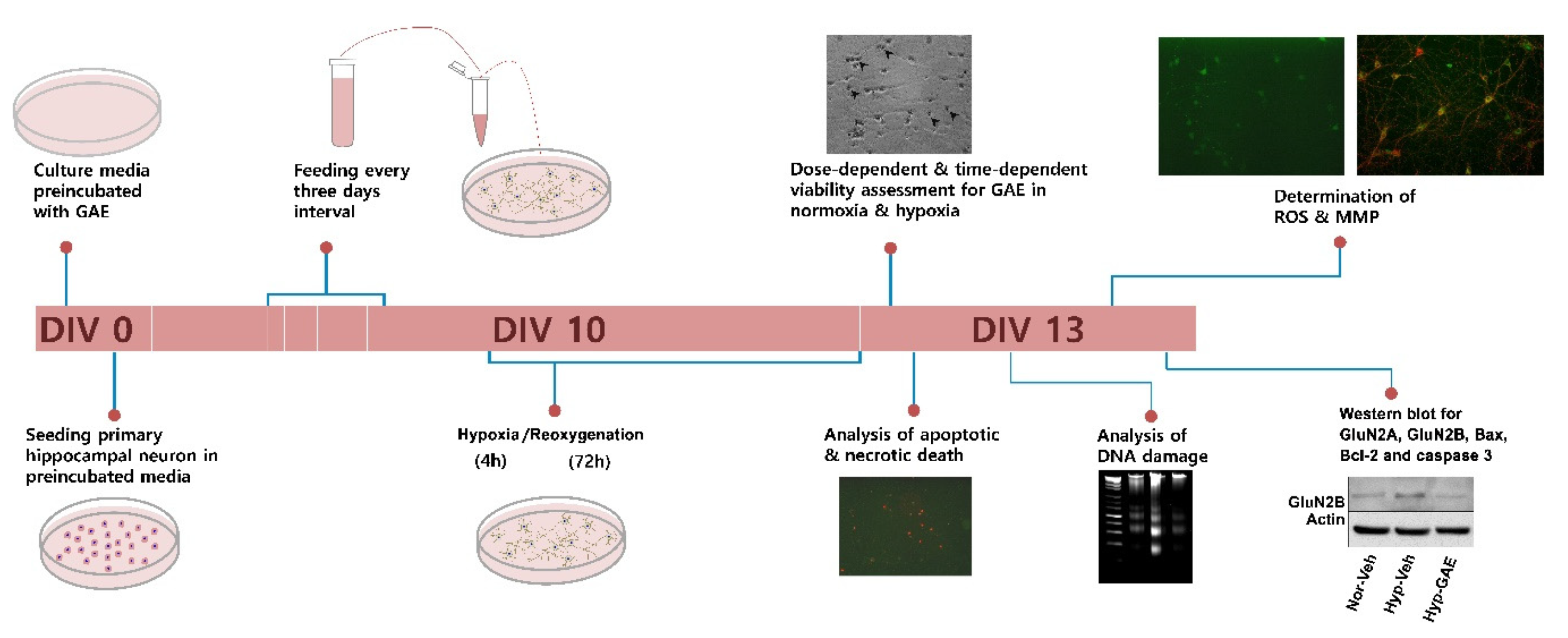
2. Materials and Methods
2.1. Sample Collection and Extract Preparation
2.2. Primary Neuronal Culture and GAE Treatment
2.3. Hypoxia/Reoxygenation (H/R) Injury
2.4. Assessment of Neuronal Viability and Cytotoxicity
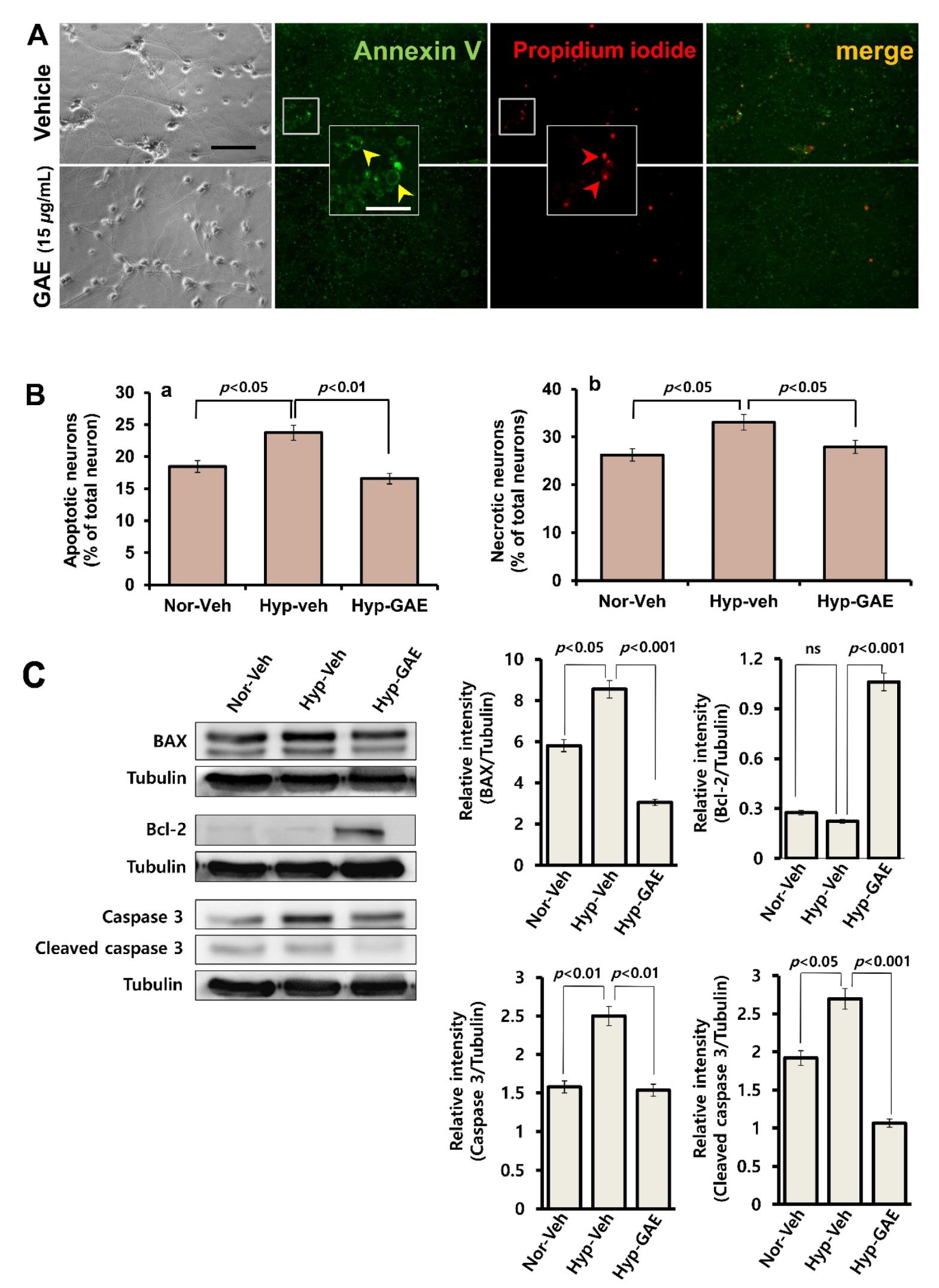
2.5. Measurement of Apoptotic Cell Death
2.6. Analysis of DNA Fragmentation by Agarose Gel Electrophoresis
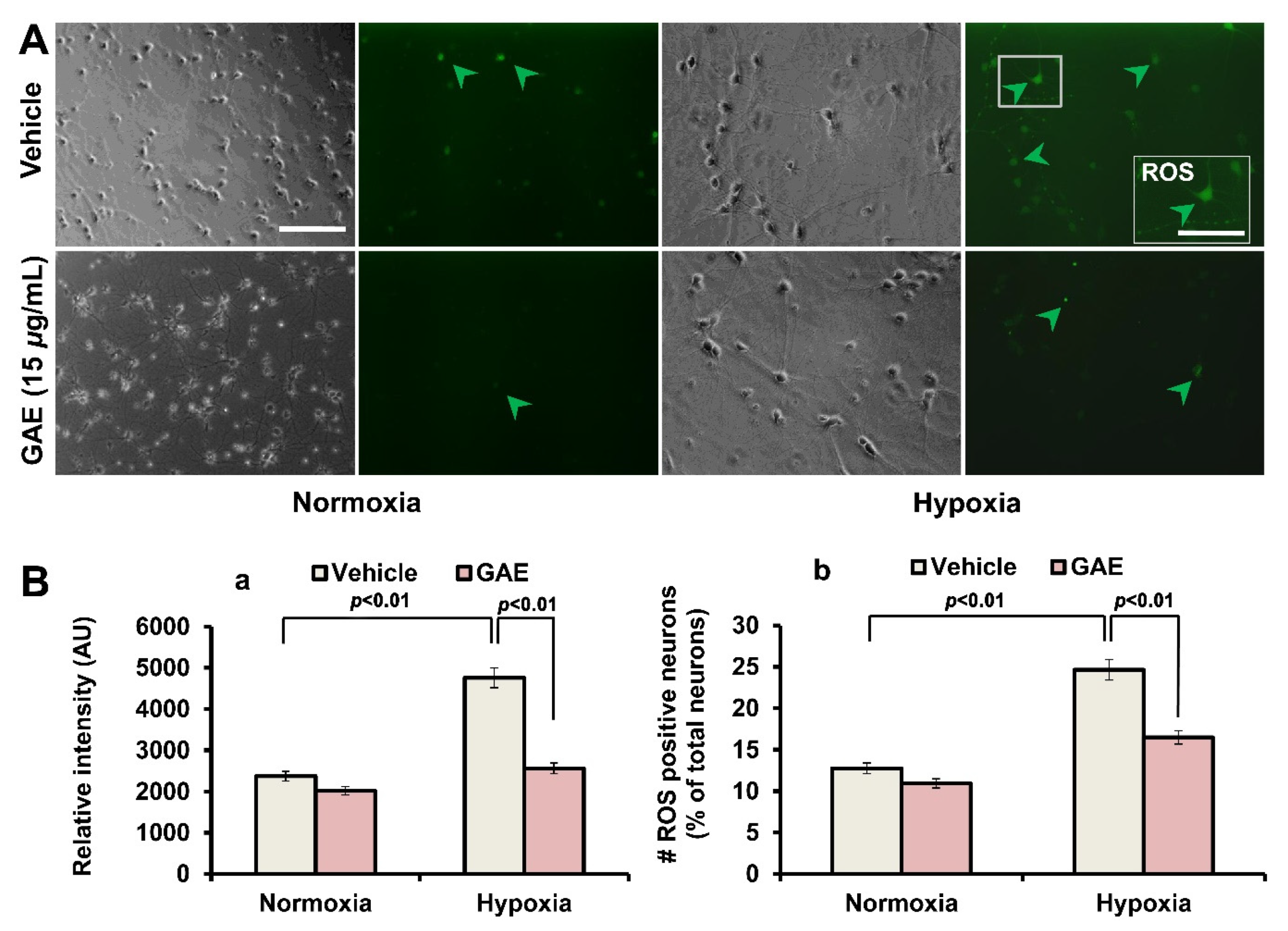
2.7. Measurement of Reactive Oxygen Species (ROS) Generation
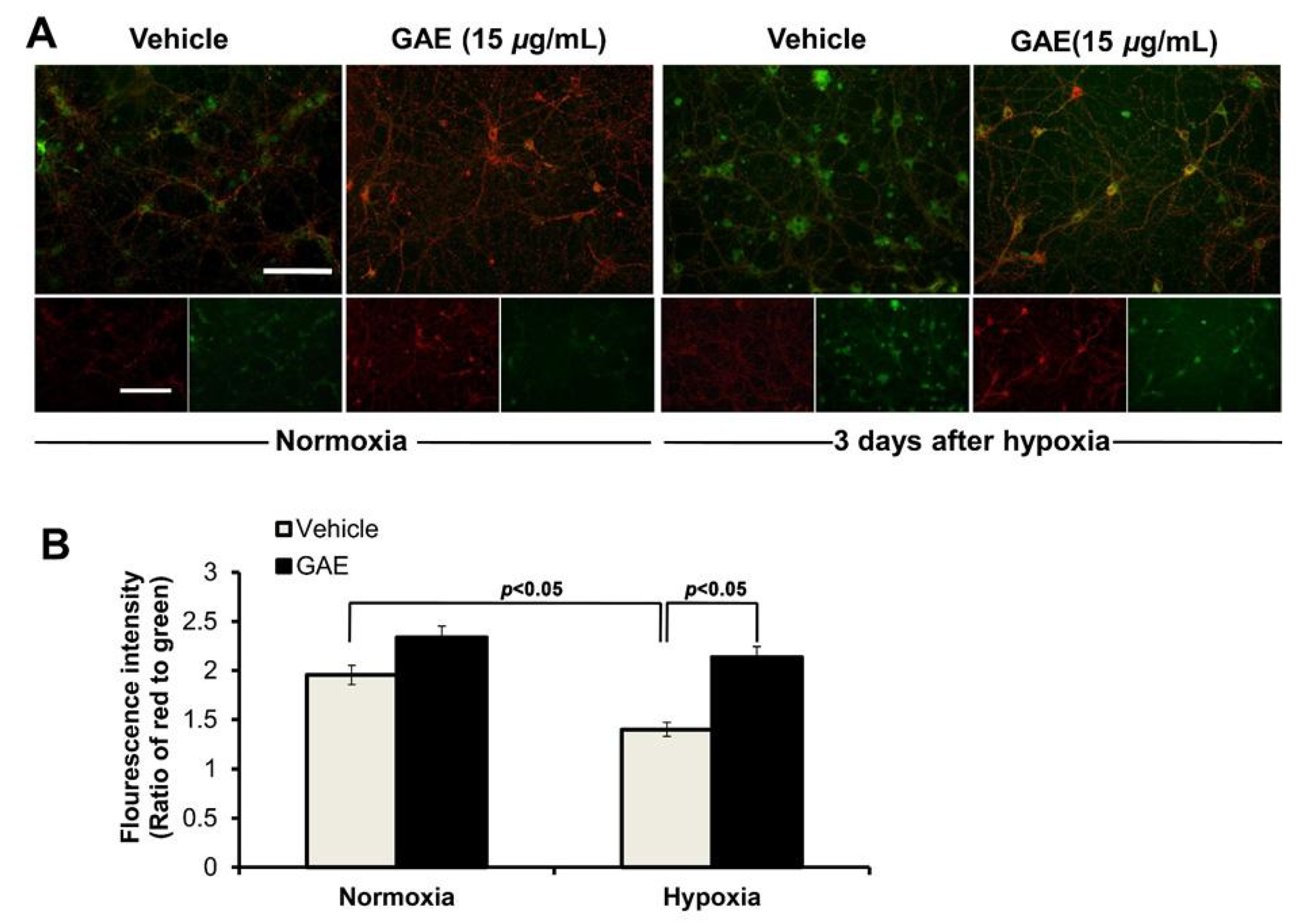
2.8. Determination of Mitochondrial Membrane Potential (ΔΨm)
2.9. Western Blot
2.10. Image Acquisition and Analysis
2.11. Statistical Analysis
3. Results
3.1. GAE Attenuates H/R-Induced Neuronal Death
3.2. GAE Reduces Apoptotic and Necrotic Death Following H/R
3.3. GAE Attenuates H/R-Induced DNA Damage
3.4. GAE Suppresses H/R-Induced ROS Generation
3.5. GAE Preserves ΔΨm
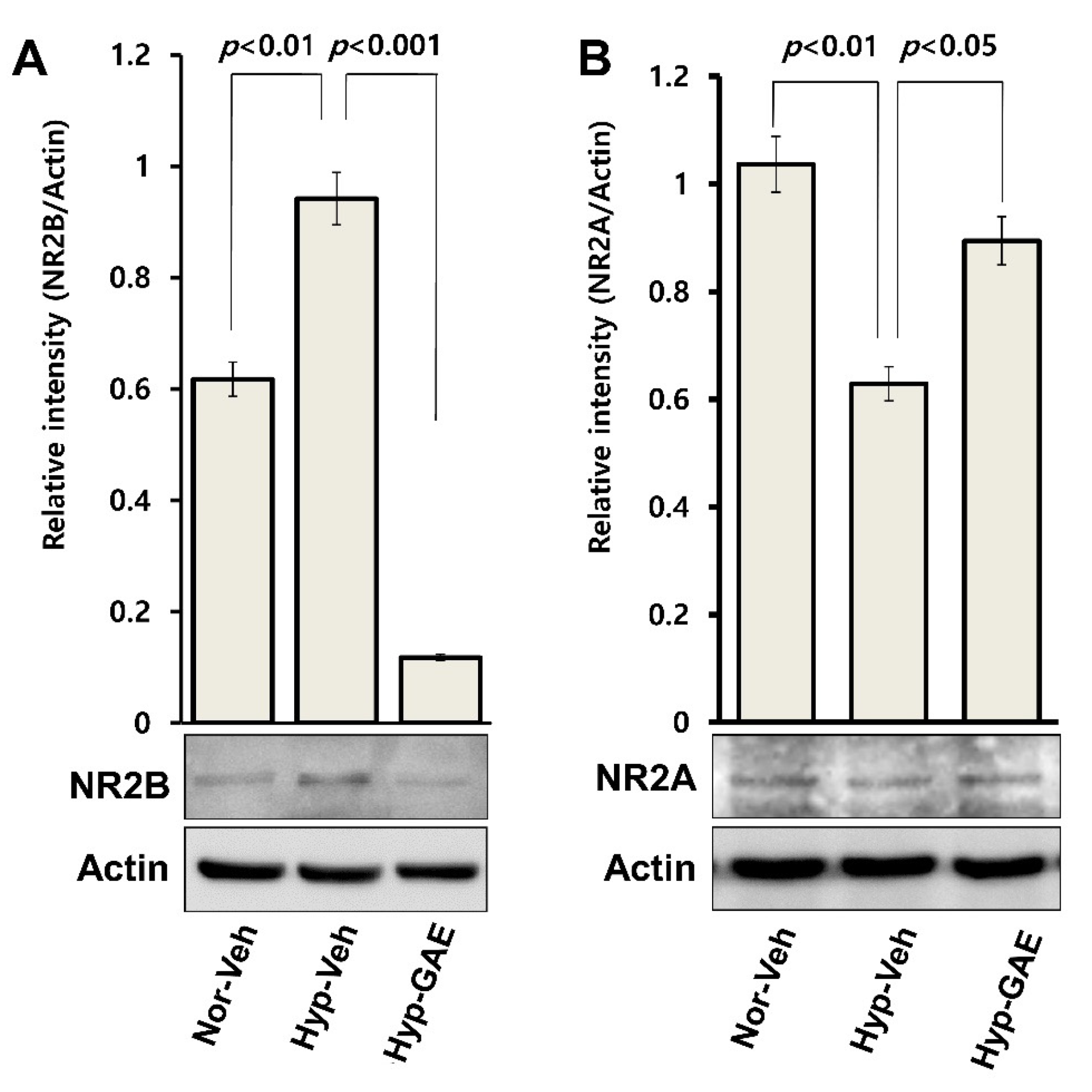
3.6. GAE Downregulates H/R-Induced Expression of GluN2B
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Li, C.; Jackson, R.M. Reactive species mechanisms of cellular hypoxia-reoxygenation injury. Am. J. Physiol. -Cell Physiol. 2002, 282, C227–C241. [Google Scholar] [CrossRef]
- Shirley, R.; Ord, E.N.J.; Work, L.M. Oxidative Stress and the Use of Antioxidants in Stroke. Antioxidants 2014, 3, 472–501. [Google Scholar] [CrossRef]
- Dotti, C.; Sullivan, C.; Banker, G. The establishment of polarity by hippocampal neurons in culture. J. Neurosci. 1988, 8, 1454–1468. [Google Scholar] [CrossRef]
- Hannan, M.A.; Kang, J.Y.; Hong, Y.K.; Lee, H.; Choi, J.S.; Choi, I.S.; Moon, I.S. The marine alga Gelidium amansii promotes the development and complexity of neuronal cytoarchitecture. Phytother. Res. 2013, 27, 21–29. [Google Scholar] [CrossRef]
- Hannan, M.A.; Mohibbullah, M.; Hong, Y.K.; Moon, I.S. Proteomic Analysis of the Neurotrophic Effect of Gelidium amansii in Primary Cultured Neurons. J. Med. Food 2017, 20, 279–287. [Google Scholar] [CrossRef]
- Hannan, M.A.; Mohibbullah, M.; Hong, Y.-K.; Nam, J.H.; Moon, I.S. Gelidium amansii promotes dendritic spine morphology and synaptogenesis, and modulates NMDA receptor-mediated postsynaptic current. In Vitro Cell. Dev. Biol. Anim. 2014, 50, 445–452. [Google Scholar] [CrossRef]
- Fu, Y.W.; Hou, W.Y.; Yeh, S.T.; Li, C.H.; Chen, J.C. The immunostimulatory effects of hot-water extract of Gelidium amansii via immersion, injection and dietary administrations on white shrimp Litopenaeus vannamei and its resistance against Vibrio alginolyticus. Fish Shellfish Immunol. 2007, 22, 673–685. [Google Scholar] [CrossRef]
- Yan, X.; Nagata, T.; Fan, X. Antioxidative activities in some common seaweeds. Plant foods Hum. Nutr. 1998, 52, 253–262. [Google Scholar] [CrossRef]
- Hannan, M.A.; Kang, J.-Y.; Hong, Y.-K.; Lee, H.; Chowdhury, M.T.H.; Choi, J.-S.; Choi, I.S.; Moon, I.S. A brown alga Sargassum fulvellum facilitates neuronal maturation and synaptogenesis. In Vitro Cell. Dev. Biol. Anim. 2012, 48, 535–544. [Google Scholar] [CrossRef]
- Mohibbullah, M.; Hannan, M.A.; Choi, J.-Y.; Bhuiyan, M.M.H.; Hong, Y.-K.; Choi, J.-S.; Choi, I.S.; Moon, I.S. The Edible Marine Alga Gracilariopsis chorda Alleviates Hypoxia/Reoxygenation-Induced Oxidative Stress in Cultured Hippocampal Neurons. J. Med. Food 2015, 18, 960–971. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Haque, M.N.; Moon, I.S. Stigmasterol promotes neuronal migration via reelin signaling in neurosphere migration assays. Nutr. Neurosci. 2018, 1–9. [Google Scholar] [CrossRef]
- Porter, A.G.; Jänicke, R.U. Emerging roles of caspase-3 in apoptosis. Cell Death Differ. 1999, 6, 99–104. [Google Scholar] [CrossRef]
- Chen, H.; Yoshioka, H.; Kim, G.S.; Jung, J.E.; Okami, N.; Sakata, H.; Maier, C.M.; Narasimhan, P.; Goeders, C.E.; Chan, P.H. Oxidative stress in ischemic brain damage: mechanisms of cell death and potential molecular targets for neuroprotection. Antioxid. Redox Signal. 2011, 14, 1505–1517. [Google Scholar] [CrossRef]
- Salim, S. Oxidative Stress and the Central Nervous System. J. Pharmacol. Exp. Ther. 2017, 360, 201–205. [Google Scholar] [CrossRef]
- Miranda, M.; Morici, J.F.; Zanoni, M.B.; Bekinschtein, P. Brain-Derived Neurotrophic Factor: A Key Molecule for Memory in the Healthy and the Pathological Brain. Front. Cell. Neurosci. 2019, 13, 363. [Google Scholar] [CrossRef]
- Kalogeris, T.; Baines, C.P.; Krenz, M.; Korthuis, R.J. Ischemia/Reperfusion. Compr. Physiol. 2016, 7, 113–170. [Google Scholar] [CrossRef]
- Naoi, M.; Wu, Y.; Shamoto-Nagai, M.; Maruyama, W. Mitochondria in Neuroprotection by Phytochemicals: Bioactive Polyphenols Modulate Mitochondrial Apoptosis System, Function and Structure. Int. J. Mol. Sci. 2019, 20, 2451. [Google Scholar]
- Webster, K.A. Mitochondrial membrane permeabilization and cell death during myocardial infarction: Roles of calcium and reactive oxygen species. Future Cardiol. 2012, 8, 863–884. [Google Scholar] [CrossRef]
- Seo, M.J.; Lee, O.H.; Choi, H.S.; Lee, B.Y. Extract from Edible Red Seaweed (Gelidium amansii) Inhibits Lipid Accumulation and ROS Production during Differentiation in 3T3-L1 Cells. Prev. Nutr. Food Sci. 2012, 17, 129–135. [Google Scholar] [CrossRef]
- Szeto, H.H. Mitochondria-targeted peptide antioxidants: novel neuroprotective agents. AAPS J. 2006, 8, E521–E531. [Google Scholar] [CrossRef] [PubMed]
- Beal, M.F. Mitochondria take center stage in aging and neurodegeneration. Ann. Neurol. 2005, 58, 495–505. [Google Scholar] [CrossRef] [PubMed]
- Martel, M.-A.; Ryan, T.J.; Bell, K.F.S.; Fowler, J.H.; McMahon, A.; Al-Mubarak, B.; Komiyama, N.H.; Horsburgh, K.; Kind, P.C.; Grant, S.G.N.; et al. The subtype of GluN2 C-terminal domain determines the response to excitotoxic insults. Neuron 2012, 74, 543–556. [Google Scholar] [CrossRef] [PubMed]
- Hardingham, G.E.; Bading, H. Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nat. Rev. Neurosci. 2010, 11, 682–696. [Google Scholar] [CrossRef]
- Dirnagl, U.; Iadecola, C.; Moskowitz, M.A. Pathobiology of ischaemic stroke: An integrated view. Trends Neurosci. 1999, 22, 391–397. [Google Scholar] [CrossRef]
- O’Donovan, S.M.; Sullivan, C.R.; McCullumsmith, R.E. The role of glutamate transporters in the pathophysiology of neuropsychiatric disorders. NPJ Schizophr. 2017, 3, 32. [Google Scholar] [CrossRef]
- Lau, A.; Tymianski, M. Glutamate receptors, neurotoxicity and neurodegeneration. Pflugers Archiv 2010, 460, 525–542. [Google Scholar] [CrossRef] [PubMed]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hannan, M.A.; Haque, M.N.; Mohibbullah, M.; Dash, R.; Hong, Y.-K.; Moon, I.S. Gelidium amansii Attenuates Hypoxia/Reoxygenation-Induced Oxidative Injury in Primary Hippocampal Neurons through Suppressing GluN2B Expression. Antioxidants 2020, 9, 223. https://doi.org/10.3390/antiox9030223
Hannan MA, Haque MN, Mohibbullah M, Dash R, Hong Y-K, Moon IS. Gelidium amansii Attenuates Hypoxia/Reoxygenation-Induced Oxidative Injury in Primary Hippocampal Neurons through Suppressing GluN2B Expression. Antioxidants. 2020; 9(3):223. https://doi.org/10.3390/antiox9030223
Chicago/Turabian StyleHannan, Md. Abdul, Md. Nazmul Haque, Md. Mohibbullah, Raju Dash, Yong-Ki Hong, and Il Soo Moon. 2020. "Gelidium amansii Attenuates Hypoxia/Reoxygenation-Induced Oxidative Injury in Primary Hippocampal Neurons through Suppressing GluN2B Expression" Antioxidants 9, no. 3: 223. https://doi.org/10.3390/antiox9030223
APA StyleHannan, M. A., Haque, M. N., Mohibbullah, M., Dash, R., Hong, Y.-K., & Moon, I. S. (2020). Gelidium amansii Attenuates Hypoxia/Reoxygenation-Induced Oxidative Injury in Primary Hippocampal Neurons through Suppressing GluN2B Expression. Antioxidants, 9(3), 223. https://doi.org/10.3390/antiox9030223