Chronic Kidney Disease as Oxidative Stress- and Inflammatory-Mediated Cardiovascular Disease



Abstract
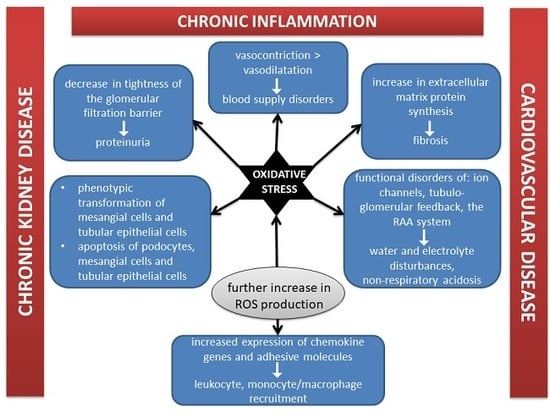
1. Introduction
2. Redox Homeostasis Disturbances Negatively Influence Kidneys Structure
3. Excessive ROS Generation in CKD
3.1. Increased Nox Activity and Role of Angiotensin II (Ang II) in CKD
3.2. Increased Xanthine Oxidase (XO) Activity
3.3. Abnormal Mitochondrial Function in CKD
4. Reduction of Antioxidative Activity in CKD
4.1. Superoxide Dismutase (SOD) Is Affected in CKD
4.2. Glutathione Peroxidase GPx Changes in CKD
4.3. Disturbances in the Management of Trace Elements
5. The Coexistence of Oxidative Stress and Inflammation in CKD
5.1. Myeloperoxidase (MPO) Activity in CKD
5.2. Nuclear Factor Kappa-Light-Chain-Enhancer of Activated B Cells (NF-κB Transcription Factor) and Superoxide Dismutase (SOD) in CKD
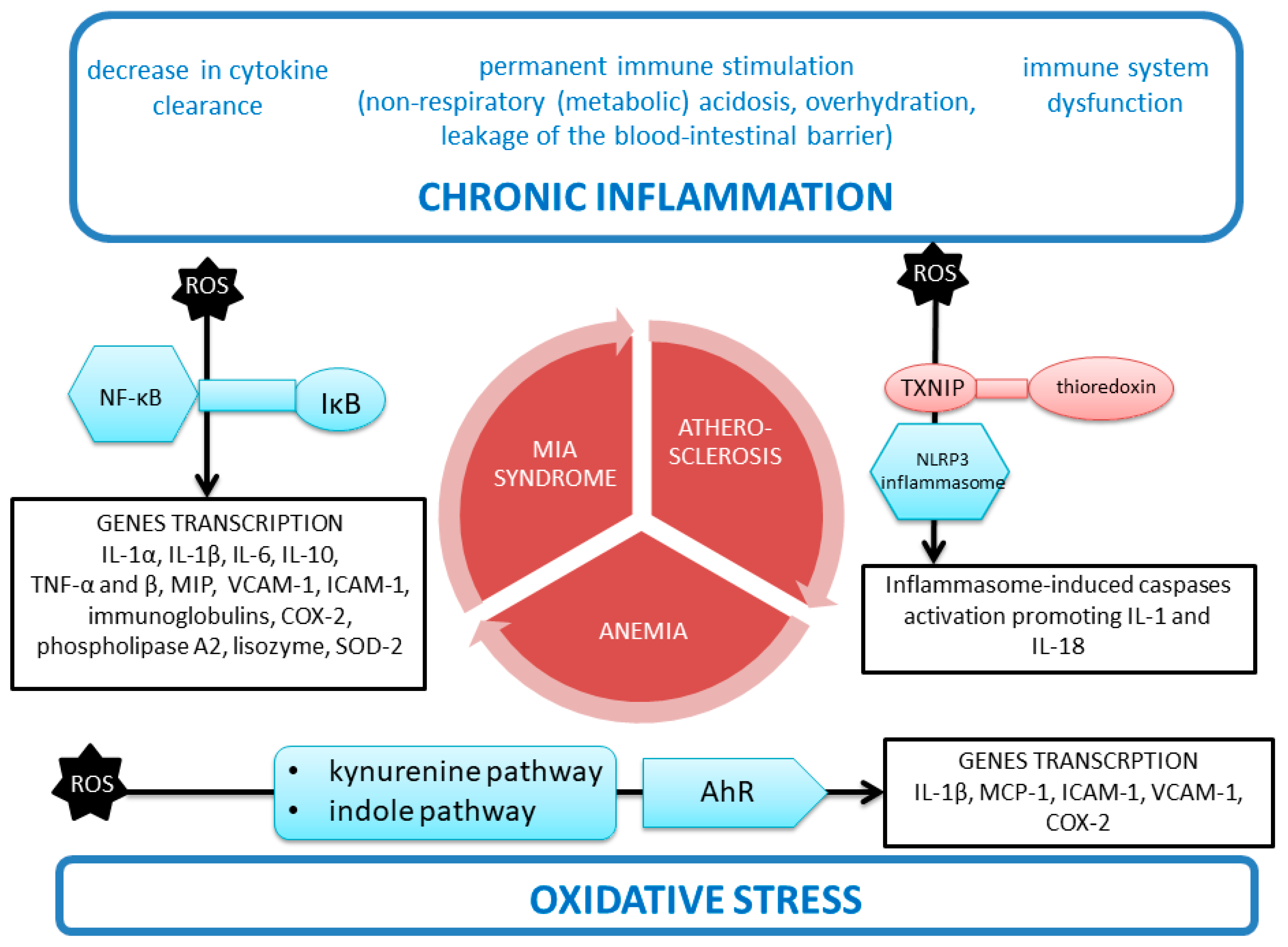
5.3. Malfunction of the Immune System and Constant Immune Stimulation in CKD Coexisting with Inflammation
5.4. Malnutrition–Inflammation–Atherosclerosis (MIA) Syndrome
5.4.1. Malnutrition as a Part of MIA Syndrome in CKD
Hormonal Derangements with Particular Emphasis on Insulin Resistance
Protein–Energy Wasting (PEW)
The Intestinal Microbial Flora Significantly Altered in CKD
Muscles Wasting in CKD
Other Malnutrition-Related Problems in CKD
5.4.2. Inflammation as a Part of MIA Syndrome in CKD
Chronic Inflammation
NLRP3 Inflammasome
Kynurenine Pathway
5.4.3. Atherosclerosis as a Part of MIA Syndrome in CKD
6. Dyslipidemia in CKD
6.1. Lipids, Lipoproteins, and Proteins Modifications
6.1.1. Oxidation
6.1.2. Glycation
6.2. Lipids Profile in CKD
6.2.1. Triglycerides (TG)
6.2.2. High-Density Lipoprotein Cholesterol (HDL-C)
Paraoxonase (PON)
Ischemia-Modified Albumin (IMA)
6.2.3. Very Low-Density Lipoprotein Cholesterol (VLDL-C)
6.2.4. Low-Density Lipoprotein Cholesterol (LDL-C) and Lipoprotein a (Lp (a))
6.2.5. Total Cholesterol (TC)
6.2.6. ApoA-IV
6.3. Disordered NO Metabolism
6.4. Uremic Toxins as Markers of Atherosclerotic Organ Damage
7. Disorders in Calcium–Phosphate Balance in CKD
7.1. Vitamin D
7.2. Parathormon (PTH)
7.3. Klotho Protein
7.4. Disturbances in Calcium–Phosphate in CKD
8. Anemia in CKD
8.1. Reduce in Erythrocytes Lifespan
8.2. Epo Deficiency in CKD
8.3. Iron Disturbances in CKD
9. Cardiovascular Risk in CKD
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- GBD Chronic Kidney Disease Collaboration. Global, regional, and national burden of chronic kidney disease, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2020, 395, 709–733. [Google Scholar] [CrossRef]
- Srivastava, A.; Kaze, A.D.; McMullan, C.J.; Isakova, T.; Waikar, S.S. Uric acid and the risks of kidney failure and death in individuals with CKD. Am. J. Kidney Dis. 2017, 71, 362–370. [Google Scholar] [CrossRef] [PubMed]
- Luczak, M.; Formanowicz, D.; Pawliczak, E.; Wanic-Kossowska, M.; Wykretowicz, A.; Figlerowicz, M. Chronic kidney disease-related atherosclerosis-proteomic studies of blood plasma. Proteome Sci. 2011, 9, 25. [Google Scholar] [CrossRef] [PubMed]
- Poulianiti, K.P.; Kaltsatou, A.; Mitrou, G.I.; Jamurtas, A.Z.; Koutedakis, Y.; Maridaki, M.; Stefanidis, I.; Sakkas, G.K.; Karatzaferi, C. Systemic redox imbalance in chronic kidney disease. Oxid. Med. Cell. Longev. 2016, 2016, 8598253. [Google Scholar] [CrossRef]
- Ling, X.C.; Kuo, K. Oxidative stress in chronic kidney disease. Ren. Replace Ther. 2018, 4, 53. [Google Scholar] [CrossRef]
- Ratliff, B.B.; Abdulmahdi, W.; Pawar, R.; Wolin, M.S. Oxidant mechanisms in renal injury and disease. Antioxid. Redox Signal. 2016, 25, 119–146. [Google Scholar] [CrossRef]
- Iglesias-De La Cruz, M.C.; Ruiz-Torres, P.; Alcamí, J.; Díez-Marqués, L.; Ortega-Velázquez, R.; Chen, S.; Rodríguez-Puyol, M.; Ziyadeh, F.N.; Rodríguez-Puyol, D. Hydrogen peroxide increases extracellular matrix mRNA through TGF-beta in human mesangial cells. Kidney Int. 2001, 59, 87–95. [Google Scholar] [CrossRef]
- Mühl, H.; Sandau, K.; Brüne, B.; Briner, V.A.; Pfeilschifter, J. Nitric oxide donors induce apoptosis in glomerular mesangial cells, epithelial cells and endothelial cells. Eur. J. Pharmacol. 1996, 317, 137–149. [Google Scholar] [CrossRef]
- Sandau, K.; Pfeilschifter, J.; Brünem, B. The balance between nitric oxide and superoxide determines apoptotic and necrotic death of rat mesangial cells. J. Immunol. 1997, 158, 4938–4946. [Google Scholar]
- Sverrisson, K.; Axelssonm, J.; Rippe, A.; Asgeirsson, D.; Rippe, B. Acute reactive oxygen species (ROS)-dependent effects of IL-1β, TNF-α, and IL-6 on the glomerular filtration barrier (GFB) in vivo. Am. J. Physiol. Renal. Physiol. 2015, 309, 800–806. [Google Scholar] [CrossRef]
- Vaziri, N.D.; Dicus, M.; Ho, N.D.; Boroujerdi-Rad, L.; Sindhu, R.K. Oxidative stress and dysregulation of superoxide dismutase and NADPH oxidase in renal insufficiency. Kidney Int. 2003, 63, 179–185. [Google Scholar] [CrossRef]
- Ortiz, P.A.; Garvin, J.L. Role of nitric oxide in the regulation of nephron transport. Am. J. Physiol. Renal. Physiol. 2002, 282, 777–784. [Google Scholar] [CrossRef] [PubMed]
- Sedeek, M.; Nasrallah, R.; Touyz, R.M.; Hébert, R.L. NADPH oxidases, reactive oxygen species, and the kidney: Friend and foe. J. Am. Soc. Nephrol. 2013, 24, 1512–1518. [Google Scholar] [CrossRef] [PubMed]
- Strutz, F.M. EMT and proteinuria as progression factors. Kidney Int. 2009, 75, 475–481. [Google Scholar] [CrossRef] [PubMed]
- Griendling, K.K.; Minieri, C.A.; Ollerenshaw, J.D.; Alexander, R.W. Angiotensin II stimulates NADH and NADPH oxidase activity in cultured vascular smooth muscle cells. Circ. Res. 1994, 74, 1141–1148. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.C.A.; Montezano, A.C.; Burger, D.; Touyz, R.M. Angiotensin II, NADPH oxidase, and redox signaling in the vasculature. Antioxid. Redox. Signal. 2013, 19, 1110–1120. [Google Scholar] [CrossRef]
- Wan, C.; Su, H.; Zhang, C. Role of NADPH oxidase in metabolic disease-related injury: An update. Oxid. Med. Cell. Longev. 2016, 2016, 7813072. [Google Scholar] [CrossRef]
- Holterman, C.E.; Read, N.C.; Kennedy, C.R.J. Nox and renal disease. Clin. Sci. 2015, 128, 465–481. [Google Scholar] [CrossRef]
- Morena, M.; Cristol, J.P.; Senécal, L.; Leray-Moragues, H.; Krieter, D.; Canaud, B. Oxidative stress in hemodialysis patients: Is NADPH oxidase complex the culprit? Kidney Int. Suppl. 2002, 61, 109–114. [Google Scholar] [CrossRef]
- Fortuño, A.; Beloqui, O.; San José, G.; Moreno, M.U.; Zalba, G.; Díez, J. Increased phagocytic nicotinamide adenine dinucleotide phosphate oxidase-dependent superoxide production in patients with early chronic kidney disease. Kidney Int. Suppl. 2005, 99, 71–75. [Google Scholar] [CrossRef]
- Yu, P.; Han, W.; Van Anthony, M.V.; Yang, Y.; Lu, Q.; Lee, H.; Li, F.; Quinn, M.T.; Gildea, J.J.; Felder, R.A.; et al. Unique role of NADPH oxidase 5 in oxidative stress in human renal proximal tubule cells. Redox Biol. 2014, 2, 570–579. [Google Scholar] [CrossRef]
- Muñoz, M.; López-Oliva, M.E.; Rodríguez, C.; Martínez, M.P.; Sáenz-Medina, J.; Sánchez, A.; Climent, B.; Benedito, S.; García-Sacristán, A.; Rivera, L.; et al. Differential contribution of Nox1, Nox2 and Nox4 to kidney vascular oxidative stress and endothelial dysfunction in obesity. Redox Biol. 2020, 28, 101330. [Google Scholar] [CrossRef] [PubMed]
- Caimi, G.; Carollo, C.; Montana, M.; Vaccaro, F.; Presti, R.L. Elastase, myeloperoxidase, nitric oxide metabolites and oxidative status in subjects with clinical stable chronic renal failure on conservative treatment. Clin. Hemorheol. Micro. 2009, 43, 253–258. [Google Scholar] [CrossRef]
- Irazabal, M.V.; Torres, V.E. Reactive oxygen species and redox signaling in chronic kidney disease. Cells 2020, 9, 1342. [Google Scholar] [CrossRef]
- Madero, M.; Sarnak, M.J.; Wang, X.; Greene, T.; Beck, G.J.; Kusek, J.W.; Collins, A.J.; Levey, A.S.; Me-non, V. Uric acid and long-term outcomes in CKD. Am. J. Kidney. Dis. 2009, 53, 796–803. [Google Scholar] [CrossRef] [PubMed]
- Ahola, A.J.; Sandholm, N.; Forsblom, C.; Harjutsalo, V.; Dahlstrom, E.; Groop, P.H.; FinnDiane Study Group. The serum uric acid concentration is not causally linked to diabetic nephropathy in type 1 diabetes. Kidney Int. 2017, 91, 1178–1185. [Google Scholar] [CrossRef] [PubMed]
- Gondouin, B.; Jourde-Chiche, N.; Sallee, M.; Dou, L.; Cerini, C.; Loundou, A.; Morgan, S.; Berland, Y.; Burtey, S.; Brunet, P.; et al. Plasma xanthine oxidase activity is predictive of cardiovascular disease in patients with chronic kidney disease, independently of uric acid levels. Nephron 2015, 131, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Chung, A.W.; Yang, H.H.; Kim, J.M.; Sigrist, M.K.; Brin, G.; Chum, E.; Gourlay, W.A.; Levin, A. Arterial stiffness and functional properties in chronic kidney disease patients on different dialysis modalities: An exploratory study. Nephrol. Dial. Transplant. 2010, 25, 4031–4041. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Granata, S.; Gassa, D.A.; Tomei, P.; Lupo, A.; Zaza, G. Mitochondria: A new therapeutic target in chronic kidney disease. Nutr. Metab. 2015, 12, 49. [Google Scholar] [CrossRef]
- Gamboa, J.L.; Billings, F.T.; Bojanowski, M.T.; Gilliam, L.A.; Yu, C.; Roshanravan, B.; Roberts, L.J.; Himmelfarb, J.; Ikizler, T.A.; Brown, N.J. Mitochondrial dysregulation and oxidative stress in patients with chronic kidney disease. Physiol. Rep. 2016, 4, 12780. [Google Scholar] [CrossRef]
- Ziegler, D.V.; Wiley, C.D.; Velarde, M.C. Mitochondrial effectors of cellular senescence: Beyond the free radical theory of aging. Aging Cell. 2015, 14, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Eirin, A.; Lerman, A.; Lerman, L.O. The emerging role of mitochondrial targeting in kidney disease. Handb. Exp. Pharmacol. 2017, 240, 229–250. [Google Scholar] [PubMed]
- Susztak, K.; Raff, A.C.; Schiffer, M.; Bottinger, E.P. Glucose-induced reactive oxygen species cause apoptosis of podocytes and podocyte depletion at the onset of diabetic nephropathy. Diabetes 2006, 55, 225–233. [Google Scholar] [CrossRef]
- Sharma, K.; Karl, B.; Mathew, A.V.; Gangoiti, J.A.; Wassel, C.L.; Saito, R.; Pu, M.; Sharma, S.; You, Y.H.; Wang, L.; et al. Metabolomics reveals signature of mitochondrial dysfunction in diabetic kidney disease. J. Am. Soc. Nephrol. 2013, 24, 1901–1912. [Google Scholar] [CrossRef] [PubMed]
- Eirin, A.; Lerman, A.; Lerman, L.O. Mitochondria: A pathogenic paradigm in hypertensive renal disease. Hypertension 2015, 65, 264–270. [Google Scholar] [CrossRef] [PubMed]
- De Cavanagh, E.M.; Toblli, J.E.; Ferder, L.; Piotrkowski, B.; Stella, I.; Inserra, F. Renal mitochondrial dysfunction in spontaneously hypertensive rats is attenuated by losartan but not by amlodipine. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2006, 290, 1616–1625. [Google Scholar] [CrossRef]
- Zheleznova, N.N.; Yang, C.; Ryan, R.P.; Halligan, B.D.; Liang, M.; Greene, A.S.; Cowley, A.W., Jr. Mitochondrial proteomic analysis reveals deficiencies in oxygen utilization in medullary thick ascending limb of Henle in the Dahl salt-sensitive rat. Physiol Genom. 2012, 44, 829–842. [Google Scholar] [CrossRef]
- Capeillère-Blandin, C.; Gausson, V.; Nguyen, A.T.; Descamps-Latscha, B.; Drüeke, T.; Witko-Sarsat, V. Respective role of uraemic toxins and myeloperoxidase in the uraemic state. Nephrol. Dial. Transplant. 2006, 21, 1555–1563. [Google Scholar] [CrossRef]
- Pawlak, K.; Mysliwiec, M.; Pawlak, D. Oxidized Low-Density Lipoprotein (OxLDL) plasma levels and OxLDL to LDL ratio—Are they real oxidative stress markers in dialyzed patients? Life Sci. 2013, 92, 253–258. [Google Scholar] [CrossRef]
- Pawlak, K.; Pawlak, D.; Mysliwiec, M. Impaired renal function and duration of dialysis therapy are associated with oxidative stress and proatherogenic cytokine levels in patients with end-stage renal disease. Clin. Biochem. 2007, 40, 81–85. [Google Scholar] [CrossRef]
- Akiyama, S.; Inagaki, M.; Tsuji, M.; Gotoh, H.; Gotoh, T.; Gotoh, Y.; Oguchi, K. mRNA study on Cu/Zn superoxide dismutase induction by hemodialysis treatment. Nephron Clin. Pract. 2005, 99, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Futenma, A.; Yamada, H.; Kitano, M.; Miyai, H.; Fukatsu, A.; Kato, K. Plasma levels of superoxide dismutase and its isomers in patients with chronic renal disease. Nippon Jinzo Gakkai Shi 1993, 35, 371–376. [Google Scholar]
- Nakamura, Y.; Inagaki, M.; Kenmotsu, S.; Yamadera, S.; Ohsawa, I.; Gotoh, H.; Goto, Y.; Sato, N.; Oguchi, T.; Tsuji, M.; et al. Significance of Cu/Zn-superoxide dismutase levels in hemodialysis patients: A mini-review. Mod. Res. Inflam. 2017, 6, 9–13. [Google Scholar] [CrossRef][Green Version]
- Ceballos-Picot, I.; Witko-Sarsat, V.; Merad-Boudia, M.; Nguyen, A.T.; Thévenin, M.; Jaudon, M.C.; Zingraff, J.; Verger, C.; Jungers, P.; Descamps-Latscha, B. Glutathione antioxidant system as a marker of oxidative stress in chronic renal failure. Free Radic. Biol. Med. 1996, 21, 845–853. [Google Scholar] [CrossRef]
- Lim, C.S.; Vaziri, N.D. Iron and oxidative stress in renal insufficiency. Am. J. Nephrol. 2004, 24, 569–575. [Google Scholar] [CrossRef] [PubMed]
- Finch, J.L.; Suarez, E.B.; Husain, K.; Ferder, L.; Cardema, M.C.; Glenn, D.J.; Gardner, D.G.; Liapis, H.; Slatopolsky, E. Effect of combining an ACE inhibitor and a VDR activator on glomerulosclerosis, proteinuria, and renal oxidative stress in uremic rats. Am. J. Physiol. Renal. Physiol. 2012, 302, 141–149. [Google Scholar] [CrossRef]
- Kim, M.S.; Ramakrishna, S.; Lim, K.H.; Kim, J.H.; Baek, K.H. Protein stability of mitochondrial superoxide dismutase SOD2 is regulated by USP36. J. Cell. Biochem. 2011, 112, 498–508. [Google Scholar] [CrossRef]
- Luo, J.; Liang, A.; Liang, M.; Xia, R.; Rizvi, Y.; Wang, Y.; Cheng, J. Serum glucocorticoid–regulated kinase 1 blocks CKD–induced muscle wasting via inactivation of FoxO3a and Smad2/3. J. Am. Soc. Nephrol. 2016, 27, 2797–2808. [Google Scholar] [CrossRef]
- Krueger, K.; Shen, J.; Maier, A.; Tepel, M.; Scholze, A. Lower Superoxide Dismutase 2 (SOD2) protein content in mononuclear cells is associated with better survival in patients with hemodialysis therapy. Oxid. Med. Cell. Longev. 2016, 2016, 7423249. [Google Scholar] [CrossRef]
- Dusso, A.S.; Tokumoto, M. Defective renal maintenance of the vitamin D endocrine system impairs vitamin D renoprotection: A downward spiral in kidney disease. Kidney Int. 2011, 79, 715–729. [Google Scholar] [CrossRef]
- Tabatabaei-Malazy, O.; Khodaeian, M.; Bitarafan, F.; Larijani, B.; Amoli, M.M. Polymorphisms of Antioxidant Genes as a Target for Diabetes Management. Int. J. Mol. Med. 2017, 6, 135–147. [Google Scholar]
- Jin, K.; Vaziri, N.D. Salt-sensitive hypertension in mitochondrial superoxide dismutase deficiency is associated with intra-renal oxidative stress and inflammation. Clin. Exp. Nephrol. 2014, 18, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Tan, R.J.; Zhou, D.; Xiao, L.; Zhou, L.; Li, Y.; Bastacky, S.I.; Oury, T.D.; Liu, Y. Extracellular superoxide dismutase protects against proteinuric kidney disease. J. Am. Soc. Nephrol. 2015, 26, 2447–2459. [Google Scholar] [CrossRef] [PubMed]
- Fujita, H.; Fujishima, H.; Chida, S.; Takahashi, K.; Qi, Z.; Kanetsuna, Y.; Breyer, M.D.; Harris, R.C.; Yamada, Y.; Takahashi, T. Reduction of renal superoxide dismutase in progressive diabetic nephropathy. J. Am. Soc. Nephrol. 2009, 20, 1303–1313. [Google Scholar] [CrossRef]
- Mohammedi, K.; Patente, T.A.; Bellili-Muñoz, N.; Driss, F.; Le Nagard, F.; Fumeron, F.; Roussel, R.; Hadjadj, S.; Corrêa-Giannella, M.L.; Marre, M.; et al. Glutathione peroxidase-1 gene (GPX1) variants, oxidative stress and risk of kidney complications in people with type 1 diabetes. Metabolism 2016, 65, 12–19. [Google Scholar] [CrossRef] [PubMed]
- De Haan, J.B.; Stefanovic, N.; Nikolic-Paterson, D.; Scurr, L.L.; Croft, K.D.; Mori, T.A.; Hertzog, P.; Kola, I.; Atkins, R.C.; Tesch, G.H. Kidney expression of glutathione peroxidase-1 is not protective against streptozotocin-induced diabetic nephropathy. Am. J. Physiol. Renal. Physiol. 2005, 289, 544–551. [Google Scholar] [CrossRef]
- De Haan, J.B.; Bladier, C.; Griffiths, P.; Kelner, M.; O’Shea, R.D.; Cheung, N.S.; Bronson, R.T.; Silvestro, M.J.; Wild, S.; Zheng, S.S.; et al. Mice with a homozygous null mutation for the most abundant glutathione peroxidase, Gpx1, show increased susceptibility to the oxidative stress-inducing agents paraquat and hydrogen peroxide. J. Biol. Chem. 1998, 273, 22528–22536. [Google Scholar] [CrossRef]
- Yoshimura, S.; Suemizu, H.; Nomoto, Y.; Sakai, H.; Katsuoka, Y.; Kawamura, N.; Moriuchi, T. Plasma glutathione peroxidase deficiency caused by renal dysfunction. Nephron 1996, 73, 207–211. [Google Scholar]
- Lubos, E.; Loscalzo, J.; Handy, D.E. Glutathione peroxidase-1 in health and disease: From molecular mechanisms to therapeutic opportunities. Antioxid. Redox Signal. 2011, 15, 1957–1997. [Google Scholar] [CrossRef]
- Zhang, J.X.; Wang, Z.M.; Zhang, J.J.; Zhu, L.L.; Gao, X.F.; Chen, S.L. Association of glutathione peroxidase-1 (GPx-1) rs1050450 Pro198Leu and Pro197Leu polymorphisms with cardiovascular risk: A meta-analysis of observational studies. J. Geriatr. Cardiol. 2014, 11, 141–150. [Google Scholar]
- Pang, P.; Abbott, M.; Abdi, M.; Fucci, Q.A.; Chauhan, N.; Mistri, M.; Proctor, B.; Chin, M.; Wang, B.; Yin, W.; et al. Pre-clinical model of severe glutathione peroxidase-3 deficiency and chronic kidney disease results in coronary artery thrombosis and depressed left ventricular function. Nephrol. Dial. Transplant. 2018, 33, 923–934. [Google Scholar] [CrossRef] [PubMed]
- Kalantar-Zadeh, K.; Block, G.; Horwich, T.; Fonarow, G.C. Reverse epidemiology of conventional cardiovascular risk factors in patients with chronic heart failure. J. Am. Coll. Cardiol. 2004, 43, 1439–1444. [Google Scholar] [CrossRef] [PubMed]
- Schnabel, R.; Lackner, K.J.; Rupprecht, H.J.; Espinola-Klein, C.; Torzewski, M.; Lubos, E.; Bickel, C.; Cambien, F.; Tiret, L.; Münzel, T.; et al. Glutathione peroxidase-1 and homocysteine for cardiovascular risk prediction: Results from the AtheroGeneStudy. J. Am. Coll. Cardiol. 2005, 45, 1631–1637. [Google Scholar] [CrossRef] [PubMed]
- Hallgren, C.; Hallmans, G.; Jansson, J.; Marklund, S.; Huhtasaari, F.; Schütz, A.; Strömberg, U.; Vessby, B.; Skerfving, S. Markers of high fish intake are associated with decreased risk of a first myocardial infarction. Br. J. Nutr. 2001, 86, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Hagmar, L.; Persson-Moschos, M.; Akesson, B.; Schütz, A. Plasma levels of selenium, selenoprotein P and glutathione peroxidase and their correlations to fish intake and serum levels of thyrotropin and thyroid hormones: A study on Latvian fish consumers. Eur. J. Clin. Nutr. 1998, 52, 796–800. [Google Scholar] [CrossRef][Green Version]
- Yang, S.; Jensen, M.K.; Rimm, E.B.; Willett, W.; Wu, T. Erythrocyte superoxide dismutase, glutathione peroxidase, and catalase activities and risk of coronary heart disease in generally healthy women: A prospective study. Am. J. Epidemiol. 2014, 180, 901–908. [Google Scholar] [CrossRef]
- Bulucu, F.; Vural, A.; Aydin, A.; Sayal, A. Oxidative stress status in adults with nephrotic syndrome. Clin. Nephrol. 2000, 53, 169–173. [Google Scholar]
- El-far, M.; Bakr, M.; Farahat, S.; El-Fattah, E.A.A. Glutathione peroxidase activity in patients with renal disorders. Clin. Exp. Nephrol. 2005, 9, 127–131. [Google Scholar] [CrossRef]
- Richard, M.J.; Arnaud, J.; Jurkovitz, C.; Hachache, T.; Meftahi, H.; Laporte, F.; Foret, M.; Favier, A.; Cordonnier, D. Trace elements and lipid peroxidation abnormalities in patients with chronic renal failure. Nephron 1991, 57, 10–15. [Google Scholar] [CrossRef]
- Roxborough, H.E.; Mercer, C.; McMaster, D.; Maxwell, A.P.; Young, I.S. Plasma glutathione peroxidase activity is reduced in haemodialysis patients. Nephron 1999, 81, 278–283. [Google Scholar] [CrossRef]
- Ozden, M.; Maral, H.; Akaydin, D.; Cetinalp, P.; Kalender, B. Erythrocyte glutathione peroxidase activity, plasma malondialdehyde and erythrocyte glutathione levels in hemodialysis and CAPD patients. Clin. Biochem. 2002, 35, 269–273. [Google Scholar] [CrossRef]
- Ninić, A.; Sopić, M.; Munjas, J.; Spasojević-Kalimanovska, V.; Kotur-Stevuljević, J.; Bogavac-Stanojević, N.; Ivanišević, J.; Simić-Ogrizović, S.; Kravljača, M.; Jelić-Ivanović, Z. Association between superoxide dismutase isoenzyme gene expression and total antioxidant status in patients with an end-stage renal disease. Balkan Med. J. 2018, 35, 431–436. [Google Scholar] [CrossRef] [PubMed]
- Crawford, A.; Fassett, R.G.; Coombes, J.S.; Kunde, D.A.; Ahuja, K.D.K.; Robertson, I.K.; Ball, M.J.; Geraghty, D.P. Relationship between antioxidant enzyme genotype and activity and kidney function: A case-control study. Clin. Nephrol. 2012, 78, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Tbahriti, H.F.; Kaddous, A.; Bouchenak, M.; Mekki, K. Effect of different stages of chronic kidney disease and renal replacement therapies on oxidant-antioxidant balance in uremic patients. Biochem. Res. Int. 2013, 2013, 358985. [Google Scholar] [CrossRef] [PubMed]
- Montazerifar, F.; Hashemi, M.; Karajibani, M.; Sanadgol, H.; Dikshit, M. Evaluation of lipid peroxidation and erythrocyte glutathione peroxidase and superoxide dismutase in hemodialysis patients. Saudi J. Kidney Dis. Transpl. 2012, 23, 274–279. [Google Scholar] [PubMed]
- Drai, J.; Bannier, E.; Chazot, C.; Hurot, J.M.; Goedert, G.; Jean, G.; Charra, B.; Laurent, G.; Baltassat, P.; Revol, A. Oxidants and antioxidants in long-term haemodialysis patients. Farmaco 2001, 56, 463–465. [Google Scholar] [CrossRef]
- Dursun, E.; Dursun, B.; Suleymanlar, G.; Ozben, T. Effect of haemodialysis on the oxidative stress and antioxidants in diabetes mellitus. Acta Diabetol. 2005, 42, 123–128. [Google Scholar] [CrossRef]
- Durak, I.; Akyol, O.; Başeşme, E.; Canbolat, O.; Kavutçu, M. Reduced erythrocyte defense mechanisms against free radical toxicity in patients with chronic renal failure. Nephron 1994, 66, 76–80. [Google Scholar] [CrossRef]
- Durak, İ.; Kavutcu, M.; Çimen, M.Y.B.; Avcı, A.; Elgün, S.; Öztürk, H.S. Oxidant/antioxidant status of erythrocytes from patients with chronic renal failure: Effects of hemodialysis. Med. Princ. Pract. 2001, 10, 187–190. [Google Scholar] [CrossRef]
- Canestrari, F.; Galli, F.; Giorgini, A.; Albertini, M.C.; Galiotta, P.; Pascucci, M.; Bossù, M. Erythrocyte redox state in uremic anemia: Effects of hemodialysis and relevance of glutathione metabolism. Acta Haematol. 1994, 91, 187–193. [Google Scholar] [CrossRef]
- Zachara, B.A.; Gromadzinska, J.; Zbrog, Z.; Swiech, R.; Wasowicz, W.; Twardowska, E.; Jablonska, E.; Sobala, W. Selenium supplementation to chronic kidney disease patients on hemodialysis does not induce the synthesis of plasma glutathione peroxidase. Acta Biochim. Pol. 2009, 56, 183–187. [Google Scholar] [CrossRef] [PubMed]
- Zachara, B.A.; Adamowicz, A.; Trafikowska, U.; Pilecki, A.; Manitius, J. Decreased plasma glutathione peroxidase activity in uraemic patients. Nephron 2000, 84, 278–281. [Google Scholar] [CrossRef] [PubMed]
- Anees, M.; Mumtaz, A.; Frooqi, S.; Ibrahim, M.; Hameed, F. Serum trace elements (aluminium, copper, zinc) in hemodialysis patients. Biomedica 2011, 27, 106–110. [Google Scholar]
- Hsieh, Y.Y.; Shen, W.S.; Lee, L.Y.; Wu, T.L.; Ning, H.C.; Sun, C.F. Long-term changes in trace elements in patients undergoing chronic hemodialysis. Biol. Trace Elem. Res. 2006, 109, 115–121. [Google Scholar] [CrossRef]
- Eljaoudi, R.; Errasfa, M.; Benyahia, M.; Bahadi, A.; Cherrah, Y.; Ibrahimi, A.; Elkabbaj, D. Copper, zinc and selenium imbalance in Moroccan haemodialysis patients and its correlation to lipid peroxidation. Int. J. Res. Med. Sci. 2015, 3, 2079–2085. [Google Scholar] [CrossRef]
- Iglesias, P.; Selgas, R.; Romero, S.; Díez, J. Selenium and kidney disease. J. Nephrol. 2013, 26, 266–272. [Google Scholar] [CrossRef]
- Tonelli, M.; Wiebe, N.; Hemmelgarn, B.; Klarenbach, S.; Field, C.; Manns, B.; Thadhani, R.; Gill, J.; Alberta Kidney Disease Network. Trace elements in haemodialysis patients: A systematic review and meta-analysis. BMC Med. 2009, 7, 25. [Google Scholar] [CrossRef]
- Milly, K.; Wit, L.; Diskin, C.; Tulley, R. Selenium in renal failure patients. Nephron 1992, 61, 139–144. [Google Scholar] [CrossRef]
- Navarro-Alarcon, M.; Reyes-Pérez, A.; Lopez-Garcia, H.; Palomares-Bayo, M.; Olalla-Herrera, M.; Lopez-Martinez, M.C. Longitudinal study of serum zinc and copper levels in haemodialysis patients and their relation to biochemical markers. Biol. Trace Elem. Res. 2006, 113, 209–222. [Google Scholar] [CrossRef]
- Richard, M.J.; Duclos, V.; Foret, M.; Arnaud, J.; Coudray, C.; Fusselier, M.; Favier, A. Reversal of selenium and zinc deficiencies in chronic hemodialysis patients by intravenous sodium selenite and zinc gluconate supplementation. Biol. Trace Elem. Res. 1993, 39, 149–159. [Google Scholar] [CrossRef]
- Montazerifar, F.; Hashemi, M.; Karajibani, M.; Dikshit, M. Hemodialysis alters lipid profiles, total antioxidant capacity, and vitamins A, E, and C concentrations in humans. J. Med. Food 2010, 13, 1490–1493. [Google Scholar] [CrossRef] [PubMed]
- Jankowska, M.; Rutkowski, B.; Dębska-Ślizień, A. Vitamins and microelement bioavailability in different stages of chronic kidney disease. Nutrients 2017, 9, 282. [Google Scholar] [CrossRef] [PubMed]
- Karamouzis, I.; Sarafidis, P.A.; Karamouzis, M.; Iliadis, S.; Haidich, A.B.; Sioulis, A.; Triantos, A.; Vavatsi-Christaki, N.; Grekas, D.M. Increase in oxidative stress but not in antioxidant capacity with advancing stages of chronic kidney disease. Am. J. Nephrol. 2008, 28, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.K. Does the interdependence between oxidative stress and inflammation explain the antioxidant paradox? Oxid. Med. Cell. Longev. 2016, 2016, 5698931. [Google Scholar] [CrossRef]
- Santhanam, A.V.; d’Uscio, L.V.; Katusic, Z.S. Erythropoietin increases bioavailability of tetrahydrobiopterin and protects cerebral icrovasculature against oxidative stress induced by eNOS uncoupling. J. Neurochem. 2014, 131, 521–529. [Google Scholar] [CrossRef]
- Kisic, B.; Miric, D.; Dragojevic, I.; Rasic, J.; Popovic, L. Role of myeloperoxidase in patients with chronic kidney disease. Oxid. Med. Cell. Longev. 2016, 2016, 1069743. [Google Scholar] [CrossRef]
- Maruyama, Y.; Lindholm, B.; Stenvinkel, P. Inflammation and oxidative stress in ESRD-the role of myeloperoxidase. J. Nephrol. 2004, 17 (Suppl. 8), 72–76. [Google Scholar]
- Madhusudhana, R.A.; Anand, U.; Anand, C.V. Myeloperoxidase in chronic kidney disease. Indian J. Clin. Biochem. 2011, 26, 28–31. [Google Scholar] [CrossRef]
- Kalantar-Zadeh, K.; Brennan, M.-L.; Hazen, S.L. Serum myeloperoxidase and mortality in maintenance hemodialysis patients. Am. J. Kidney Dis. 2006, 48, 59–68. [Google Scholar] [CrossRef]
- Miric, D.; Kisic, B.; Stolic, R.; Miric, B.; Mitic, R.; Janicijevic-Hudomal, S. The role of xanthine oxidase in hemodialysis-induced oxidative injury: Relationship with nutritional status. Oxid. Med. Cell. Longev. 2013, 2013, 245253. [Google Scholar] [CrossRef]
- Honda, H.; Ueda, M.; Kojima, S.; Mashiba, S.; Hirai, Y.; Hosaka, N.; Suzuki, H.; Mukai, M.; Watanabe, M.; Takahashi, K.; et al. Assessment of myeloperoxidase and oxidative α 1-antitrypsin in patients on hemodialysis. Clin. J. Am. Soc. Nephrol. 2009, 4, 142–151. [Google Scholar] [CrossRef] [PubMed]
- Lehners, A.; Lange, S.; Niemann, G.; Rosendahl, A.; Meyer-Schwesinger, C.; Oh, J.; Stahl, R.; Ehmke, H.; Benndorf, R.; Kinke, A.; et al. Myeloperoxidase deficiency ameliorates progression of chronic kidney disease in mice. Am. J. Physiol. Renal. Physiol. 2014, 307, 407–417. [Google Scholar] [CrossRef] [PubMed]
- Himmelfarb, J.; McMenamin, M.E.; Loseto, G.; Heinecke, J.W. Myeloperoxidase-catalyzed 3-chlorotyrosine formation in dialysis patients. Free Radic. Biol. Med. 2001, 31, 1163–1169. [Google Scholar] [CrossRef]
- Morgan, M.J.; Liu, Z.G. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. 2011, 21, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Stenvinkel, P.; Ketteler, M.; Johnson, R.J.; Lindholm, B.; Pecoits-Filho, R.; Riella, M.; Heimbürger, O.; Cederholm, T.; Girndt, M. IL-10, IL-6, and TNF-α: Central factors in the altered cytokine network of uremia—The good, the bad, and the ugly. Kidney Int. 2005, 67, 1216–1233. [Google Scholar] [CrossRef] [PubMed]
- Kato, S.; Chmielewski, M.; Honda, H.; Pecoits-Filho, R.; Matsuo, S.; Yuzawa, Y.; Tranaeus, A.; Stenvinkel, P.; Lindholm, B. Aspects of immune dysfunction in end-stage renal disease. Clin. J. Am. Soc. Nephrol. 2008, 3, 1526–1533. [Google Scholar] [CrossRef]
- Formanowicz, D.; Gutowska, K.; Formanowicz, P. Theoretical studies on the engagement of interleukin 18 in the immuno-inflammatory processes underlying atherosclerosis. Int. J. Mol. Sci. 2018, 19, 3476. [Google Scholar] [CrossRef]
- Formanowicz, D.; Wanic-Kossowska, M.; Pawliczak, E.; Radom, M.; Formanoiwcz, P. Usefulness of serum interleukin-18 in predicting cardiovascular mortality in patients with chronic kidney disease—Systems and clinical approach. Sci. Rep. 2015, 5, 18332. [Google Scholar] [CrossRef]
- Akchurin, O.M.; Kaskel, F. Update on inflammation in chronic kidney disease. Blood Purif. 2015, 39, 84–92. [Google Scholar] [CrossRef]
- Kosmas, C.E.; Silverio, D.; Tsomidou, C.; Salcedo, M.D.; Montan, P.D.; Guzman, E. The impact of insulin resistance and chronic kidney disease on inflammation and cardiovascular disease. Clin. Med. Insights Endocrinol. Diabetes 2018, 11, 1179551418792257. [Google Scholar] [CrossRef]
- Spoto, B.; Pisano, A.; Zoccali, C. Insulin resistance in chronic kidney disease: A systematic review. Am. J. Physiol. Renal. Physiol. 2016, 311, 1087–1108. [Google Scholar] [CrossRef] [PubMed]
- Kopple, J.D. Pathophysiology of protein-energy wasting in chronic renal failure. J. Nutr. 1994, 129, 147–251. [Google Scholar] [CrossRef] [PubMed]
- Fouque, D.; Kalantar-Zadeh, K.; Kopple, J.; Cano, N.; Chauveau, P.; Cuppari, L.; Franch, H.; Guarnieri, G.; Ikizler, T.A.; Kaysen, G.; et al. A proposed nomenclature and diagnostic criteria for protein-energy wasting in acute and chronic kidney disease. Kidney Int. 2008, 73, 391–398. [Google Scholar] [CrossRef] [PubMed]
- Jankowska, M.; Cobo, G.; Lindholm, B.; Stenvinkel, P. Inflammation and protein-energy wasting in the uremic milieu. Contrib. Nephrol. 2017, 191, 58–71. [Google Scholar]
- Jadeja, Y.P.; Kher, V. Protein energy wasting in chronic kidney disease: An update with focus on nutritional interventions to improve outcomes. Indian J. Endocrinol. Metab. 2012, 16, 246–251. [Google Scholar] [CrossRef]
- Iorember, F.M. Malnutrition in chronic kidney disease. Front. Pediatr. 2018, 6, 161. [Google Scholar] [CrossRef]
- Evenepoel, P.; Poesen, R.; Meijers, B. The gut-kidney axis. Pediatr. Nephrol. 2016, 32, 2005–2014. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, D.; Chen, L.; Liu, J.R.; Vaziri, N.D.; Guo, Y.; Zhao, Y.Y. Microbiome-metabolome reveals the contribution of gut-kidney axis on kidney disease. J. Transl. Med. 2019, 17, 5. [Google Scholar] [CrossRef]
- Zha, Y.; Qian, Q. Protein nutrition and malnutrition in CKD and ESRD. Nutrients 2017, 27, 208. [Google Scholar] [CrossRef]
- Wesson, D.E.; Buysse, J.M.; Bushinsky, D.A. Mechanisms of Metabolic Acidosis–Induced Kidney Injury in Chronic Kidney Disease. J. Am. Soc. Nephrol. 2020, 31, 469–482. [Google Scholar] [CrossRef]
- Junior, S.L.D.; Santos, P.R.; dos Santos, A.A.; de Souza, M.H.L.P. Dyspepsia and gastric emptying in end-stage renal disease patients on hemodialysis. BMC Nephrol. 2013, 14, 275. [Google Scholar]
- Eustace, J.A.; Astor, B.; Muntner, P.M.; Ikizler, T.A.; Coresh, J. Prevalence of acidosis and inflammation and their association with low serum albumin in chronic kidney disease. Kidney Int. 2004, 65, 1031–1040. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Axelsson, J.; Machowska, A.; Heimbürger, O.; Bárány, P.; Lindholm, B.; Lindström, K.; Stenvinkel, P.; Qureshiet, A.R. Biomarkers of cardiovascular disease and mortality risk in patients with advanced CKD. Clin. J. Am. Soc. Nephrol. 2016, 11, 1163–1172. [Google Scholar] [CrossRef] [PubMed]
- Cobo, G.; Lindholm, B.; Stenvinkel, P. Chronic inflammation in end-stage renal disease and dialysis. Nephrol. Dial. Transplant. 2018, 33 (Suppl. 3), 35–40. [Google Scholar] [CrossRef] [PubMed]
- Gupta, J.; Mitra, N.; Kanetsky, P.A.; Devaney, J.; Wing, M.R.; Reilly, M.; Shah, V.O.; Balakrishnan, V.S.; Guzman, N.J.; CRIC Study Investigators. Association between albuminuria, kidney function, and inflammatory biomarker profile in CKD in CRIC. Clin. J. Am. Soc. Nephrol. 2012, 7, 1938–1946. [Google Scholar] [CrossRef]
- Mun, K.C.; Golper, T.A. Impaired biological activity of erythropoietin by cyanate carbamylation. Blood Purif. 2000, 18, 13–17. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.D.; Phillips, T.M.; Khetpal, P.; Kimmel, P.L. Cytokine patterns and survival in haemodialysis patients. Nephrol. Dial. Transplant. 2010, 25, 1239–1243. [Google Scholar] [CrossRef]
- Granata, S.; Masola, V.; Zoratti, E.; Scupoli, M.T.; Baruzzi, A.; Messa, M.; Sallustio, F.; Gesualdo, L.; Lupo, A.; Zaza, G. NLRP3 inflammasome activation in dialyzed chronic kidney disease patients. PLoS ONE 2015, 10, e0122272. [Google Scholar] [CrossRef]
- Yilmaz, N.; Ustundag, Y.; Kivrak, S.; Kahvecioglu, S.; Celik, H.; Kivrak, I.; Huysal, K. Serum indoleamine 2,3 dioxygenase and tryptophan and kynurenine ratio using the UPLC-MS/MS method, in patients undergoing peritoneal dialysis, hemodialysis, and kidney transplantation. Ren. Fail. 2016, 38, 1300–1309. [Google Scholar] [CrossRef]
- Eleftheriadis, T.; Antoniadi, G.; Liakopoulos, V.; Stefanidis, I.; Galaktidou, G. Plasma indoleamine 2,3-dioxygenase concentration is increased in hemodialysis patients and may contribute to the pathogenesis of coronary heart disease. Ren. Fail. 2012, 34, 68–72. [Google Scholar] [CrossRef]
- Schefold, J.C.; Zeden, J.P.; Fotopoulou, C.; von Haehling, S.; Psychowski, R.; Hasper, D.; Volk, H.D.; Schett, C.; Reinke, P. Increased indoleamine 2,3-dioxygenase (IDO) activity and elevated serum levels of tryptophan catabolites in patients with chronic kidney disease: A possible link between chronic inflammation and uraemic symptoms. Nephrol. Dial. Transplant. 2009, 24, 1901–1908. [Google Scholar] [CrossRef] [PubMed]
- Lano, G.; Burtey, S.; Sallée, M. Indoxyl sulfate, a uremic endotheliotoxin. Toxins 2020, 12, 229. [Google Scholar] [CrossRef] [PubMed]
- Muntner, P.; He, J.; Astor, B.C.; Folsom, A.R.; Coresh, J. Traditional and nontraditional risk factors predict coronary heart disease in chronic kidney disease: Results from the atherosclerosis risk in communities study. J. Am. Soc. Nephrol. 2005, 16, 529–538. [Google Scholar] [CrossRef] [PubMed]
- Beckman, J.S.; Koppenol, W.H. Nitric oxide, superoxide, and peroxynitrite: The good, the bad, and ugly. Am. J. Physiol. 1996, 271, 1424–1437. [Google Scholar] [CrossRef] [PubMed]
- Kazancioğlu, R. Risk factors for chronic kidney disease: An update. Kidney Int. Suppl. 2013, 3, 368–371. [Google Scholar] [CrossRef] [PubMed]
- Mathew, R.O.; Bangalore, S.; Lavelle, M.P.; Pellikka, P.A.; Sidhu, M.S.; Boden, W.E.; Asif, A. Diagnosis and management of atherosclerotic cardiovascular disease in chronic kidney disease: A review. Kidney Int. 2017, 91, 797–807. [Google Scholar] [CrossRef]
- Vanholder, R.; Massy, Z.; Argiles, A.; Spasovski, G.; Verbeke, F.; Lameire, N.; European Uremic Toxin Work Group. Chronic kidney disease as cause of cardiovascular morbidity and mortality. Dial Transplant. 2005, 20, 1048–1056. [Google Scholar] [CrossRef]
- Luczak, M.; Suszynska-Zajczyk, J.; Marczak, L.; Formanowicz, D.; Pawliczak, E.; Wanic-Kossowska, M.; Stobiecki, M. Label-free quantitative proteomics reveals differences in molecular mechanism of atherosclerosis related and non-related to chronic kidney disease. Int. J. Mol. Sci. 2016, 17, 631. [Google Scholar] [CrossRef]
- Muntner, P.; Coresh, J.; Smith, J.C.; Eckfeldt, J.; Klag, M.J. Plasma lipids and risk of developing renal dysfunction: The atherosclerosis risk in communities study. Kidney Int. 2000, 58, 293–301. [Google Scholar] [CrossRef]
- Florens, N.; Calzada, C.; Lyasko, E.; Juillard, L.; Soulage, C.O. Modified lipids and lipoproteins in chronic kidney disease: A new class of uremic toxins. Toxins 2016, 8, 376. [Google Scholar] [CrossRef]
- Murphy, R.C.; Johnson, K.M. Cholesterol, reactive oxygen species, and the formation of biologically active mediators. J. Biol. Chem. 2008, 283, 15521–15525. [Google Scholar] [CrossRef] [PubMed]
- Berliner, J.A.; Leitinger, N.; Tsimikas, S. The role of oxidized phospholipids in atherosclerosis. J Lipid Res. 2009, 50, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Radi, R. Nitric oxide, oxidants, and protein tyrosine nitration. Proc. Natl. Acad. Sci. USA 2004, 101, 4003–4008. [Google Scholar] [CrossRef] [PubMed]
- Levitan, I.; Volkov, S.; Subbaiah, P.V. Oxidized LDL: Diversity, patterns of recognition, and pathophysiology. Antioxid. Redox Signal. 2010, 13, 39–75. [Google Scholar] [CrossRef] [PubMed]
- Kwan, B.C.H.; Kronenberg, F.; Beddhu, S.; Cheung, A.K. Lipoprotein metabolism and lipid management in chronic kidney disease. J. Am. Soc. Nephrol. 2007, 18, 1246–1261. [Google Scholar] [CrossRef] [PubMed]
- Pirillo, A.; Norata, G.D.; Catapano, A.L. LOX-1, OxLDL, and atherosclerosis. Mediat. Inflamm. 2013, 2013, 152786. [Google Scholar] [CrossRef] [PubMed]
- Exner, M.; Hermann, M.; Hofbauer, R.; Hartmann, B.; Kapiotis, S.; Gmeiner, B. Thiocyanate catalyzes myeloperoxidase-initiated lipid oxidation in LDL. Free Radic. Biol. Med. 2004, 37, 146–155. [Google Scholar] [CrossRef]
- Podrez, E.A.; Febbraio, M.; Sheibani, N.; Schmitt, D.; Silverstein, R.L.; Hajjar, D.P.; Cohen, P.A.; Frazier, W.A.; Hoff, H.F.; Hazen, S.L. Macrophage scavenger receptor CD36 is the major receptor for LDL modified by monocyte-generated reactive nitrogen species. J. Clin. Investig. 2000, 105, 1095–1108. [Google Scholar] [CrossRef]
- Chmielewski, M.; Bryl, E.; Marzec, L.; Aleksandrowicz, E.; Witkowski, J.M.; Rutkowski, B. Expression of scavenger receptor CD36 in chronic renal failure patients. Artif. Organs 2005, 29, 608–614. [Google Scholar] [CrossRef]
- Kon, V.; Linton, M.F.; Fazio, S. Atherosclerosis in chronic kidney disease: The role of macrophages. Nat. Rev. Nephrol. 2011, 7, 45–54. [Google Scholar] [CrossRef]
- Estruch, M.; Sánchez-Quesada, J.L.; Ordóñez Llanos, J.; Benítez, S. Electronegative LDL: A circulating modified LDL with a role in inflammation. Mediat. Inflamm. 2013, 2013, 181324. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.C.; Lee, A.; Chen, W.Y.; Lin, Y.N.; Hsu, J.F.; Chan, H.C.; Chang, C.M.; Chang, S.S.; Pan, C.C.; Sawamura, T.; et al. Increased LDL electronegativity in chronic kidney disease disrupts calcium homeostasis resulting in cardiac dysfunction. J. Mol. Cell. Cardiol. 2015, 84, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Witko-Sarsat, V.; Gausson, V.; Descamps-Latscha, B. Are advanced oxidation protein products potential uremic toxins? Kidney Int. Suppl. 2003, 84, 11–14. [Google Scholar] [CrossRef] [PubMed]
- Marsche, G.; Frank, S.; Hrzenjak, A.; Holzer, M.; Dirnberger, S.; Wadsack, C.; Scharnagl, H.; Stojakovic, T.; Heinemann, A.; Oettl, K. Plasma-advanced oxidation protein products are potent high-density lipoprotein receptor antagonists in vivo. Circ. Res. 2009, 104, 750–757. [Google Scholar] [CrossRef]
- Conti, G.; Caccamo, D.; Siligato, R.; Dembillo, G.; Satta, E.; Pazzano, D.; Carucci, N.; Carella, A.; Del Campo, G.; Salvo, A.; et al. Association of higher Advanced Oxidation Protein Products (AOPPs) levels in patients with diabetic and hypertensive nephropathy. Medicina 2019, 55, 675. [Google Scholar] [CrossRef]
- Singh, R.; Barden, A.; Mori, T.; Beilin, L. Advanced glycation end-products: A review. Diabetologia 2001, 44, 129–146. [Google Scholar] [CrossRef]
- Younis, N.; Soran, H.; Sharma, R.; Charlton-Menys, V.; Durrington, D. Lipoprotein glycation in atherogenesis. Clin. Lipidol. 2009, 4, 781–790. [Google Scholar] [CrossRef]
- Sobal, G.; Menzel, J.; Sinzinger, H. Why is glycated LDL more sensitive to oxidation than native LDL? A comparative study. Prostaglandins Leukot. Essent. Fatty Acids 2000, 63, 177–186. [Google Scholar] [CrossRef]
- Prasad, K.; Mishra, M. Do advanced glycation end products and its receptor play a role in pathophysiology of hypertension? Int. J. Angiol. 2017, 26, 1–11. [Google Scholar]
- Chen, Q.; Dong, L.; Wang, L.; Kang, L.; Xu, B. Advanced glycation end products impair function of late endothelial progenitor cells through effects on protein kinase Akt and cyclooxygenase-2. Biochem. Biophys. Res. Commun. 2009, 381, 192–197. [Google Scholar] [CrossRef]
- Soran, H.; Durrington, P.N. Susceptibility of LDL and its subfractions to glycation. Curr. Opin. Lipidol. 2011, 22, 254–261. [Google Scholar] [CrossRef] [PubMed]
- Younis, N.; Sharma, R.; Soran, H.; Charlton-Menys, V.; Elseweidy, M.; Durrington, P.N. Glycation as an atherogenic modification of LD. Curr. Opin. Lipidol. 2008, 19, 378–384. [Google Scholar] [CrossRef] [PubMed]
- Chu, M.; Wang, A.Y.; Chan, I.H.; Chui, S.H.; Lam, C.W.K. Serum small-dense LDLA abnormalities in chronic renal disease patients. Br. J. Biomed. Sci. 2012, 69, 99–102. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Xu, Y.; Lu, J.; Ma, C.; Zhou, Y.; Li, Q.; Chen, X.; Zhu, A.; Shen, G. Small dense low-density lipoprotein cholesterol was associated with future cardiovascular events in chronic kidney disease patients. BMC Nephrol. 2016, 17, 134. [Google Scholar] [CrossRef]
- Filler, G.; Taheri, S.; McIntyre, C.; Smith, C.; Subramanian, L.; Fusch, G.; Fusch, G. Chronic kidney disease stage affect small, dense low-density lipoprotein but not glycated low-density lipoprotein in younger chronic kidney disease patients: A cross-sectional study. Clin. Kidney J. 2018, 11, 383–388. [Google Scholar] [CrossRef]
- Vaziri, N.D.; Norris, K. Lipid disorders and their relevance to outcomes in chronic kidney disease. Blood Purif. 2011, 31, 189–196. [Google Scholar] [CrossRef]
- Moradi, H.; Pahl, M.V.; Elahimehr, R.; Vaziri, N.D. Impaired antioxidant activity of high-density lipoprotein in chronic kidney disease. Transl. Res. 2009, 153, 77–85. [Google Scholar] [CrossRef]
- Batista, M.C.; Welty, F.K.; Diffenderfer, M.R.; Sarnak, M.J.; Schaefer, E.J.; Lamon-Fava, S.; Asztalos, B.F.; Dolnikowski, G.G.; Brousseau, M.E.; Marsh, J.B. Apolipoprotein A-I, B-100, and B-48 metabolism in subjects with chronic kidney disease, obesity, and the metabolic syndrome. Metabolism 2004, 53, 1255–1261. [Google Scholar] [CrossRef]
- Duni, A.; Liakopoulos, V.; Rapsomanikis, K.P.; Dounousi, E. Kidney disease and disproportionally increased cardiovascular damage: Does oxidative stress explain the burden? Oxid. Med. Cell. Longev. 2017, 2017, 9036450. [Google Scholar] [CrossRef]
- Aviram, M.; Rosenblat, M. Paraoxonases and cardiovascular diseases: Pharmacological and nutritional influences. Curr. Opin. Lipidol. 2005, 16, 393–399. [Google Scholar] [CrossRef]
- Gugliucci, A.; Kotani, K.; Kimura, S. Paraoxonase 1 in chronic kidney failure. J. Lipids. 2012, 2012, 726048. [Google Scholar] [CrossRef] [PubMed]
- Kotani, K.; Kimura, S.; Gugliucci, A. Paraoxonase-1 and ischemia-modified albumin in patients with end-stage renal disease. J. Physiol. Biochem. 2011, 67, 437–441. [Google Scholar] [CrossRef] [PubMed]
- Savic, J.; Zeljkovic, A.; Bogavac-Stanojevic, N.; Simic-Ogrizovic, S.; Kravljaca, M.; Stosovic, M.; Vekic, J.; Spasojevic-Kalimanovska, V.; Jelic-Ivanovic, Z.; Gojkovic, T.; et al. Association of small, dense low-density lipoprotein cholesterol and galectin-3 in patients with chronic kidney disease. Scand. J. Clin. Lab. Investig. 2014, 74, 637–643. [Google Scholar] [CrossRef] [PubMed]
- Mikolasevic, I.; Žutelija, M.; Mavrinac, V.; Orlic, L. Dyslipidemia in patients with chronic kidney disease: Etiology and management. Int. J. Nephrol. Renovasc. Dis. 2017, 10, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Ikewaki, K.; Schaefer, J.R.; Frischmann, M.E.; Okubo, K.; Hosoya, T.; Mochizuki, S.; Dieplinger, B.; Trenkwalder, E.; Schweer, H.; Kronenberg, F.; et al. Delayed in vivo catabolism of intermediate-density lipoprotein and low-density lipoprotein in hemodialysis patients as potential cause of premature atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 2615–2622. [Google Scholar] [CrossRef] [PubMed]
- Longenecker, J.C.; Coresh, J.; Powe, N.R.; Levey, A.S.; Fink, N.E.; Martin, A.; Klag, M.J. Traditional cardiovascular disease risk factors in dialysis patients compared with the general population: The CHOICE study. J. Am. Soc. Nephrol. 2002, 13, 1918–1927. [Google Scholar] [CrossRef]
- Qu, J.; Ko, C.W.; Tso, P.; Bhargava, A. Apolipoprotein A-IV: A multifunctional protein involved in protection against atherosclerosis and diabetes. Cells 2019, 8, 319. [Google Scholar] [CrossRef]
- Boes, E.; Fliser, D.; Ritz, E.; König, P.; Lhotta, K.; Mann, J.F.E.; Müller, G.A.; Neyer, U.; Riegel, W.; Riegler, P.; et al. Apolipoprotein A-IV predicts progression of chronic kidney disease: The mild to moderate kidney disease study. J. Am. Soc. Nephrol. 2006, 17, 528–536. [Google Scholar] [CrossRef]
- Rasool, M.; Ashraf, M.A.; Malik, A.; Waquar, S.; Khan, S.A.; Qazi, M.H.; Ahmad, W.; Asif, M.; Khan, S.U.; Zaheer, A.; et al. Comparative study of extrapolative factors linked with oxidative injury and anti-inflammatory status in chronic kidney disease patients experiencing cardiovascular distress. PLoS ONE 2017, 12, e0171561. [Google Scholar] [CrossRef]
- Feron, O.; Balligand, J.L. Caveolins and the regulation of endothelial nitric oxide synthase in the heart. Cardiovasc. Res. 2006, 69, 788–797. [Google Scholar] [CrossRef]
- Reddy, Y.S.; Kiranmayi, V.S.; Bitla, A.R.; Krishna, G.S.; Rao, P.V.; Sivakumar, V. Nitric oxide status in patients with chronic kidney disease. Indian J. Nephrol. 2015, 25, 287–291. [Google Scholar] [PubMed]
- Dobrian, A.D. ADMA and NOS regulation in chronic renal disease: Beyond the old rivalry for l-arginine. Kidney Int. 2012, 81, 722–724. [Google Scholar] [CrossRef] [PubMed]
- De Gennaro Colonna, V.; Bianchi, M.; Pascale, V.; Ferrario, P.; Morelli, F.; Pascale, W.; Tomasoni, L.; Turiel, M. Asymmetric dimethylarginine (ADMA): An endogenous inhibitor of nitric oxide synthase and a novel cardiovascular risk molecule. Med. Sci. Monit. 2009, 15, 91–101. [Google Scholar]
- Yilmaz, M.I.; Saglam, M.; Caglar, K.; Cakir, E.; Sonmez, A.; Ozgurtas, T.; Aydin, A.; Eyileten, T.; Ozcan, O.; Acikel, C.; et al. The determinants of endothelial dysfunction in CKD: Oxidative stress and asymmetric dimethylarginine. Am. J. Kidney Dis. 2006, 47, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Gryszczyńska, B.; Budzyń, M.; Formanowicz, D.; Wanic-Kossowska, M.; Formanowicz, P.; Majewski, W.; Iskra, M.; Kasprzak, M.P. Selected atherosclerosis-related diseases may differentially affect the relationship between plasma advanced glycation end products, receptor sRAGE, and uric acid. J. Clin. Med. 2020, 9, 1416. [Google Scholar] [CrossRef]
- Gryszczyńska, B.; Formanowicz, D.; Budzyń, M.; Wanic-Kossowska, M.; Pawliczak, E.; Formanowicz, P.; Iskra, M. Advanced oxidation protein products and carbonylated proteins as biomarkers of oxidative stress in selected atherosclerosis-mediated diseases. BioMed Res. Int. 2017, 2017, 4975264. [Google Scholar] [CrossRef]
- Donato, A.J.; Machin, D.R.; Lesniewski, L.A. Mechanisms of dysfunction in the aging vasculature and role in age-related disease. Circ. Res. 2018, 123, 825–848. [Google Scholar] [CrossRef]
- Vanholder, R.; Glorieux, G. The intestine and the kidneys: A bad marriage can be hazardous. Clin. Kidney J. 2015, 8, 168–179. [Google Scholar] [CrossRef]
- Kato, A.; Suzuki, Y.; Suda, T.; Suzuki, M.; Fujie, M.; Takita, T.; Furuhashi, M.; Maruyama, Y.; Chida, K.; Hishida, A. Relationship between an increased serum kynurenine/tryptophan ratio and atherosclerotic parameters in hemodialysis patients. Hemodial Int. 2010, 14, 418–424. [Google Scholar] [CrossRef]
- Sato, B.; Yoshikawa, D.; Ishii, H.; Suzuki, S.; Inoue, Y.; Takeshita, K.; Tanaka, M.; Kumagai, S.; Matsumoto, M.; Okumura, S.; et al. Relation of plasma indoxyl sulfate levels and estimated glomerular filtration rate to left ventricular diastolic dysfunction. Am. J. Cardiol. 2013, 111, 712–716. [Google Scholar] [CrossRef]
- Sallée, M.; Dou, L.; Cerini, C.; Poitevin, S.; Brunet, P.; Burtey, S. The aryl hydrocarbon receptor-activating effect of uremic toxins from tryptophan metabolism: A new concept to understand cardiovascular complications of chronic kidney disease. Toxins 2014, 6, 934–949. [Google Scholar] [CrossRef] [PubMed]
- Glorieux, G.; Gryp, T.; Perna, A. Gut-derived metabolites and their role in immune dysfunction in chronic kidney disease. Toxins 2020, 12, 245. [Google Scholar] [CrossRef] [PubMed]
- Von Leitner, E.C.; Klinke, A.; Atzler, D.; Slocum, J.L.; Lund, N.; Kielstein, J.T.; Maas, R.; Schmidt-Haupt, R.; Pekarova, M.; Hellwinkel, O.; et al. Pathogenic cycle between the endogenous nitric oxide synthase inhibitor asymmetrical dimethylarginine and the leukocyte-derived hemoprotein myeloperoxidase. Circulation 2011, 124, 2735–2745. [Google Scholar] [CrossRef] [PubMed]
- Vanholder, R.; Pletinck, A.; Schepers, E.; Glorieux, G. Biochemical and clinical impact of organic uremic retention solutes: A comprehensive update. Toxins 2018, 10, 33. [Google Scholar] [CrossRef] [PubMed]
- Sinha-Hikim, I.; Shen, R.; Paul Lee, W.N.; Crum, A.; Vaziri, N.D.; Norris, K.C. Effects of a novel cystine-based glutathione precursor on oxidative stress in vascular smooth muscle cells. Am. J. Physiol. Cell. Physiol. 2010, 299, 638–642. [Google Scholar] [CrossRef]
- Kalim, S.; Karumanchi, S.A.; Thadhani, R.I.; Berg, A.H. Protein carbamylation in kidney disease: Pathogenesis and clinical implications. Am. J. Kidney Dis. 2014, 64, 793–803. [Google Scholar] [CrossRef]
- Roumeliotis, S.; Roumeliotis, A.; Dounousi, E.; Eleftheriadis, T.; Liakopoulos, V. Dietary antioxidant supplements and uric acid in chronic kidney disease: A review. Nutrients 2019, 11, 1911. [Google Scholar] [CrossRef]
- Motojima, M.; Hosokawa, A.; Yamato, H.; Muraki, T.; Yoshioka, T. Uraemic toxins induce proximal tubular injury via organic anion transporter 1-mediated uptake. Br. J. Pharmacol. 2002, 135, 555–563. [Google Scholar] [CrossRef]
- Fujii, H.; Goto, S.; Fukagawa, M. Role of uremic toxins for kidney, cardiovascular, and bone dysfunction. Toxins 2018, 10, 202. [Google Scholar] [CrossRef]
- Gambardella, J.; De Rosa, M.; Sorriento, D.; Prevete, N.; Fiordelisi, A.; Ciccarelli, M.; Trimarco, B.; de Luca, N.; Iaccarino, G. Parathyroid hormone causes endothelial dysfunction by inducing mitochondrial ROS and specific oxidative signal transduction modifications. Oxid. Med. Cell. Longev. 2018, 2018, 9582319. [Google Scholar] [CrossRef]
- De Oliveira, R.B.; Liabeuf, S.; Okazaki, H.; Lenglet, A.; Desjardins, L.; Lemke, H.D.; Vanholder, R.; Choukroun, G.; Massy, Z.A.; European Uremic Toxin Work Group (EUTox). The clinical impact of plasma leptin levels in a cohort of chronic kidney disease patients. Clin. Kidney. J. 2013, 6, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Mao, S.; Fang, L.; Liu, F.; Jiang, S.; Wu, L.; Zhang, J. Leptin and chronic kidney diseases. J. Recept. Sig. Transd. 2018, 38, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Kuczera, P.; Adamczak, M.; Wiecek, A. Fibroblast growth factor-23-A potential uremic toxin. Toxins 2016, 8, 369. [Google Scholar] [CrossRef] [PubMed]
- Franca Gois, P.H.; Wolley, M.; Ranganathan, D.; Seguro, A.C. Vitamin D deficiency in chronic kidney disease: Recent evidence and controversies. Int. J. Environ. Res. Public Health 2018, 15, 1773. [Google Scholar] [CrossRef]
- Eftekhari, M.H.; Akbarzadeh, M.; Dabbaghmanesh, M.H.; Hassanzadeh, J. The effect of calcitriol on lipid profile and oxidative stress in hyperlipidemic patients with type 2 diabetes mellitus. ARYA Atheroscler. 2014, 10, 82–88. [Google Scholar]
- Wiseman, H. Vitamin D is a membrane antioxidant. Ability to inhibit iron-dependent lipid peroxidation in liposomes compared to cholesterol, ergosterol and tamoxifen and relevance to anticancer action. FEBS Lett. 1993, 326, 285–288. [Google Scholar] [CrossRef]
- Lin, A.M.; Chen, K.; Chao, P. Antioxidative effect of vitamin D3 on zinc-induced oxidative stress in CNS. Ann. N. Y. Acad. Sci. 2005, 1053, 319–329. [Google Scholar] [CrossRef]
- Zhong, W.; Gu, B.; Gu, Y.; Groome, L.J.; Sun, J.; Wang, Y. Activation of vitamin D receptor promotes VEGF and CuZn-SOD expression in endothelial cells. J. Steroid. Biochem. Mol. Biol. 2014, 140, 56–62. [Google Scholar] [CrossRef]
- Kanikarla-Marie, P.; Jain, S.K. 1,25(OH)2D3 inhibits oxidative stress and monocyte adhesion by mediating the upregulation of GCLC and GSH in endothelial cells treated with acetoacetate (ketosis). J. Steroid. Biochem. Mol. Biol. 2016, 159, 94–101. [Google Scholar] [CrossRef]
- Mokhtari, Z.; Hekmatdoost, A.; Nourian, M. Antioxidant efficacy of vitamin D. J. Parathyr. Dis. 2017, 5, 11–16. [Google Scholar]
- Pedruzzi, L.M.; Stockler-Pinto, M.B.; Leite, M., Jr.; Mafra, D. Nrf2-keap1 system versus NF-κB: The good and the evil in chronic kidney disease? Biochimie 2012, 94, 2461–2466. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, T.M.; da Costa, D.C.; Resende, A.C.; Soulage, C.O.; Bezerra, F.F.; Daleprane, J.B. Activation of Nrf2-antioxidant signaling by 1,25-dihydroxycholecalciferol prevents leptin-induced oxidative stress and inflammation in human endothelial cells. J. Nutr. 2017, 147, 506–513. [Google Scholar] [CrossRef] [PubMed]
- Nakai, K.; Fujii, H.; Kono, K.; Goto, S.; Kitazawa, R.; Kitazawa, S.; Hirata, M.; Shinohara, M.; Fukagawa, M.; Nishi, S. Vitamin D activates the Nrf2-Keap1 antioxidant pathway and ameliorates nephropathy in diabetic rats. Am. J. Hypertens. 2014, 27, 586–595. [Google Scholar] [CrossRef] [PubMed]
- Binswanger, U. Calcium metabolism and kidney disease. Ergeb. Inn. Med. Kinderheilkd. 1974, 34, 105–153. [Google Scholar]
- Bricker, N.S.; Slatopolsky, E.; Reiss, E.; Avioli, L.V. Calcium, phosphorus, and bone in renal disease and transplantation. Arch. Intern. Med. 1969, 123, 543–553. [Google Scholar] [CrossRef]
- Williams, S.; Malatesta, K.; Norris, K. Vitamin D and chronic kidney disease. Ethn. Dis. 2009, 19 (Suppl. 5), 5–11. [Google Scholar]
- Jones, G. Expanding role of vitamin D in chronic kidney disease: Importance of blood 25-OH-D levels and extra-renal 1α-hydroxylase in the classical and nonclassical actions of 1α,25-dihydroxyvitamin D3. Semin. Dial. 2007, 20, 316–324. [Google Scholar] [CrossRef]
- Canaff, L.; Hendy, G.N. Human calcium-sensing receptor gene. Vitamin D response elements in promoters P1 and P2 confer transcriptional responsiveness to 1,25-dihydroxyvitamin D. J. Biol. Chem. 2002, 277, 30337–30350. [Google Scholar] [CrossRef]
- Jean, G.; Souberbielle, J.C.; Chazot, C. Vitamin D in chronic kidney disease and dialysis Patients. Nutrients 2017, 9, 328. [Google Scholar] [CrossRef]
- Wolf, M.; Shah, A.; Gutierrez, O.; Ankers, E.; Monroy, M.; Tamez, H.; Steele, D.; Chang, Y.; Camargo, C.A.; Tonelli, M.; et al. Vitamin D levels and early mortality among incident hemodialysis patients. Kidney Int. 2007, 72, 1004–1013. [Google Scholar] [CrossRef]
- Rodriguez, M.; Lorenzo, V. Parathyroid hormone, a uremic toxin. Semin. Dial. 2009, 22, 363–368. [Google Scholar] [CrossRef] [PubMed]
- Feinfeld, D.A.; Sherwood, L.M. Parathyroid hormone and 1,25(OH)2D3 in chronic renal failure. Kidney Int. 1988, 33, 1049–1058. [Google Scholar] [CrossRef][Green Version]
- Hocher, B.; Armbruster, F.P.; Stoeva, S.; Reichetzeder, C.; Grön, H.J.; Lieker, I.; Khadzhynov, D.; Slowinski, T.; Roth, H.J. Measuring Parathyroid Hormone (PTH) in patients with oxidative stress—Do we need a fourth generation parathyroid hormone assay? PLoS ONE 2012, 7, e40242. [Google Scholar] [CrossRef] [PubMed]
- Jaqueto, M.; Delfino, V.D.; Bortolasci, C.C.; Barbosa, D.S.; Morimoto, H.K.; Frange, R.F.; Ferreira, L.F.; Guimarães, F.B. Are PTH levels related to oxidative stress and inflammation in chronic kidney disease patients on hemodialysis? J. Bras. Nefrol. 2016, 38, 288–295. [Google Scholar] [CrossRef] [PubMed]
- Noyan, T.; Avci, G.; Sekeroglu, M.R.; Erkoc, R. The investigation of relationship between secondary hyperparathyroidism and oxidative stress in patients with chronic kidney disease. Turk. Neph. Dial. Transpl. J. 2009, 18, 69–75. [Google Scholar]
- Nakatani, S.; Yasukawa, K.; Ishimura, E.; Nakatani, A.; Toi, N.; Uedono, H.; Tsuda, A.; Yamada, S.; Ikeda, H.; Mori, K.; et al. Non-mercaptalbumin, oxidized form of serum albumin, significantly associated with renal function and anemia in chronic kidney disease patients. Sci. Rep. 2018, 8, 16796. [Google Scholar] [CrossRef]
- Lishmanov, A.; Dorairajan, S.; Pak, Y.; Chaudhary, K.; Chockalingam, A. Elevated serum parathyroid hormone is a cardiovascular risk factor in moderate chronic kidney disease. Int. Urol. Nephrol. 2012, 44, 541–547. [Google Scholar] [CrossRef]
- Perkovic, V.; Hewitson, T.; Kelynack, K.; Martic, M.; Tait, M.; Becker, G. Parathyroid hormone has a prosclerotic effect on vascular smooth muscle cells. Kidney Blood Press. Res. 2003, 26, 27–33. [Google Scholar] [CrossRef]
- Kuro-o, M. Klotho and aging. Biochim. Biophys. Acta 2009, 1790, 1049–1058. [Google Scholar] [CrossRef]
- Wolf, M.T.; An, S.W.; Nie, M.; Bal, M.S.; Huang, C.L. Klotho up-regulates renal calcium channel transient receptor potential vanilloid 5 (TRPV5) by intra- and extracellular N-glycosylation-dependent mechanisms. J. Biol. Chem. 2014, 289, 35849–35857. [Google Scholar] [CrossRef]
- Jüppner, H. Phosphate and FGF-23. Kidney Int. Suppl. 2011, 121, 24–27. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhou, Y.; Graves, D.T. FOXO transcription factors: Their clinical significance and regulation. BioMed Res. Int. 2014, 2014, 925350. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Clark, J.D.; Pastor, J.V.; Gurnani, P.; Nandi, A.; Kurosu, H.; Miyoshi, M.; Ogawa, Y.; Castrillon, D.H.; Rosenblatt, K.P.; et al. Regulation of oxidative stress by the anti-aging hormone klotho. J. Biol. Chem. 2005, 280, 38029–38034. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Castañeda, J.R.; Rodelo-Haad, C.; Pendon-Ruiz de Mier, M.V.; Martin-Malo, A.; Santamaria, R.; Rodriguez, M. Klotho/FGF23 and Wnt signaling as important players in the comorbidities associated with chronic kidney disease. Toxins 2020, 12, 185. [Google Scholar] [CrossRef] [PubMed]
- Christman, M.A.; Goetz, D.J.; Dickerson, E.; McCall, K.D.; Lewis, C.J.; Benencia, F.; Silver, M.J.; Kohn, L.D.; Malgor, R. Wnt5a is expressed in murine and human atherosclerotic lesions. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, 2864–2870. [Google Scholar] [CrossRef]
- Milovanova, L.Y.; Dobrosmyslov, I.A.; Milovanov, Y.S.; Taranova, M.V.; Kozlov, V.V.; Milovanova, S.Y.; Kozevnikova, E.I. Fibroblast growth factor-23 (FGF-23)/soluble Klotho protein (sKlotho)/sclerostin glycoprotein ratio disturbance is a novel risk factor for cardiovascular complications in ESRD patients receiving treatment with regular hemodialysis or hemodiafiltration. Ter. Arkh. 2018, 90, 48–55. [Google Scholar] [CrossRef]
- Wang, Q.; Su, W.; Shen, Z.; Wang, R. Correlation between soluble α-klotho and renal function in patients with chronic kidney disease: A review and meta-analysis. BioMed Res. Int. 2018, 2018, 9481475. [Google Scholar] [CrossRef]
- Liu, Q.F.; Yu, L.X.; Feng, J.H.; Sun, Q.; Li, S.S.; Ye, J.M. The prognostic role of klotho in patients with chronic kidney disease: A systematic review and meta-analysis. Dis. Markers 2019, 2019, 6468729. [Google Scholar] [CrossRef]
- Dalton, G.D.; Xie, J.; An, S.W.; Huang, C.L. New insights into the mechanism of action of soluble klotho. Front. Endocrinol. 2017, 8, 323. [Google Scholar] [CrossRef]
- Raeisi, S.; Ghorbanihaghjo, A.; Argani, H.; Dastmalchi, S.; Ghasemi, B.; Ghazizadeh, T.; Rashtchizadeh, N.; Nemati, M.; Mesgari Abbasi, M.; Bargahi, N.; et al. Effects of angiotensin II receptor blockade on soluble klotho and oxidative stress in calcineurin inhibitor nephrotoxicity in rats. Iran. J. Kidney Dis. 2016, 10, 358–363. [Google Scholar]
- Oh, H.J.; Nam, B.Y.; Lee, M.J.; Kim, C.H.; Koo, H.M.; Doh, F.M.; Han, J.H.; Kim, E.J.; Han, J.S.; Park, J.T.; et al. Decreased circulating klotho levels in patients undergoing dialysis and relationship to oxidative stress and inflammation. Perit. Dial. Int. 2015, 35, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Nitta, K.; Nagano, N.; Tsuchiya, K. Fibroblast growth factor 23/klotho axis in chronic kidney disease. Nephron Clin. Pract. 2014, 128, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, O.; Isakova, T.; Rhee, E.; Shah, A.; Holmes, J.; Collerone, G.; Jüppner, H.; Wolf, M. Fibroblast growth factor-23 mitigates hyperphosphatemia but accentuates calcitriol deficiency in chronic kidney disease. J. Am. Soc. Nephrol. 2005, 16, 2205–2215. [Google Scholar] [CrossRef] [PubMed]
- Freemont, T. Histological diagnosis of renal osteodystrophy. Kidney Int. Suppl. 1999, 73, 26–30. [Google Scholar] [CrossRef] [PubMed]
- Martin, K.J.; González, E.A. Metabolic bone disease in chronic kidney disease. J. Am. Soc. Nephrol. 2007, 18, 875–885. [Google Scholar] [CrossRef]
- Brandenburg, V.M.; Floege, J. Adynamic bone disease-bone and beyond. NDT Plus 2008, 1, 135–147. [Google Scholar] [CrossRef][Green Version]
- Kendrick, J.; Chonchol, M. The role of phosphorus in the development and progression of vascular calcification. Am. J. Kidney Dis. 2011, 58, 826–834. [Google Scholar] [CrossRef]
- Schlieper, G.; Schurgers, L.; Brandenburg, V.; Reutelingsperger, C.; Floege, J. Vascular calcification in chronic kidney disease: An update. Nephrol. Dial. Transplant. 2016, 31, 31–39. [Google Scholar] [CrossRef]
- Westenfeld, R.; Jahnen-Dechent, W.; Ketteler, M. Vascular calcification and fetuin-a deficiency in chronic kidney disease. Trends Cardiovasc. Med. 2007, 17, 124–128. [Google Scholar] [CrossRef]
- Westenfeld, R.; Schäfer, C.; Krüger, T.; Haarmann, C.; Schurgers, L.J.; Reutelingsperger, C.; Ivanovski, O.; Drueke, T.; Massy, Z.A.; Ketteler, M.; et al. Fetuin-A protects against atherosclerotic calcification in CKD. J. Am. Soc. Nephrol. 2009, 20, 1264–1274. [Google Scholar] [CrossRef]
- Sage, A.P.; Lu, J.; Tintut, Y.; Demer, L.L. Hyperphosphatemia-induced nanocrystals upregulate the expression of bone morphogenetic protein-2 and osteopontin genes in mouse smooth muscle cells in vitro. Kidney Int. 2011, 79, 414–422. [Google Scholar] [CrossRef] [PubMed]
- Virani, S.A.; Khosla, A.; Levin, A. Chronic kidney disease, heart failure and anemia. Can. J. Cardiol. 2008, 24 (Suppl. B), 22–24. [Google Scholar] [CrossRef]
- Besarab, A.; Ayyoub, F. Anemia in renal disease. In Diseases of the Kidney and Urinary Tract, 8th ed.; Schrier, R.W., Ed.; Lippincott Williams and Wilkins: Philadelphia, PA, USA, 2007; pp. 2406–2430. [Google Scholar]
- Bissinger, R.; Bhuyan, A.A.M.; Qadri, S.M.; Lang, F. Oxidative stress, eryptosis and anemia: A pivotal mechanistic nexus in systemic diseases. FEBS J. 2019, 286, 826–854. [Google Scholar] [CrossRef] [PubMed]
- Pieniążek, A.; Gwoździński, K. Carbamylation of proteins leads to alterations in the membrane structure of erythrocytes. Cell. Mol. Biol. Lett. 2003, 8, 127–131. [Google Scholar]
- Bonan, N.B.; Steiner, T.M.; Kuntsevich, V.; Virzi, G.M.; Azevedo, M.; Nakao, L.S.; Barreto, F.C.; Ronco, C.; Thijssen, S.; Kotanko, P.; et al. Uremic toxicity-induced eryptosis and monocyte modulation: The erythrophagocytosis as a novel pathway to renal anemia. Blood Purif. 2016, 41, 317–323. [Google Scholar] [CrossRef]
- Wagner, M.; Alam, A.; Zimmermann, J.; Rauh, K.; Koljaja-Batzner, A.; Raff, U.; Wanner, C.; Schramm, L. Endogenous erythropoietin and the association with inflammation and mortality in diabetic chronic kidney disease. Clin. J. Am. Soc. Nephrol. 2011, 6, 1573–1579. [Google Scholar] [CrossRef]
- Van der Putten, K.; Braam, B.; Jie, K.E.; Gaillard, C.A. Mechanisms of disease: Erythropoietin resistance in patients with both heart and kidney failure. Nat. Clin. Pract. Nephrol. 2008, 4, 47–57. [Google Scholar] [CrossRef]
- Khalil, S.K.; Amer, H.A.; El Behairy, A.M.; Warda, M. Oxidative stress during erythropoietin hyporesponsiveness anemia at end stage renal disease: Molecular and biochemical studies. J. Adv. Res. 2016, 7, 348–358. [Google Scholar] [CrossRef][Green Version]
- Morceau, F.; Dicato, M.; Diederich, M. Pro-inflammatory cytokine-mediated anemia: Regarding molecular mechanisms of erythropoiesis. Mediat. Inflamm. 2009, 2009, 405016. [Google Scholar] [CrossRef]
- Ikonomi, P.; Rivera, C.E.; Riordan, M.; Washington, G.; Schechter, A.N.; Noguchi, C.T. Overexpression of GATA-2 inhibits erythroid and promotes megakaryocyte differentiation. Exp. Hematol. 2000, 28, 1423–1431. [Google Scholar] [CrossRef]
- Ganz, T.; Nemeth, E. Iron balance and the role of hepcidin in chronic kidney disease. Semin. Nephrol. 2016, 36, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, E.; Rivera, S.; Gabayan, V.; Keller, C.; Taudorf, S.; Pedersen, B.K.; Ganz, T. IL-6 mediates hypoferremia of inflammation by inducing the synthesis of the iron regulatory hormone hepcidin. J. Clin. Investig. 2004, 113, 1271–1276. [Google Scholar] [PubMed]
- Nicolas, G.; Chauvet, C.; Viatte, L.; Danan, J.L.; Bigard, X.; Devaux, I.; Beaumont, C.; Kahn, A.; Vaulont, S. The gene encoding the iron regulatory peptide hepcidin is regulated by anemia, hypoxia, and inflammation. J. Clin. Investig. 2002, 110, 1037–1044. [Google Scholar] [PubMed]
- Maruyama, Y.; Yokoyama, K.; Yamamoto, H.; Nakayama, M.; Hosoya, T. Do serum hepcidin-25 levels correlate with oxidative stress in patients with chronic kidney disease not receiving dialysis? Clin. Nephrol. 2012, 78, 281–286. [Google Scholar]
- Kumagai, T.; Matsukawa, N.; Kaneko, Y.; Kusumi, Y.; Mitsumata, M.; Uchida, K. A lipid peroxidation-derived inflammatory mediator: Identification of 4-hydroxy-2-nonenal as a potential inducer of cyclooxygenase-2 in macrophages. J. Biol. Chem. 2004, 279, 48389–48396. [Google Scholar]
- Formanowicz, D.; Radom, M.; Rybarczyk, A.; Formanowicz, P. The role of Fenton reaction in ROS-induced toxicity underlying atherosclerosis—Modeled and analyzed using a Petri net-based approach. Biosystems 2018, 165, 71–87. [Google Scholar] [CrossRef]
- Liakopoulos, V.; Roumeliotis, S.; Gorny, X.; Dounousi, E.; Mertens, P.R. Oxidative stress in hemodialysis patients: A review of the literature. Oxid. Med. Cell. Longev. 2017, 2017, 3081856. [Google Scholar] [CrossRef]
- Alhamdani, M.S.; Al-Kassir, A.H.; Jaleel, N.A.; Hmood, A.M.; Ali, H.M. Elevated levels of alkanals, alkenals and 4-HO-alkenals in plasma of hemodialysis patients. Am. J. Nephrol. 2006, 26, 299–303. [Google Scholar] [CrossRef]
- Malindretos, P.; Sarafidis, P.A.; Rudenco, I.; Raptis, V.; Makedou, K.; Makedou, A.; Grekas, D.M. Slow intravenous iron administration does not aggravate oxidative stress and inflammatory biomarkers during hemodialysis: A comparative study between iron sucrose and iron dextran. Am. J. Nephrol. 2007, 27, 572–579. [Google Scholar] [CrossRef]
- Toblli, J.E.; Cao, G.; Giani, J.F.; Dominici, F.P.; Angerosa, M. Markers of oxidative/nitrosative stress and inflammation in lung tissue of rats exposed to different intravenous iron compounds. Drug Des. Devel. Ther. 2017, 11, 2251–2263. [Google Scholar] [CrossRef]
- Toblli, J.E.; Cao, G.; Giani, J.F.; Dominici, F.P.; Angerosa, M. Nitrosative stress and apoptosis by intravenous ferumoxytol, iron isomaltoside 1000, iron dextran, iron sucrose, and ferric carboxymaltose in a nonclinical model. Drug Res. 2015, 65, 354–360. [Google Scholar] [CrossRef] [PubMed]
- Toblli, J.E.; Cao, G.; Oliveri, L.; Angerosa, M. Assessment of the extent of oxidative stress induced by intravenous ferumoxytol, ferric carboxymaltose, iron sucrose and iron dextran in a dextran in a nonclinical model. Arzneimittelforschung 2011, 61, 399–410. [Google Scholar] [CrossRef] [PubMed]
- Culleton, B.F.; Larson, M.G.; Wilson, P.W.; Evans, J.C.; Parfrey, P.S.; Levy, D. Cardiovascular disease and mortality in a community-based cohort with mild renal insufficiency. Kidney Int. 1999, 56, 2214–2219. [Google Scholar] [CrossRef] [PubMed]
- Manjunath, G.; Tighiouart, H.; Ibrahim, H.; MacLeod, B.; Salem, D.N.; Griffith, J.L.; Coresh, J.; Levey, A.S.; Sarnak, M.L. Level of kidney function as a risk factor for atherosclerotic cardiovascular outcomes in the community. J. Am. Coll. Cardiol. 2003, 41, 47–55. [Google Scholar] [CrossRef]
- Garg, A.X.; Clark, W.F.; Haynes, R.B.; House, A.A. Moderate renal insufficiency and the risk of cardiovascular mortality: Results from the NHANES I. Kidney Int. 2002, 61, 1486–1494. [Google Scholar] [CrossRef]
- Muntner, P.; He, J.; Hamm, L.; Loria, C.; Whelton, P.K. Renal insufficiency and subsequent death resulting from cardiovascular disease in the United States. J. Am. Soc. Nephrol. 2002, 13, 745–753. [Google Scholar]
- Weiner, D.E.; Tighiouart, H.; Amin, M.G.; Stark, P.C.; MacLeod, B.; Griffith, J.L.; Salem, D.N.; Levey, A.S.; Sarnak, M.J. Chronic kidney disease as a risk factor for cardiovascular disease and all-cause mortality: A pooled analysis of community-based studies. J. Am. Soc. Nephrol. 2004, 15, 1307–1315. [Google Scholar] [CrossRef]
- Schillaci, G.; Reboldi, G.; Verdecchia, P. High-normal serum creatinine concentration is a predictor of cardiovascular risk in essential hypertension. Arch. Intern. Med. 2001, 161, 886–891. [Google Scholar] [CrossRef]
- Go, A.S.; Chertow, G.M.; Fan, D.; McCulloch, C.E.; Hsu, C.Y. Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. N. Engl. J. Med. 2004, 351, 1296–1305. [Google Scholar] [CrossRef]
- Zoccali, C. Traditional and emerging cardiovascular and renal risk factors: An epidemiologic perspective. Kidney Int. 2006, 70, 26–33. [Google Scholar] [CrossRef]
- Tripepi, G.; Mallamaci, F.; Zoccali, C. Inflammation markers, adhesion molecules, and all-cause and cardiovascular mortality in patients with ESRD: Searching for the best risk marker by multivariate modeling. J. Am. Soc. Nephrol. 2005, 16 (Suppl. 1), 83–88. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Study Group | ||||
|---|---|---|---|---|---|
| Non-Dialyzed CKD vs. HV | HD | before HD vs. after HD | PD | Nephrotic Syndrome | |
| E-GPx | ↑ [44,73] ↓ [68,69,71,74,79] N [58,76,78] | ↑ [44] ↓↓ [69,70,74,75,78,79] N [58,68,71,80] | no change [68,70,71,80] ↘ [75] | ↓ [74,78] N [58,71,78] | ↓ [67,68] |
| P-GPx | ↘ [44] ↓ [67,68,69,73,76] | ↓↓ [44,58,68,69,80,81,82] | ↗ [68,80] | ↓ [44,58] | ↓ [67] N [68] |
| SOD-1 | ↑ [38,41,72,73] ↗ [74] ↓ [77] N [27,44,76,78,79] | ↑ [41,72,80] ↓ [27,38,74,75,77,78] N [44] | ↘ [80] | N [44] ↓ [74,78] | ↓ [67] |
| CAT | ↘ [74,78] | ↑ [69] ↓↓ [74,77,78,79] | ↗ [77] | ↓↓ [74,78] | |
| Parameter | Trends of Changes in the Course of Deterioration of Kidney Function (CKD Stages from 1 to 5) | HD | PD |
|---|---|---|---|
| Total cholesterol | ↗ | N or↓ | ↑ |
| LDL cholesterol | ↗ | N or ↓ | ↑ |
| Lp (a) | ↑ | ↑↑ | ↑ |
| HDL cholesterol | ↓ | ↓ | ↓ |
| Non–HDL cholesterol (includes cholesterol in LDL, VLDL, IDL, and chylomicron and its remnant) | ↗ | N or↓ | ↑ |
| ApoA-I | ↘ | ↓ | ↓ |
| ApoA-IV | ↗ | ↑ | ↑ |
| ApoB | ↗ | N or ↓ | ↑ |
| Triglycerides | ↗ | ↑ | ↑↑ |
| Uremic Toxin | Impact of the Uremic Toxin on Complications Observed in CKD | Relations to Oxidative Stress in CKD | References |
|---|---|---|---|
| Low Water-Soluble Molecules (Molecular Weight < 500 Da) | |||
| ADMA | hypertension; atherosclerosis | MPO secretion | [181,193] |
| Polyamine | anemia; suppression of the immune system coagulation disorders | glutathione metabolism (VSMC) | [194,195] |
| Carbamylated compounds | kidney damage; anemia; atherosclerosis; calciphylaxis | respiratory burst (neutrophils) | [196] |
| Uric acid | kidney damage; chronic inflammation; hypertension; atherosclerosis | ROS release from mitochondria | [194,197] |
| Protein-Bound Molecules | |||
| AGEs | kidney damage; chronic inflammation; hypertension; atherosclerosis; dialysis amyloidosis | activity of NADPH oxidase; ROS release from mitochondria | [158,193] |
| AOPPs | kidney damage; chronic inflammation; hypertension; atherosclerosis; cardiomyopathy | activity of NADPH oxidase; ROS release from mitochondria | [153,154] |
| CMPF—a metabolite of furan fatty acid and a marker of fish oil intake | metabolism disruption; hepatopathy | glutathione metabolism (hepatocytes) | [194] |
| O-hydroxy hippuric acid | kidney damage | ROS generation (renal tubules) | [198] |
| P-cresol sulfate | kidney damage; chronic inflammation | ROS generation (leucocytes) | [199] |
| Indoxyl sulfate; Indole 3-acetic acid; Indoxyl-β-D-glucuronide | kidney damage; hypertension; atherosclerosis; calciphylaxis; prothrombotic effect | activity of NADPH oxidase | [132,191,199] |
| Kynurenine; 3-hydroxy kynurenine; Kynurenic acid; Quinolinic acid | anemia; hypertension; atherosclerosis; calciphylaxis; prothrombotic effect | activity of NADPH oxidase | [191,199] |
| Phenylacetic acid | chronic inflammation; hypertension; atherosclerosis; renal osteodystrophy | ROS generation (VSMC) | [194] |
| Larger Middle Molecules (Molecular Weight > 500 Da) | |||
| IL-1β, IL-6, IL-8, IL-18, TNF-α | chronic inflammation; atherosclerosis | respiratory burst (neutrophils) | [105,200] |
| Ghrelin | water and electrolyte disturbances endocrine disorders atherosclerosis | ROS release from mitochondria | [194] |
| PTH | secondary hyperparathyroidism | ROS release from mitochondria | [94] |
| Leptin | chronic inflammation anemia hypertension; atherosclerosis; suppression of the immune system | ROS release from mitochondria | [201,202] |
| FGF-23 | chronic inflammation hypertension atherosclerosis calciphylaxis cardiomyopathy | ROS release from mitochondria | [203] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Podkowińska, A.; Formanowicz, D. Chronic Kidney Disease as Oxidative Stress- and Inflammatory-Mediated Cardiovascular Disease. Antioxidants 2020, 9, 752. https://doi.org/10.3390/antiox9080752
Podkowińska A, Formanowicz D. Chronic Kidney Disease as Oxidative Stress- and Inflammatory-Mediated Cardiovascular Disease. Antioxidants. 2020; 9(8):752. https://doi.org/10.3390/antiox9080752
Chicago/Turabian StylePodkowińska, Alina, and Dorota Formanowicz. 2020. "Chronic Kidney Disease as Oxidative Stress- and Inflammatory-Mediated Cardiovascular Disease" Antioxidants 9, no. 8: 752. https://doi.org/10.3390/antiox9080752
APA StylePodkowińska, A., & Formanowicz, D. (2020). Chronic Kidney Disease as Oxidative Stress- and Inflammatory-Mediated Cardiovascular Disease. Antioxidants, 9(8), 752. https://doi.org/10.3390/antiox9080752