The BACH1/Nrf2 Axis in Brain in Down Syndrome and Transition to Alzheimer Disease-Like Neuropathology and Dementia

 ,
,  and
and
Abstract
:1. Genetics of Oxidative Stress in Down Syndrome
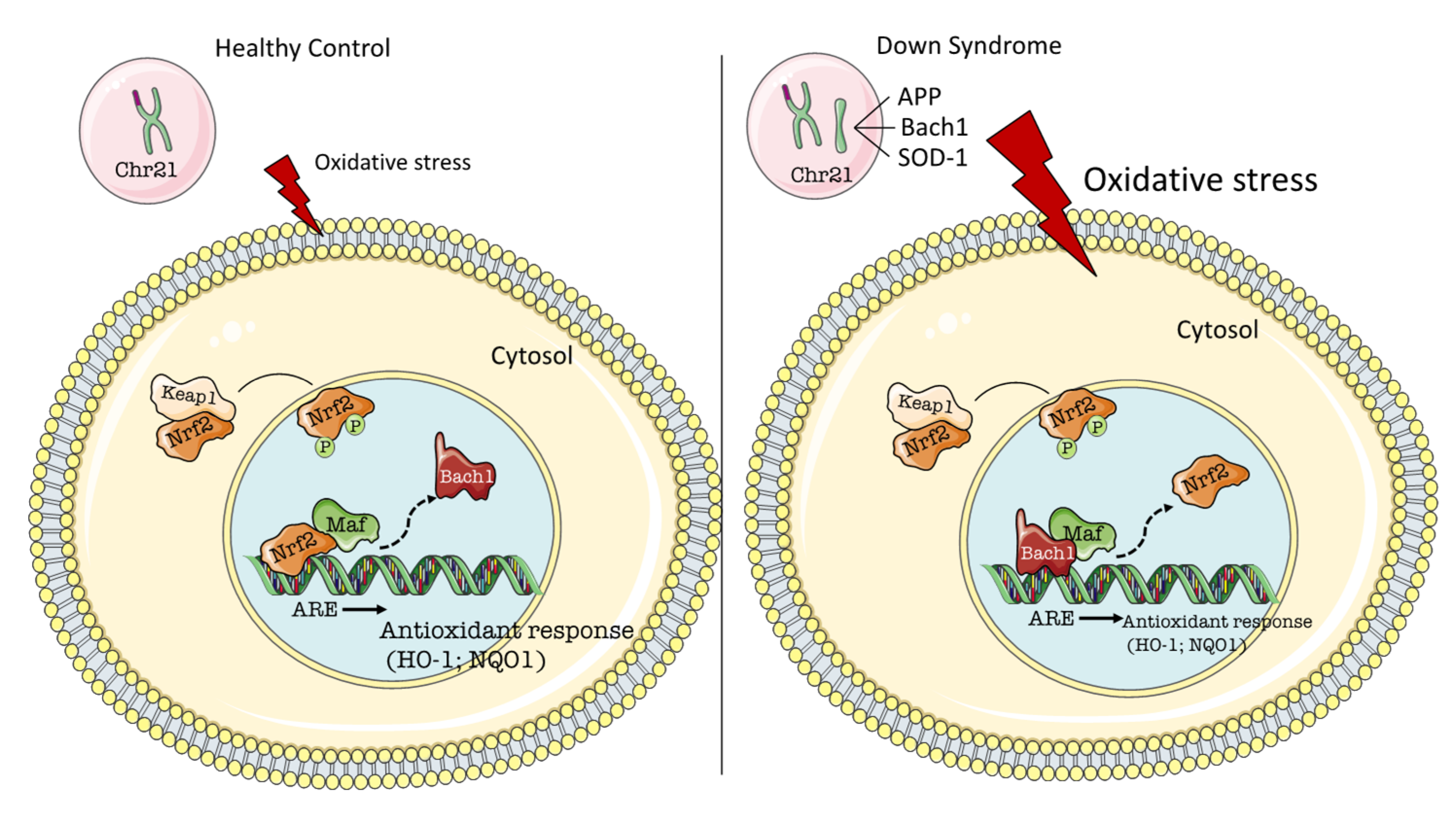
2. BACH1/Nrf2 Signaling
3. Involvement of BACH1 in AD and DS
4. Concluding Remarks
Funding
Conflicts of Interest
Abbreviations
| Aβ | amyloid beta-peptide |
| AD | Alzheimer disease |
| APP | amyloid precursor protein |
| ARE | antioxidant response element |
| BACH1 | the transcription factor BTB and CNC homology 1 |
| BVR-A | biliverdin reductase A |
| CAT | catalase |
| CBR | carbonyl reductase |
| CO | carbon monoxide |
| DS | Down syndrome |
| DSAD | Down syndrome with Alzheimer disease |
| ETS2 | the Protein C-ets-2 |
| HNE | 4-hydroxy-2-nonenal |
| HO-1 | heme oxygenase 1 |
| HSA21 | human chromosome 21 |
| Keap1 | Kelch-like ECH-Associating protein 1 |
| MAPK | mitogen-activated protein kinase |
| MAPT | microtubule associated protein tau |
| NFT | neurofibrillary tangles |
| NO | nitric oxide |
| NQO1 | NADPH quinone oxidoreductase 1 |
| Nrf2 | nuclear factor erythroid 2 related factor 2 |
| OXOPHOS | oxidative phosphorylation |
| OS | oxidative stress |
| ROS | reactive oxygen species |
| SOD1 | Cu, Zn superoxide dismutase 1 |
| MnSOD | manganese superoxide dismutase |
| SP | senile plaques |
| S100B | S100 calcium-binding protein B |
References
- Cenini, G.; Dowling, A.-L.; Beckett, T.-L.; Barone, E.; Mancuso, C.; Murphy, M.-P.; Levine, H.; Lott, I.-T.; Schmitt, F.-A.; Butterfield, D.A.; et al. Association between frontal cortex oxidative damage and beta-amyloid as a function of age in Down syndrome. Biochim. Biophys. Acta 2012, 1822, 130–138. [Google Scholar] [CrossRef]
- Lott, I.T. Neurological phenotypes for Down syndrome across the life span. Prog. Brain Res. 2012, 197, 101–121. [Google Scholar] [PubMed] [Green Version]
- Lu, J.; McCarter, M.; Lian, G.; Esposito, G.; Capoccia, E.; Delli-Bovi, L.C.; Hecht, J.; Sheen, V. Global hypermethylation in fetal cortex of Down syndrome due to DNMT3L overexpression. Hum. Mol. Genet. 2016, 25, 1714–1727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conti, A.; Fabbrini, F.; D’Agostino, P.; Negri, R.; Greco, D.; Genesio, R.; D’Armiento, M.; Olla, C.; Paladini, D.; Zannini, M.; et al. Altered expression of mitochondrial and extracellular matrix genes in the heart of human fetuses with chromosome 21 trisomy. BMC Genom. 2007, 8, 268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Domenico, F.; Pupo, G.; Tramutola, A.; Giorgi, A.; Schinina, M.E.; Coccia, R.; Head, E.; Butterfield, D.A.; Perluigi, M. Redox proteomics analysis of HNE-modified proteins in Down syndrome brain: Clues for understanding the development of Alzheimer disease. Free Radic. Biol. Med. 2014, 71, 270–280. [Google Scholar] [CrossRef] [Green Version]
- Di Domenico, F.; Pupo, G.; Mancuso, C.; Barone, E.; Paolini, F.; Arena, A.; Blarzino, C.; Schmitt, F.-A.; Head, E.; Butterfield, D.-A.; et al. Bach1 overexpression in Down syndrome correlates with the alteration of the HO-1/BVR-a system: Insights for transition to Alzheimer’s disease. J. Alzheimer’s Dis. 2015, 44, 1107–1120. [Google Scholar] [CrossRef] [Green Version]
- Montine, T.-J.; Neely, M.-D.; Quinn, J.-F.; Beal, M.-F.; Markesbery, W.R.; Roberts, L.-J.; Morrow, J.-D. Lipid peroxidation in aging brain and Alzheimer’s disease. Free Radic. Biol. Med. 2002, 33, 620–626. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Halliwell, B. Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat. Rev. Neurosci. 2019, 20, 148–160. [Google Scholar] [CrossRef]
- Halliwell, B. Reactive oxygen species in living systems: Source, biochemistry, and role in human disease. Am. J. Med. 1991, 91, 14S–22S. [Google Scholar] [CrossRef]
- Cenini, G.; Lloret, A.; Cascella, R. Oxidative Stress in Neurodegenerative Diseases: From a Mitochondrial Point of View. Oxid. Med. Cell. Longev. 2019, 2019, 2105607. [Google Scholar] [CrossRef] [Green Version]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003, 552, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valenti, D.; Manente, G.-A.; Moro, L.; Marra, E.; Vacca, R.A. Deficit of complex I activity in human skin fibroblasts with chromosome 21 trisomy and overproduction of reactive oxygen species by mitochondria: Involvement of the cAMP/PKA signalling pathway. Biochem. J. 2011, 435, 679–688. [Google Scholar] [CrossRef] [Green Version]
- Zana, M.; Janka, Z.; Kalman, J. Oxidative stress: A bridge between Down’s syndrome and Alzheimer’s disease. Neurobiol. Aging 2007, 28, 648–676. [Google Scholar] [CrossRef]
- Capone, G.T. Down syndrome: Advances in molecular biology and the neurosciences. J. Dev. Behav. Pediatr. 2001, 22, 40–59. [Google Scholar] [CrossRef] [Green Version]
- Pallardo, F.V.; Degan, P.; d’Ischia, M.; Kelly, F.J.; Zatterale, A.; Calzone, R.; Castello, G.; Fernandez-Delgado, R.; Dunster, C.; Lloret, A.; et al. Multiple evidence for an early age pro-oxidant state in Down Syndrome patients. Biogerontology 2006, 7, 211–220. [Google Scholar] [CrossRef]
- Nunomura, A.; Perry, G.; Aliev, G.; Hirai, K.; Takeda, A.; Balraj, E.K.; Jones, P.K.; Ghanbari, H.; Wataya, T.; Shimohama, S.; et al. Oxidative damage is the earliest event in Alzheimer disease. J. Neuropathol. Exp. Neurol. 2001, 60, 759–767. [Google Scholar] [CrossRef] [Green Version]
- Barone, E.; Head, E.; Butterfield, D.-A. HNE-modified proteins in Down syndrome: Involvement in development of Alzheimer disease neuropathology. Free Radic. Biol. Med. 2017, 111, 262–269. [Google Scholar] [CrossRef] [Green Version]
- Perluigi, M.; Butterfield, D.A. Oxidative Stress and Down Syndrome: A Route toward Alzheimer-Like Dementia. Curr. Gerontol. Geriatr. Res. 2012, 2012, 724904. [Google Scholar] [CrossRef] [Green Version]
- Butterfield, D.-A.; Perluigi, M.; Reed, T.; Muharib, T.; Hughes, C.-P.; Robinson, R.-A.; Sultana, R. Redox proteomics in selected neurodegenerative disorders: From its infancy to future applications. Antioxid. Redox Signal. 2012, 17, 1610–1655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Head, E.; Lott, I.-T.; Wilcock, D.-M.; Lemere, C.A. Aging in Down Syndrome and the Development of Alzheimer’s Disease Neuropathology. Curr. Alzheimer Res. 2016, 13, 18–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perluigi, M.; di Domenico, F.; Buttterfield, D.A. Unraveling the complexity of neurodegeneration in brains of subjects with Down syndrome: Insights from proteomics. Proteom. Clin. Appl. 2014, 8, 73–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barone, E.; Arena, A.; Head, E.; Butterfield, D.-A.; Perluigi, M. Disturbance of redox homeostasis in Down Syndrome: Role of iron dysmetabolism. Free Radic. Biol. Med. 2018, 114, 84–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valenti, D.; Braidy, N.; De Rasmo, D.; Signorile, A.; Rossi, L.; Atanasov, A.G.; Volpicella, M.; Henrion-Caude, A.; Nabavi, S.M.; Vacca, R.A. Mitochondria as pharmacological targets in Down syndrome. Free Radic. Biol. Med. 2018, 114, 69–83. [Google Scholar] [CrossRef] [PubMed]
- Busciglio, J.; Yankner, B.A. Apoptosis and increased generation of reactive oxygen species in Down’s syndrome neurons in vitro. Nature 1995, 378, 776–779. [Google Scholar] [CrossRef]
- Pratico, D.; Iuliano, L.; Amerio, G.; Tang, L.-X.; Rokach, J.; Sabatino, G.; Violi, F. Down’s syndrome is associated with increased 8,12-iso-iPF2alpha-VI levels: Evidence for enhanced lipid peroxidation in vivo. Ann. Neurol. 2000, 48, 795–798. [Google Scholar] [CrossRef]
- Valenti, D.; de Bari, L.; De Filippis, B.; Henrion-Caude, A.; Vacca, R. Mitochondrial dysfunction as a central actor in intellectual disability-related diseases: An overview of Down syndrome, autism, Fragile X and Rett syndrome. Neurosci. Biobehav. Rev. 2014, 46, 202–217. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Swomley, A.M.; Sultana, R. Amyloid beta-peptide (1-42)-induced oxidative stress in Alzheimer disease: Importance in disease pathogenesis and progression. Antioxid. Redox Signal. 2013, 19, 823–835. [Google Scholar] [CrossRef] [Green Version]
- Mao, P.; Reddy, P.H. Aging and amyloid beta-induced oxidative DNA damage and mitochondrial dysfunction in Alzheimer’s disease: Implications for early intervention and therapeutics. Biochim. Biophys. Acta 2011, 1812, 1359–1370. [Google Scholar] [CrossRef] [Green Version]
- Butterfield, D.A.; Boyd-Kimball, D. Redox proteomics and amyloid beta-peptide: Insights into Alzheimer disease. J. Neurochem. 2019, 151, 459–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Head, E.; Lott, I.T. Down syndrome and beta-amyloid deposition. Curr. Opin. Neurol. 2004, 17, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Mehta, P.D.; Capone, G.; Jewell, A. Increased amyloid beta protein levels in children and adolescents with Down syndrome. J. Neurol. Sci. 2007, 254, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Anandatheerthavarada, H.-K.; Biswas, G.; Robin, M.-A.; Avadhani, N.G. Mitochondrial targeting and a novel transmembrane arrest of Alzheimer’s amyloid precursor protein impairs mitochondrial function in neuronal cells. J. Cell Biol. 2003, 161, 41–54. [Google Scholar] [CrossRef]
- Simon, A.M.; Schiapparelli, L.; Salazar-Colocho, P.; Cuadrado-Tejedor, M.; Escribano, L.; Lopez de Maturana, R.; Del Rio, J.; Perez-Mediavilla, A.; Frechilla, D. Overexpression of wild-type human APP in mice causes cognitive deficits and pathological features unrelated to Abeta levels. Neurobiol. Dis. 2009, 33, 369–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Guo, J.; Wei, X.; Niu, C.; Jia, M.; Li, Q.; Meng, D. Bach1: Function, Regulation, and Involvement in Disease. Oxid. Med. Cell. Longev. 2018, 2018, 1347969. [Google Scholar] [CrossRef]
- Balcz, B.; Kirchner, L.; Cairns, N.; Fountoulakis, M.; Lubec, G. Increased brain protein levels of carbonyl reductase and alcohol dehydrogenase in Down syndrome and Alzheimer’s disease. J. Neural Transm. 2001, 61, 193–201. [Google Scholar]
- Di Domenico, F.; Coccia, R.; Cocciolo, A.; Murphy, M.P.; Cenini, G.; Head, E.; Butterfield, D.A.; Giorgi, A.; Schinina, M.E.; Mancuso, C.; et al. Impairment of proteostasis network in Down syndrome prior to the development of Alzheimer’s disease neuropathology: Redox proteomics analysis of human brain. Biochim. Biophys. Acta 2013, 1832, 1249–1259. [Google Scholar] [CrossRef]
- Butterfield, D.-A.; Di Domenico, F.; Swomley, A.M.; Head, E.; Perluigi, M. Redox proteomics analysis to decipher the neurobiology of Alzheimer-like neurodegeneration: Overlaps in Down’s syndrome and Alzheimer’s disease brain. Biochem. J. 2014, 463, 177–189. [Google Scholar] [CrossRef] [Green Version]
- Butterfield, D.A.; Boyd-Kimball, D. Oxidative Stress, Amyloid-beta Peptide, and Altered Key Molecular Pathways in the Pathogenesis and Progression of Alzheimer’s Disease. J. Alzheimer’s Dis. 2018, 62, 1345–1367. [Google Scholar] [CrossRef] [Green Version]
- Sultana, R.; Perluigi, M.; Butterfield, D.A. Oxidatively modified proteins in Alzheimer’s disease (AD), mild cognitive impairment and animal models of AD: Role of Abeta in pathogenesis. Acta Neuropathol. 2009, 118, 131–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMahon, M.; Thomas, N.; Itoh, K.; Yamamoto, M.; Hayes, J.-D. Dimerization of substrate adaptors can facilitate cullin-mediated ubiquitylation of proteins by a “tethering” mechanism: A two-site interaction model for the Nrf2-Keap1 complex. J. Biol. Chem. 2006, 281, 24756–24768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Davies, K.J.A.; Forman, H.J. Oxidative stress response and Nrf2 signaling in aging. Free Radic. Biol. Med. 2015, 88, 314–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itoh, K.; Mimura, J.; Yamamoto, M. Discovery of the negative regulator of Nrf2, Keap1: A historical overview. Antioxid. Redox Signal. 2010, 13, 1665–1678. [Google Scholar] [CrossRef]
- Helguera, P.; Seiglie, J.; Rodriguez, J.; Hanna, M.; Helguera, G.; Busciglio, J. Adaptive downregulation of mitochondrial function in down syndrome. Cell Metab. 2013, 17, 132–140. [Google Scholar] [CrossRef] [Green Version]
- Swatton, J.-E.; Sellers, L.-A.; Faull, R.-L.; Holland, A.; Iritani, S.; Bahn, S. Increased MAP kinase activity in Alzheimer’s and Down syndrome but not in schizophrenia human brain. Eur. J. Neurosci. 2004, 19, 2711–2719. [Google Scholar] [CrossRef]
- Oyake, T.; Itoh, K.; Motohashi, H.; Hayashi, N.; Hoshino, H.; Nishizawa, M.; Yamamoto, M.; Igarashi, K. Bach proteins belong to a novel family of BTB-basic leucine zipper transcription factors that interact with MafK and regulate transcription through the NF-E2 site. Mol. Cell. Biol. 1996, 16, 6083–6095. [Google Scholar] [CrossRef] [Green Version]
- Ogawa, K.; Sun, J.; Taketani, S.; Nakajima, O.; Nishitani, C.; Sassa, S.; Hayashi, N.; Yamamoto, M.; Shibahara, S.; Fujita, H.; et al. Heme mediates derepression of Maf recognition element through direct binding to transcription repressor Bach1. EMBO J. 2001, 20, 2835–2843. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Brand, M.; Zenke, Y.; Tashiro, S.; Groudine, M.; Igarashi, K. Heme regulates the dynamic exchange of Bach1 and NF-E2-related factors in the Maf transcription factor network. Proc. Natl. Acad. Sci. USA 2004, 101, 1461–1466. [Google Scholar] [CrossRef] [Green Version]
- Kaspar, J.W.; Jaiswal, A.K. Antioxidant-induced phosphorylation of tyrosine 486 leads to rapid nuclear export of Bach1 that allows Nrf2 to bind to the antioxidant response element and activate defensive gene expression. J. Biol. Chem. 2010, 285, 153–162. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, H.; Tashiro, S.; Sun, J.; Doi, H.; Satomi, S.; Igarashi, K. Cadmium induces nuclear export of Bach1, a transcriptional repressor of heme oxygenase-1 gene. J. Biol. Chem. 2003, 278, 49246–49253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, H.; Tashiro, S.; Hira, S.; Sun, J.; Yamazaki, C.; Zenke, Y.; Ikeda-Saito, M.; Yoshida, M.; Igarashi, K. Heme regulates gene expression by triggering Crm1-dependent nuclear export of Bach1. EMBO J. 2004, 23, 2544–2553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, J.; Hoshino, H.; Takaku, K.; Nakajima, O.; Muto, A.; Suzuki, H.; Tashiro, S.; Takahashi, S.; Shibahara, S.; Alam, J.; et al. Hemoprotein Bach1 regulates enhancer availability of heme oxygenase-1 gene. EMBO J. 2002, 21, 5216–5224. [Google Scholar] [CrossRef] [PubMed]
- Dhakshinamoorthy, S.; Jain, A.-K.; Bloom, D.-A.; Jaiswal, A.-K. Bach1 competes with Nrf2 leading to negative regulation of the antioxidant response element (ARE)-mediated NAD(P)H:quinone oxidoreductase 1 gene expression and induction in response to antioxidants. J. Biol. Chem. 2005, 280, 16891–16900. [Google Scholar] [CrossRef] [Green Version]
- Ka, S.-O.; Bang, I.-H.; Bae, E.-J.; Park, B.-H. Hepatocyte-specific sirtuin 6 deletion predisposes to nonalcoholic steatohepatitis by up-regulation of Bach1, an Nrf2 repressor. FASEB J. 2017, 31, 3999–4010. [Google Scholar] [CrossRef] [PubMed]
- Sakoda, E.; Igarashi, K.; Sun, J.; Kurisu, K.; Tashiro, S. Regulation of heme oxygenase-1 by transcription factor Bach1 in the mouse brain. Neurosci. Lett. 2008, 440, 160–165. [Google Scholar] [CrossRef]
- Kanno, H.; Ozawa, H.; Dohi, Y.; Sekiguchi, A.; Igarashi, K.; Itoi, E. Genetic ablation of transcription repressor Bach1 reduces neural tissue damage and improves locomotor function after spinal cord injury in mice. J. Neurotrauma 2009, 26, 31–39. [Google Scholar] [CrossRef]
- Shim, K.S.; Ferrando-Miguel, R.; Lubec, G. Aberrant protein expression of transcription factors BACH1 and ERG, both encoded on chromosome 21, in brains of patients with Down syndrome and Alzheimer’s disease. J. Neural Transm. 2003, 67, 39–49. [Google Scholar]
- Ferrando-Miguel, R.; Cheon, M.S.; Yang, J.W.; Lubec, G. Overexpression of transcription factor BACH1 in fetal Down syndrome brain. J. Neural Transm. 2003, 67, 193–205. [Google Scholar]
- Schipper, H.-M.; Song, W.; Tavitian, A.; Cressatti, M. The sinister face of heme oxygenase-1 in brain aging and disease. Prog. Neurobiol. 2019, 172, 40–70. [Google Scholar] [CrossRef]
- Barone, E.; Di Domenico, F.; Mancuso, C.; Butterfield, D.-A. The Janus face of the heme oxygenase/biliverdin reductase system in Alzheimer disease: It’s time for reconciliation. Neurobiol. Dis. 2014, 62, 144–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sferrazzo, G.; Di Rosa, M.; Barone, E.; Li Volti, G.; Musso, N.; Tibullo, D.; Barbagallo, I. Heme Oxygenase-1 in Central Nervous System Malignancies. J. Clin. Med. 2020, 9, 1562. [Google Scholar] [CrossRef] [PubMed]
- Baranano, D.E.; Snyder, S.H. Neural roles for heme oxygenase: Contrasts to nitric oxide synthase. Proc. Natl. Acad. Sci. USA 2001, 98, 10996–11002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergeron, M.; Ferriero, D.M.; Sharp, F.R. Developmental expression of heme oxygenase-1 (HSP32) in rat brain: An immunocytochemical study. Brain Res. Dev. Brain Res. 1998, 105, 181–194. [Google Scholar] [CrossRef]
- Matz, P.; Turner, C.; Weinstein, P.-R.; Massa, S.-M.; Panter, S.-S.; Sharp, F.-R. Heme-oxygenase-1 induction in glia throughout rat brain following experimental subarachnoid hemorrhage. Brain Res. 1996, 713, 211–222. [Google Scholar] [CrossRef]
- Nakaso, K.; Kitayama, M.; Fukuda, H.; Kimura, K.; Yanagawa, T.; Ishii, T.; Nakashima, K.; Yamada, K. Oxidative stress-related proteins A170 and heme oxygenase-1 are differently induced in the rat cerebellum under kainate-mediated excitotoxicity. Neurosci. Lett. 2000, 282, 57–60. [Google Scholar] [CrossRef]
- Vincent, S.R.; Das, S.; Maines, M.D. Brain heme oxygenase isoenzymes and nitric oxide synthase are co-localized in select neurons. Neuroscience 1994, 63, 223–231. [Google Scholar] [CrossRef]
- Maines, M.D. The heme oxygenase system: A regulator of second messenger gases. Annu. Rev. Pharmacol. Toxicol. 1997, 37, 517–554. [Google Scholar] [CrossRef]
- Maines, M.D. The heme oxygenase system and its functions in the brain. Cell. Mol. Biol. 2000, 46, 573–585. [Google Scholar]
- Mancuso, C. Heme oxygenase and its products in the nervous system. Antioxid. Redox Signal. 2004, 6, 878–887. [Google Scholar]
- Tramutola, A.; Di Domenico, F.; Barone, E.; Perluigi, M.; Butterfield, D.-A. It Is All about (U)biquitin: Role of Altered Ubiquitin-Proteasome System and UCHL1 in Alzheimer Disease. Oxid. Med. Cell. Longev. 2016, 2016, 2756068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tramutola, A.; Di Domenico, F.; Barone, E.; Arena, A.; Giorgi, A.; di Francesco, L.; Schinina, M.-E.; Coccia, R.; Head, E.; Butterfield, D.-A.; et al. Polyubiquitinylation Profile in Down Syndrome Brain Before and After the Development of Alzheimer Neuropathology. Antioxid. Redox Signal. 2017, 26, 280–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapitulnik, J.; Maines, M.D. Pleiotropic functions of biliverdin reductase: Cellular signaling and generation of cytoprotective and cytotoxic bilirubin. Trends Pharmacol. Sci. 2009, 30, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Tudor, C.; Lerner-Marmarosh, N.; Engelborghs, Y.; Gibbs, P.-E.; Maines, M.-D. Biliverdin reductase is a transporter of haem into the nucleus and is essential for regulation of HO-1 gene expression by haematin. Biochem. J. 2008, 413, 405–416. [Google Scholar] [CrossRef] [Green Version]
- Barone, E.; Butterfield, D.A. Insulin resistance in Alzheimer disease: Is heme oxygenase-1 an Achille’s heel? Neurobiol. Dis. 2015, 84, 69–77. [Google Scholar] [CrossRef]
- Fao, L.; Mota, S.I.; Rego, A.C. Shaping the Nrf2-ARE-related pathways in Alzheimer’s and Parkinson’s diseases. Ageing Res. Rev. 2019, 54, 100942. [Google Scholar] [CrossRef]
- Ramsey, C.-P.; Glass, C.A.; Montgomery, M.-B.; Lindl, K.-A.; Ritson, G.-P.; Chia, L.-A.; Hamilton, R.-L.; Chu, C.-T.; Jordan-Sciutto, K.-L. Expression of Nrf2 in neurodegenerative diseases. J. Neuropathol. Exp. Neurol. 2007, 66, 75–85. [Google Scholar] [CrossRef]
- Mota, S.I.; Costa, R.O.; Ferreira, I.L.; Santana, I.; Caldeira, G.L.; Padovano, C.; Fonseca, A.C.; Baldeiras, I.; Cunha, C.; Letra, L.; et al. Oxidative stress involving changes in Nrf2 and ER stress in early stages of Alzheimer’s disease. Biochim. Biophys. Acta 2015, 1852, 1428–1441. [Google Scholar] [CrossRef] [Green Version]
- Torres-Lista, V.; Parrado-Fernandez, C.; Alvarez-Monton, I.; Frontinan-Rubio, J.; Duran-Prado, M.; Peinado, J.R.; Johansson, B.; Alcain, F.J.; Gimenez-Llort, L. Neophobia, NQO1 and SIRT1 as premorbid and prodromal indicators of AD in 3xTg-AD mice. Behav. Brain Res. 2014, 271, 140–146. [Google Scholar] [CrossRef]
- Resende, R.; Moreira, P.-I.; Proenca, T.; Deshpande, A.; Busciglio, J.; Pereira, C.; Oliveira, C.R. Brain oxidative stress in a triple-transgenic mouse model of Alzheimer disease. Free Radic. Biol. Med. 2008, 44, 2051–2057. [Google Scholar] [CrossRef] [Green Version]
- Kanninen, K.; Malm, T.M.; Jyrkkanen, H.K.; Goldsteins, G.; Keksa-Goldsteine, V.; Tanila, H.; Yamamoto, M.; Yla-Herttuala, S.; Levonen, A.L.; Koistinaho, J. Nuclear factor erythroid 2-related factor 2 protects against beta amyloid. Mol. Cell. Neurosci. 2008, 39, 302–313. [Google Scholar] [CrossRef] [PubMed]
- Buee, L. Dementia Therapy Targeting Tau. Adv. Exp. Med. Biol. 2019, 1184, 407–416. [Google Scholar] [PubMed]
- Warnatz, H.J.; Schmidt, D.; Manke, T.; Piccini, I.; Sultan, M.; Borodina, T.; Balzereit, D.; Wruck, W.; Soldatov, A.; Vingron, M.; et al. The BTB and CNC homology 1 (BACH1) target genes are involved in the oxidative stress response and in control of the cell cycle. J. Biol. Chem. 2011, 286, 23521–23532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koglsberger, S.; Cordero-Maldonado, M.-L.; Antony, P.; Forster, J.-I.; Garcia, P.; Buttini, M.; Crawford, A.; Glaab, E. Gender-Specific Expression of Ubiquitin-Specific Peptidase 9 Modulates Tau Expression and Phosphorylation: Possible Implications for Tauopathies. Mol. Neurobiol. 2017, 54, 7979–7993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piras, S.; Furfaro, A.-L.; Brondolo, L.; Passalacqua, M.; Marinari, U.-M.; Pronzato, M.-A.; Nitti, M. Differentiation impairs Bach1 dependent HO-1 activation and increases sensitivity to oxidative stress in SH-SY5Y neuroblastoma cells. Sci. Rep. 2017, 7, 7568. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [Green Version]
- Tili, E.; Mezache, L.; Michaille, J.J.; Amann, V.; Williams, J.; Vandiver, P.; Quinonez, M.; Fadda, P.; Mikhail, A.; Nuovo, G. microRNA 155 up regulation in the CNS is strongly correlated to Down’s syndrome dementia. Ann. Diagn. Pathol. 2018, 34, 103–109. [Google Scholar] [CrossRef]
- Sun, X.; Li, X.; Ma, S.; Guo, Y.; Li, Y. MicroRNA-98-5p ameliorates oxygen-glucose deprivation/reoxygenation (OGD/R)-induced neuronal injury by inhibiting Bach1 and promoting Nrf2/ARE signaling. Biochem. Biophys. Res. Commun. 2018, 507, 114–121. [Google Scholar] [CrossRef]

{kind=link}
| Gene on Hsa21 | Molecular Function | Biological Process | Relevance in Down Syndrome |
|---|---|---|---|
| SOD-1 (Cu,Zn-superoxide dismutase 1) | Oxidoreductase | Antioxidant Response | Triplication of SOD-1 in DS brain results in an imbalance in the ratio of SOD-1 to CAT and GPX (two enzymes involved in its metabolism), thus leading to the accumulation of H2O2 in the cells. |
| APP (amyloid precursor protein) | Heparin-binding, Protease inhibitor | Apoptosis, Cell adhesion, Endocytosis, Notch signaling pathway and Aβ processing | Triplication of APP causes the over-production of Aβ (1-40/42) in DS brain. Deposition of senile plaques of Aβ is observed in post-mortem brain and plasma from DS compared with non-DS individuals. |
| BACH1 (BTB Domain and CNC Homolog 1) | DNA-binding, Transcription regulation | Antioxidant Response | Triplication of BACH1, as a negative transcription regulator, in DS brain could block the induction of antioxidant genes, therefore promoting increased OS in the cell. |
| CBR(Carbonyl reductase) | Oxidoreductase | Oxidative stress Response | Triplication of CBR in DS play a role in exacerbating OS. Carbonyls are toxic metabolic intermediates that are mainly detoxified by aldehyde dehydrogenase or reduced by CBR and/or alcohol dehydrogenase to their corresponding alcohols. Increased levels of these enzymes were detected in the brain of DS patients, likely in response to elevated carbonyls production in DS. |
| ETS2(Protein C-ets-2) | Transcription regulation | Cell differentiation, maturation and signaling | Triplication of Ets-2 could play a role in the increased susceptibility of DS cells to undergo apoptosis given common pathophysiological features shared between Ets-2 overexpressing transgenic mice and individuals with DS. |
| S100B(S100 calcium-binding protein B) | Ca2+-binding protein | Neurotrophic factor | Triplication of S100β in DS corresponds to an increase of its expression levels in astrocytes in association with neuritic plaques. In addition, chronic overexpression of S100β contributes to increased neuronal and neuritic βAPP expression with consequent accelerated amyloid deposition, as well as abnormal growth of neurites in β-amyloid plaques, similar to observations in middle-aged DS patients. |
| Pathology | BACH1 Changes | References |
|---|---|---|
| Down Syndrome (DS) | ↑ BACH1 protein levels in fetal cortical specimens of human DS | Ferrando-Miguel R.J. Neural Transm Suppl, 2003(67): p. 193-205 Tili, E., et al., Ann Diagn Pathol, 2018. 34: p. 103-109. |
| ↑ BACH1 protein levels in human DS subjects, either before or after the development of AD | Di Domenico, F., et al., J.Alzheimers Dis, 2015. 44(4): p. 1107–20. | |
| Changes in post-translational modifications of BACH-1: | Di Domenico, F., et al., J.Alzheimers Dis, 2015. 44(4): p. 1107–20. | |
| ↓ mono-ubiquitination of BACH1 in young DS human brain | ||
| ↑ poly-ubiquitinylation of BACH1 only in DSAD subjects | ||
| ↑ BACH1 protein levels in brain of Ts65Dn mice. | Di Domenico, F., et al., J.Alzheimers Dis, 2015. 44(4): p. 1107–20. | |
| No changes were observed in BACH1 ubiquitination in Ts65Dn mice compared to euploid mice. | ||
| Alzheimer Disease (AD) | NO changes were observed in BACH1 protein levels in AD brain. | Shim, K.S., R. Ferrando-Miguel, and G. Lubec, J Neural Transm Suppl, 2003(67): p. 39–49. |
| ↑ BACH1 protein levels in AD Brain using an immunohistochemistry approach. | Tili, E., et al., Ann Diagn Pathol, 2018. 34: p. 103–109. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perluigi, M.; Tramutola, A.; Pagnotta, S.; Barone, E.; Butterfield, D.A. The BACH1/Nrf2 Axis in Brain in Down Syndrome and Transition to Alzheimer Disease-Like Neuropathology and Dementia. Antioxidants 2020, 9, 779. https://doi.org/10.3390/antiox9090779
Perluigi M, Tramutola A, Pagnotta S, Barone E, Butterfield DA. The BACH1/Nrf2 Axis in Brain in Down Syndrome and Transition to Alzheimer Disease-Like Neuropathology and Dementia. Antioxidants. 2020; 9(9):779. https://doi.org/10.3390/antiox9090779
Chicago/Turabian StylePerluigi, Marzia, Antonella Tramutola, Sara Pagnotta, Eugenio Barone, and D. Allan Butterfield. 2020. "The BACH1/Nrf2 Axis in Brain in Down Syndrome and Transition to Alzheimer Disease-Like Neuropathology and Dementia" Antioxidants 9, no. 9: 779. https://doi.org/10.3390/antiox9090779