Methylglyoxal-Induced Dysfunction in Brain Endothelial Cells via the Suppression of Akt/HIF-1α Pathway and Activation of Mitophagy Associated with Increased Reactive Oxygen Species


Abstract
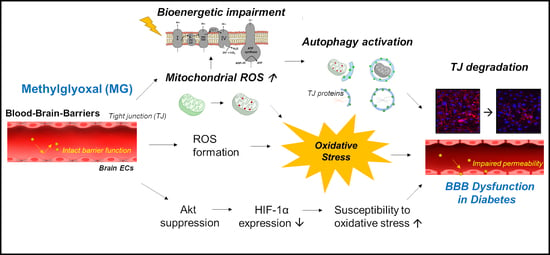
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Culture
2.3. PI Fluorscence and MTT Reduction Assay
2.4. In Vitro Permeability Assay
2.5. Immunofluorescence Staining
2.6. Cellular ROS Detection Assay
2.7. Mitochondrial ROS Detection Assay
2.8. Mitochondrial Fractionation
2.9. Measurement of Bioenergetic Function
2.10. Mitochondrial Mass Analysis
2.11. Western Blot Analysis
2.12. Statistical Analysis
3. Results
3.1. MG induced Accumulation of MG-Adducts and Deterioration of the Glyoxalase System in bEND.3 Cells
3.2. Mitochondrial and Total Cellular ROS Production Increases after MG Exposure with the Suppression of the Akt/HIF-1α Pathway
3.3. Disturbnace of Bioenergetic Mitochondrial Function and Activates Parkin-Mediated Mitophagy by MG in Brain ECs
3.4. Restoration of Mitochondrial Function and Mitophagy by NAC
3.5. Activated Autophagy Might Contribute to Occludin Degradation
3.6. MG Induces Endothelial Dysfunction and Degradation of Tight Junction Proteins in bEND.3 Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- De Ferranti, S.D.; de Boer, I.H.; Fonseca, V.; Fox, C.S.; Golden, S.H.; Lavie, C.J.; Magge, S.N.; Marx, N.; McGuire, D.K.; Orchard, T.J.; et al. Type 1 diabetes mellitus and cardiovascular disease: A scientific statement from the American Heart Association and American Diabetes Association. Circulation 2014, 130, 1110–1130. [Google Scholar] [CrossRef]
- Jansen, F.; Yang, X.; Franklin, B.S.; Hoelscher, M.; Schmitz, T.; Bedorf, J.; Nickenig, G.; Werner, N. High glucose condition increases nadph oxidase activity in endothelial microparticles that promote vascular inflammation. Cardiovasc. Res. 2013, 98, 94–106. [Google Scholar] [CrossRef]
- Kim, K.A.; Shin, Y.J.; Akram, M.; Kim, E.S.; Choi, K.W.; Suh, H.; Lee, C.H.; Bae, O.N. High glucose condition induces autophagy in endothelial progenitor cells contributing to angiogenic impairment. Biol. Pharm. Bull. 2014, 37, 1248–1252. [Google Scholar] [CrossRef]
- Peng, C.; Ma, J.; Gao, X.; Tian, P.; Li, W.; Zhang, L. High glucose induced oxidative stress and apoptosis in cardiac microvascular endothelial cells are regulated by FoxO3a. PLoS ONE 2013, 8, e79739. [Google Scholar] [CrossRef]
- Lu, J.; Randell, E.; Han, Y.; Adeli, K.; Krahn, J.; Meng, Q.H. Increased plasma methylglyoxal level, inflammation, and vascular endothelial dysfunction in diabetic nephropathy. Clin. Biochem. 2011, 44, 307–311. [Google Scholar] [CrossRef]
- Thornalley, P.J.; Hooper, N.I.; Jennings, P.E.; Florkowski, C.M.; Jones, A.F.; Lunec, J.; Barnett, A.H. The human red blood cell glyoxalase system in diabetes mellitus. Diabetes Res. Clin. Pract. 1989, 7, 115–120. [Google Scholar] [CrossRef]
- Wang, H.; Meng, Q.H.; Gordon, J.R.; Khandwala, H.; Wu, L. Proinflammatory and proapoptotic effects of methylglyoxal on neutrophils from patients with type 2 diabetes mellitus. Clin. Biochem. 2007, 40, 1232–1239. [Google Scholar] [CrossRef]
- Kim, J.H.; Kim, K.A.; Shin, Y.J.; Kim, H.; Majid, A.; Bae, O.N. Methylglyoxal induced advanced glycation end products (AGE)/receptor for AGE (RAGE)-mediated angiogenic impairment in bone marrow-derived endothelial progenitor cells. J. Toxicol. Environ. Health A 2018, 81, 266–277. [Google Scholar] [CrossRef]
- Santini, S.J.; Cordone, V.; Mijit, M.; Bignotti, V.; Aimola, P.; Dolo, V.; Falone, S.; Amicarelli, F. SIRT1-Dependent Upregulation of Antiglycative Defense in HUVECs Is Essential for Resveratrol Protection against High Glucose Stress. Antioxidants 2019, 8, 346. [Google Scholar] [CrossRef]
- Vulesevic, B.; McNeill, B.; Giacco, F.; Maeda, K.; Blackburn, N.J.; Brownlee, M.; Milne, R.W.; Suuronen, E.J. Methylglyoxal-Induced Endothelial Cell Loss and Inflammation Contribute to the Development of Diabetic Cardiomyopathy. Diabetes 2016, 65, 1699–1713. [Google Scholar] [CrossRef]
- Yuan, J.; Zhu, C.; Hong, Y.; Sun, Z.; Fang, X.; Wu, B.; Li, S. The role of cPLA2 in Methylglyoxal-induced cell apoptosis of HUVECs. Toxicol. Appl. Pharmacol. 2017, 323, 44–52. [Google Scholar] [CrossRef]
- Figarola, J.L.; Singhal, J.; Rahbar, S.; Awasthi, S.; Singhal, S.S. LR-90 prevents methylglyoxal-induced oxidative stress and apoptosis in human endothelial cells. Apoptosis 2014, 19, 776–788. [Google Scholar] [CrossRef]
- Lv, Q.; Gu, C.; Chen, C. Venlafaxine protects methylglyoxal-induced apoptosis in the cultured human brain microvascular endothelial cells. Neurosci. Lett. 2014, 569, 99–103. [Google Scholar] [CrossRef]
- Phalitakul, S.; Okada, M.; Hara, Y.; Yamawaki, H. Vaspin prevents methylglyoxal-induced apoptosis in human vascular endothelial cells by inhibiting reactive oxygen species generation. Acta Physiol. (Oxf.) 2013, 209, 212–219. [Google Scholar] [CrossRef]
- Hawkins, B.T.; Davis, T.P. The blood-brain barrier/neurovascular unit in health and disease. Pharmacol. Rev. 2005, 57, 173–185. [Google Scholar] [CrossRef]
- Obermeier, B.; Daneman, R.; Ransohoff, R.M. Development, maintenance and disruption of the blood-brain barrier. Nat. Med. 2013, 19, 1584–1596. [Google Scholar] [CrossRef]
- Abbott, N.J.; Patabendige, A.A.; Dolman, D.E.; Yusof, S.R.; Begley, D.J. Structure and function of the blood-brain barrier. Neurobiol. Dis. 2010, 37, 13–25. [Google Scholar] [CrossRef]
- Kim, J.H.; Byun, H.M.; Chung, E.C.; Chung, H.Y.; Bae, O.N. Loss of Integrity: Impairment of the Blood-brain Barrier in Heavy Metal-associated Ischemic Stroke. Toxicol. Res. 2013, 29, 157–164. [Google Scholar] [CrossRef][Green Version]
- Kim, K.A.; Kim, D.; Kim, J.H.; Shin, Y.J.; Kim, E.S.; Akram, M.; Kim, E.H.; Majid, A.; Baek, S.H.; Bae, O.N. Autophagy-mediated occludin degradation contributes to blood-brain barrier disruption during ischemia in bEnd.3 brain endothelial cells and rat ischemic stroke models. Fluids Barriers CNS 2020, 17, 21. [Google Scholar] [CrossRef]
- Kim, K.A.; Shin, D.; Kim, J.H.; Shin, Y.J.; Rajanikant, G.K.; Majid, A.; Baek, S.H.; Bae, O.N. Role of Autophagy in Endothelial Damage and Blood-Brain Barrier Disruption in Ischemic Stroke. Stroke 2018, 49, 1571–1579. [Google Scholar] [CrossRef]
- Ighodaro, O.M. Molecular pathways associated with oxidative stress in diabetes mellitus. Biomed. Pharmacother. 2018, 108, 656–662. [Google Scholar] [CrossRef] [PubMed]
- Rolo, A.P.; Palmeira, C.M. Diabetes and mitochondrial function: Role of hyperglycemia and oxidative stress. Toxicol. Appl. Pharmacol. 2006, 212, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Enciu, A.M.; Gherghiceanu, M.; Popescu, B.O. Triggers and effectors of oxidative stress at blood-brain barrier level: Relevance for brain ageing and neurodegeneration. Oxid. Med. Cell Longev. 2013, 2013, 297512. [Google Scholar] [CrossRef]
- Doll, D.N.; Hu, H.; Sun, J.; Lewis, S.E.; Simpkins, J.W.; Ren, X. Mitochondrial crisis in cerebrovascular endothelial cells opens the blood-brain barrier. Stroke 2015, 46, 1681–1689. [Google Scholar] [CrossRef]
- Do, V.Q.; Park, K.H.; Park, J.M.; Lee, M.Y. Comparative In Vitro Toxicity Study of Docetaxel and Nanoxel, a Docetaxel-Loaded Micellar Formulation Using Cultured and Blood Cells. Toxicol. Res. 2019, 35, 201–207. [Google Scholar] [CrossRef]
- Rellick, S.L.; Hu, H.; Simpkins, J.W.; Ren, X. Evaluation of Bioenergetic Function in Cerebral Vascular Endothelial Cells. J. Vis. Exp. 2016. [Google Scholar] [CrossRef]
- Doherty, E.; Perl, A. Measurement of Mitochondrial Mass by Flow Cytometry during Oxidative Stress. React. Oxyg. Species (Apex) 2017, 4, 275–283. [Google Scholar] [CrossRef]
- Rabbani, N.; Thornalley, P.J. Methylglyoxal, glyoxalase 1 and the dicarbonyl proteome. Amino Acids 2012, 42, 1133–1142. [Google Scholar] [CrossRef]
- Stepanenko, A.A.; Dmitrenko, V.V. Pitfalls of the MTT assay: Direct and off-target effects of inhibitors can result in over/underestimation of cell viability. Gene 2015, 574, 193–203. [Google Scholar] [CrossRef]
- Semenza, G.L. Hypoxia-inducible factor 1: Regulator of mitochondrial metabolism and mediator of ischemic preconditioning. Biochim. Biophys. Acta Mol. Cell Res. 2011, 1813, 1263–1268. [Google Scholar] [CrossRef]
- Kietzmann, T.; Mennerich, D.; Dimova, E. Hypoxia-Inducible Factors (HIFs) and Phosphorylation: Impact on Stability, Localization, and Transactivity. Front. Cell Dev. Biol. 2016, 4, 11. [Google Scholar] [CrossRef] [PubMed]
- Brand, M.D.; Nicholls, D.G. Assessing mitochondrial dysfunction in cells. Biochem. J. 2011, 435, 297–312. [Google Scholar] [CrossRef] [PubMed]
- Dafre, A.L.; Schmitz, A.E.; Maher, P. Methylglyoxal-induced AMPK activation leads to autophagic degradation of thioredoxin 1 and glyoxalase 2 in HT22 nerve cells. Free Radic. Biol. Med. 2017, 108, 270–279. [Google Scholar] [CrossRef]
- Masterjohn, C.; Park, Y.; Lee, J.; Noh, S.K.; Koo, S.I.; Bruno, R.S. Dietary fructose feeding increases adipose methylglyoxal accumulation in rats in association with low expression and activity of glyoxalase-2. Nutrients 2013, 5, 3311–3328. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Li, H.; Xi, H.S.; Li, S. HIF1alpha is required for survival maintenance of chronic myeloid leukemia stem cells. Blood 2012, 119, 2595–2607. [Google Scholar] [CrossRef] [PubMed]
- Baek, S.H.; Noh, A.R.; Kim, K.A.; Akram, M.; Shin, Y.J.; Kim, E.S.; Yu, S.W.; Majid, A.; Bae, O.N. Modulation of mitochondrial function and autophagy mediates carnosine neuroprotection against ischemic brain damage. Stroke 2014, 45, 2438–2443. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef]
- Rai, Y.; Pathak, R.; Kumari, N.; Sah, D.K.; Pandey, S.; Kalra, N.; Soni, R.; Dwarakanath, B.S.; Bhatt, A.N. Mitochondrial biogenesis and metabolic hyperactivation limits the application of MTT assay in the estimation of radiation induced growth inhibition. Sci. Rep. 2018, 8, 1531. [Google Scholar] [CrossRef]
- Nigro, C.; Leone, A.; Raciti, G.A.; Longo, M.; Mirra, P.; Formisano, P.; Beguinot, F.; Miele, C. Methylglyoxal-Glyoxalase 1 Balance: The Root of Vascular Damage. Int. J. Mol. Sci. 2017, 18, 188. [Google Scholar] [CrossRef]
- Maessen, D.E.; Stehouwer, C.D.; Schalkwijk, C.G. The role of methylglyoxal and the glyoxalase system in diabetes and other age-related diseases. Clin. Sci. (Lond.) 2015, 128, 839–861. [Google Scholar] [CrossRef]
- Rabbani, N.; Thornalley, P.J. Glyoxalase in diabetes, obesity and related disorders. Semin. Cell Dev. Biol. 2011, 22, 309–317. [Google Scholar] [CrossRef]
- Miyazawa, N.; Abe, M.; Souma, T.; Tanemoto, M.; Abe, T.; Nakayama, M.; Ito, S. Methylglyoxal augments intracellular oxidative stress in human aortic endothelial cells. Free Radic. Res. 2010, 44, 101–107. [Google Scholar] [CrossRef]
- Chu, P.; Han, G.; Ahsan, A.; Sun, Z.; Liu, S.; Zhang, Z.; Sun, B.; Song, Y.; Lin, Y.; Peng, J.; et al. Phosphocreatine protects endothelial cells from Methylglyoxal induced oxidative stress and apoptosis via the regulation of PI3K/Akt/eNOS and NF-kappaB pathway. Vasc. Pharmacol. 2017, 91, 26–35. [Google Scholar] [CrossRef]
- Do, M.; Kim, S.; Seo, S.Y.; Yeo, E.J.; Kim, S.Y. Delta-Tocopherol prevents methylglyoxal-induced apoptosis by reducing ROS generation and inhibiting apoptotic signaling cascades in human umbilical vein endothelial cells. Food Funct. 2015, 6, 1568–1577. [Google Scholar] [CrossRef]
- Pang, N.; Chen, T.; Deng, X.; Chen, N.; Li, R.; Ren, M.; Li, Y.; Luo, M.; Hao, H.; Wu, J.; et al. Polydatin Prevents Methylglyoxal-Induced Apoptosis through Reducing Oxidative Stress and Improving Mitochondrial Function in Human Umbilical Vein Endothelial Cells. Oxid. Med. Cell Longev. 2017, 2017, 7180943. [Google Scholar] [CrossRef]
- Liu, C.; Cao, B.; Zhang, Q.; Zhang, Y.; Chen, X.; Kong, X.; Dong, Y. Inhibition of thioredoxin 2 by intracellular methylglyoxal accumulation leads to mitochondrial dysfunction and apoptosis in INS-1 cells. Endocrine 2020, 68, 103–115. [Google Scholar] [CrossRef]
- Forrester, S.J.; Kikuchi, D.S.; Hernandes, M.S.; Xu, Q.; Griendling, K.K. Reactive Oxygen Species in Metabolic and Inflammatory Signaling. Circ. Res. 2018, 122, 877–902. [Google Scholar] [CrossRef]
- Kluge, M.A.; Fetterman, J.L.; Vita, J.A. Mitochondria and endothelial function. Circ. Res. 2013, 112, 1171–1188. [Google Scholar] [CrossRef]
- Pickles, S.; Vigie, P.; Youle, R.J. Mitophagy and Quality Control Mechanisms in Mitochondrial Maintenance. Curr. Biol. 2018, 28, R170–R185. [Google Scholar] [CrossRef]
- Thomas, L.W.; Ashcroft, M. Exploring the molecular interface between hypoxia-inducible factor signalling and mitochondria. Cell. Mol. Life Sci. 2019, 76, 1759–1777. [Google Scholar] [CrossRef]
- Bento, C.F.; Fernandes, R.; Ramalho, J.; Marques, C.; Shang, F.; Taylor, A.; Pereira, P. The chaperone-dependent ubiquitin ligase CHIP targets HIF-1alpha for degradation in the presence of methylglyoxal. PLoS ONE 2010, 5, e15062. [Google Scholar] [CrossRef]
- Costanzini, A.; Sgarbi, G.; Maresca, A.; Del Dotto, V.; Solaini, G.; Baracca, A. Mitochondrial Mass Assessment in a Selected Cell Line under Different Metabolic Conditions. Cells 2019, 8, 1454. [Google Scholar] [CrossRef]
- Monaco, C.M.F.; Hughes, M.C.; Ramos, S.V.; Varah, N.E.; Lamberz, C.; Rahman, F.A.; McGlory, C.; Tarnopolsky, M.A.; Krause, M.P.; Laham, R.; et al. Altered mitochondrial bioenergetics and ultrastructure in the skeletal muscle of young adults with type 1 diabetes. Diabetologia 2018, 61, 1411–1423. [Google Scholar] [CrossRef]
- Pinti, M.V.; Fink, G.K.; Hathaway, Q.A.; Durr, A.J.; Kunovac, A.; Hollander, J.M. Mitochondrial dysfunction in type 2 diabetes mellitus: An organ-based analysis. Am. J. Physiol. Endocrinol. Metab. 2019, 316, E268–E285. [Google Scholar] [CrossRef]
- Twig, G.; Shirihai, O.S. The interplay between mitochondrial dynamics and mitophagy. Antioxid. Redox. Signal. 2011, 14, 1939–1951. [Google Scholar] [CrossRef]
- Shefa, U.; Jeong, N.Y.; Song, I.O.; Chung, H.J.; Kim, D.; Jung, J.; Huh, Y. Mitophagy links oxidative stress conditions and neurodegenerative diseases. Neural Regen. Res. 2019, 14, 749–756. [Google Scholar] [CrossRef]
- Majmundar, A.J.; Wong, W.J.; Simon, M.C. Hypoxia-inducible factors and the response to hypoxic stress. Mol. Cell 2010, 40, 294–309. [Google Scholar] [CrossRef]
- Chen, R.; Lai, U.H.; Zhu, L.; Singh, A.; Ahmed, M.; Forsyth, N.R. Reactive Oxygen Species Formation in the Brain at Different Oxygen Levels: The Role of Hypoxia Inducible Factors. Front. Cell Dev. Biol. 2018, 6, 132. [Google Scholar] [CrossRef]
- Xue, J.; Ray, R.; Singer, D.; Bohme, D.; Burz, D.S.; Rai, V.; Hoffmann, R.; Shekhtman, A. The receptor for advanced glycation end products (RAGE) specifically recognizes methylglyoxal-derived AGEs. Biochemistry 2014, 53, 3327–3335. [Google Scholar] [CrossRef]
- Han, Y.; Randell, E.; Vasdev, S.; Gill, V.; Gadag, V.; Newhook, L.A.; Grant, M.; Hagerty, D. Plasma methylglyoxal and glyoxal are elevated and related to early membrane alteration in young, complication-free patients with Type 1 diabetes. Mol. Cell. Biochem. 2007, 305, 123–131. [Google Scholar] [CrossRef]
- Bierhaus, A.; Fleming, T.; Stoyanov, S.; Leffler, A.; Babes, A.; Neacsu, C.; Sauer, S.K.; Eberhardt, M.; Schnolzer, M.; Lasitschka, F.; et al. Methylglyoxal modification of Nav1.8 facilitates nociceptive neuron firing and causes hyperalgesia in diabetic neuropathy. Nat. Med. 2012, 18, 926–933. [Google Scholar] [CrossRef]
- Fleming, T.; Cuny, J.; Nawroth, G.; Djuric, Z.; Humpert, P.M.; Zeier, M.; Bierhaus, A.; Nawroth, P.P. Is diabetes an acquired disorder of reactive glucose metabolites and their intermediates? Diabetologia 2012, 55, 1151–1155. [Google Scholar] [CrossRef]
- Brouwers, O.; Niessen, P.M.; Haenen, G.; Miyata, T.; Brownlee, M.; Stehouwer, C.D.; De Mey, J.G.; Schalkwijk, C.G. Hyperglycaemia-induced impairment of endothelium-dependent vasorelaxation in rat mesenteric arteries is mediated by intracellular methylglyoxal levels in a pathway dependent on oxidative stress. Diabetologia 2010, 53, 989–1000. [Google Scholar] [CrossRef]
- Oguri, G.; Nakajima, T.; Yamamoto, Y.; Takano, N.; Tanaka, T.; Kikuchi, H.; Morita, T.; Nakamura, F.; Yamasoba, T.; Komuro, I. Effects of methylglyoxal on human cardiac fibroblast: Roles of transient receptor potential ankyrin 1 (TRPA1) channels. Am. J. Physiol. Heart Circ. Physiol. 2014, 307, H1339–H1352. [Google Scholar] [CrossRef]
- De Meyer, G.R.; Grootaert, M.O.; Michiels, C.F.; Kurdi, A.; Schrijvers, D.M.; Martinet, W. Autophagy in vascular disease. Circ. Res. 2015, 116, 468–479. [Google Scholar] [CrossRef]
- Fang, L.; Li, X.; Zhong, Y.; Yu, J.; Yu, L.; Dai, H.; Yan, M. Autophagy protects human brain microvascular endothelial cells against methylglyoxal-induced injuries, reproducible in a cerebral ischemic model in diabetic rats. J. Neurochem. 2015, 135, 431–440. [Google Scholar] [CrossRef]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, D.; Kim, K.-A.; Kim, J.-H.; Kim, E.-H.; Bae, O.-N. Methylglyoxal-Induced Dysfunction in Brain Endothelial Cells via the Suppression of Akt/HIF-1α Pathway and Activation of Mitophagy Associated with Increased Reactive Oxygen Species. Antioxidants 2020, 9, 820. https://doi.org/10.3390/antiox9090820
Kim D, Kim K-A, Kim J-H, Kim E-H, Bae O-N. Methylglyoxal-Induced Dysfunction in Brain Endothelial Cells via the Suppression of Akt/HIF-1α Pathway and Activation of Mitophagy Associated with Increased Reactive Oxygen Species. Antioxidants. 2020; 9(9):820. https://doi.org/10.3390/antiox9090820
Chicago/Turabian StyleKim, Donghyun, Kyeong-A Kim, Jeong-Hyeon Kim, Eun-Hye Kim, and Ok-Nam Bae. 2020. "Methylglyoxal-Induced Dysfunction in Brain Endothelial Cells via the Suppression of Akt/HIF-1α Pathway and Activation of Mitophagy Associated with Increased Reactive Oxygen Species" Antioxidants 9, no. 9: 820. https://doi.org/10.3390/antiox9090820
APA StyleKim, D., Kim, K.-A., Kim, J.-H., Kim, E.-H., & Bae, O.-N. (2020). Methylglyoxal-Induced Dysfunction in Brain Endothelial Cells via the Suppression of Akt/HIF-1α Pathway and Activation of Mitophagy Associated with Increased Reactive Oxygen Species. Antioxidants, 9(9), 820. https://doi.org/10.3390/antiox9090820