
Measuring Cellular Immunity to Influenza: Methods of Detection, Applications and Challenges

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. T-Cell Immunity Cellular Immunity to Influenza Virus
3. Cellular Immunity in Human Infection
4. Current Influenza Vaccines
5. T-Cell Vaccination Approaches
6. Techniques Used to Quantify Cellular Immunity
6.1. Cytokine Based Assays
6.1.1. ELISA and Multiplex Cytokine Assays
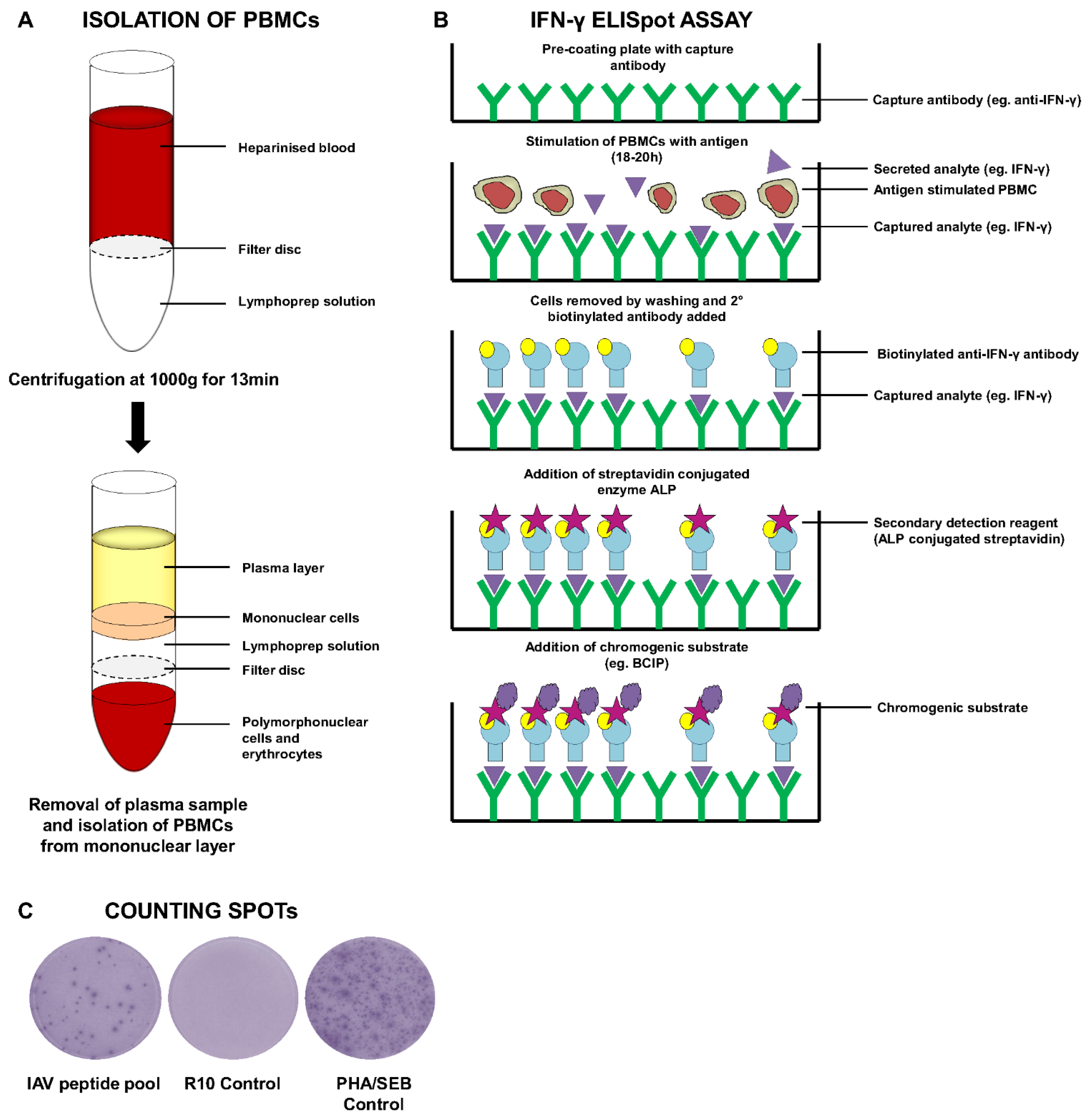
6.1.2. ELISPOT Assays

6.2. Flow Cytometry/Cell Phenotyping Assays
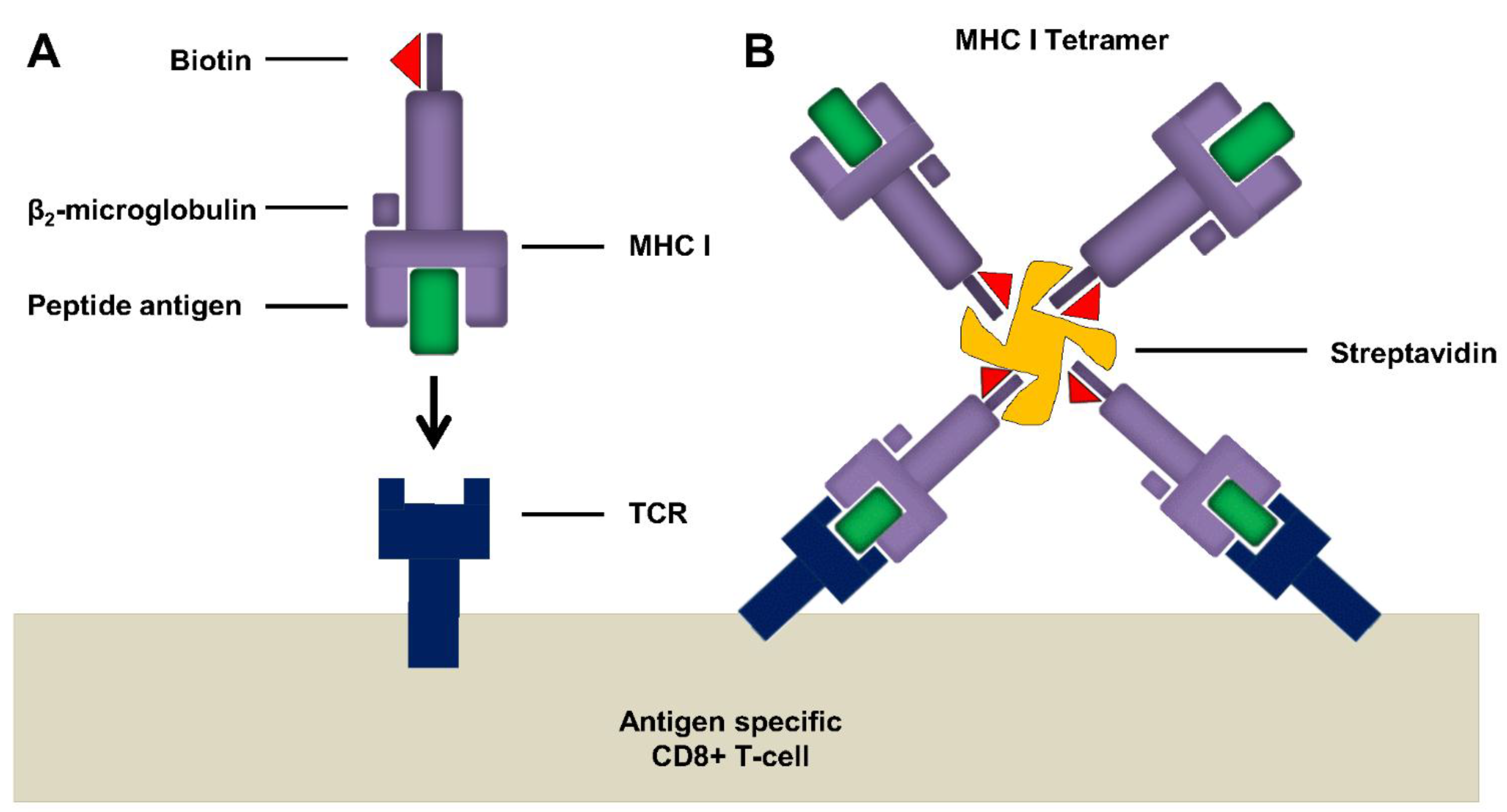
6.2.1. Tetramer Assay

6.2.2. ICS Intracellular Cytokine Staining
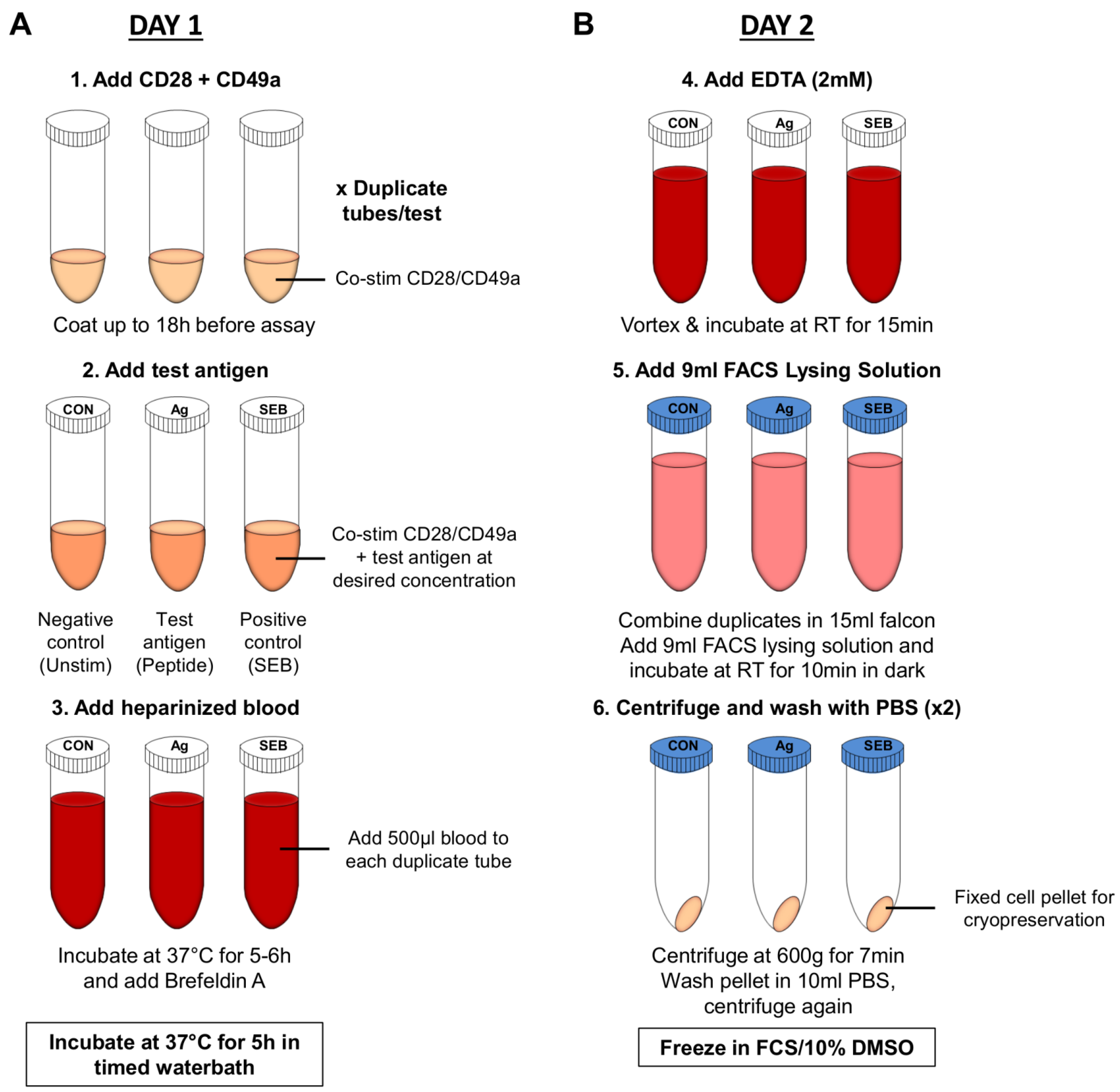
6.2.3. Whole Blood Assay
6.3. Proliferation Assays

6.4. Cell Killing or Cytotoxicity Assays
6.5. Systems Biology Approaches to Assessing Cellular Immunity
6.5.1. DNA Microarray
6.5.2. microRNA Profiling
7. Summary of Differences in T-Cell Assays Used in Clinical Trials
8. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lambert, L.C.; Fauci, A.S. Influenza vaccines for the future. N. Engl. J. Med. 2010, 363, 2036–2044. [Google Scholar] [CrossRef] [PubMed]
- Molinari, N.A.; Ortega-Sanchez, I.R.; Messonnier, M.L.; Thompson, W.W.; Wortley, P.M.; Weintraub, E.; Bridges, C.B. The annual impact of seasonal influenza in the US: Measuring disease burden and costs. Vaccine 2007, 25, 5086–5096. [Google Scholar] [CrossRef] [PubMed]
- Krammer, F.; Palese, P. Influenza virus hemagglutinin stalk-based antibodies and vaccines. Curr. Opin. Virol. 2013, 3, 521–530. [Google Scholar] [CrossRef] [PubMed]
- Margine, I.; Hai, R.; Albrecht, R.A.; Obermoser, G.; Harrod, A.C.; Banchereau, J.; Palucka, K.; Garcia-Sastre, A.; Palese, P.; Treanor, J.J.; et al. H3N2 influenza virus infection induces broadly reactive hemagglutinin stalk antibodies in humans and mice. J. Virol. 2013, 87, 4728–4737. [Google Scholar] [CrossRef] [PubMed]
- Taubenberger, J.K.; Kash, J.C. Influenza virus evolution, host adaptation, and pandemic formation. Cell Host Microbe 2010, 7, 440–451. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Holmes, E.C. Avian influenza virus exhibits rapid evolutionary dynamics. Mol. Biol. Evol. 2006, 23, 2336–2341. [Google Scholar] [CrossRef] [PubMed]
- McMichael, A.J.; Gotch, F.M.; Noble, G.R.; Beare, P.A. Cytotoxic T-cell immunity to influenza. N. Engl. J. Med. 1983, 309, 13–17. [Google Scholar] [CrossRef] [PubMed]
- McCullers, J.A.; van de Velde, L.A.; Allison, K.J.; Branum, K.C.; Webby, R.J.; Flynn, P.M. Recipients of vaccine against the 1976 “swine flu” have enhanced neutralization responses to the 2009 novel H1N1 influenza virus. Clin. Infect. Dis. 2010, 50, 1487–1492. [Google Scholar] [CrossRef] [PubMed]
- Throsby, M.; van den Brink, E.; Jongeneelen, M.; Poon, L.L.; Alard, P.; Cornelissen, L.; Bakker, A.; Cox, F.; van Deventer, E.; Guan, Y.; et al. Heterosubtypic neutralizing monoclonal antibodies cross-protective against H5N1 and H1N1 recovered from human IgM+ memory B cells. PLOS ONE 2008, 3, e3942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coughlan, L.; Mullarkey, C.; Gilbert, S. Adenoviral vectors as novel vaccines for influenza. J. Pharm. Pharmacol. 2015. [Google Scholar] [CrossRef]
- Yewdell, J.W.; Schubert, U.; Bennink, J.R. At the crossroads of cell biology and immunology: DRiPs and other sources of peptide ligands for MHC class I molecules. J. Cell Sci. 2001, 114, 845–851. [Google Scholar] [PubMed]
- Pamer, E.; Cresswell, P. Mechanisms of MHC class I—restricted antigen processing. Annu. Rev. Immunol. 1998, 16, 323–358. [Google Scholar] [CrossRef] [PubMed]
- Leone, P.; Shin, E.C.; Perosa, F.; Vacca, A.; Dammacco, F.; Racanelli, V. MHC class I antigen processing and presenting machinery: Organization, function, and defects in tumor cells. J. Natl. Cancer Inst. 2013, 105, 1172–1187. [Google Scholar] [CrossRef] [PubMed]
- Germain, R.N.; Margulies, D.H. The biochemistry and cell biology of antigen processing and presentation. Annu. Rev. Immunol. 1993, 11, 403–450. [Google Scholar] [CrossRef] [PubMed]
- Weinfurter, J.T.; Brunner, K.; Capuano, S.V., 3rd; Li, C.; Broman, K.W.; Kawaoka, Y.; Friedrich, T.C. Cross-reactive T cells are involved in rapid clearance of 2009 pandemic H1N1 influenza virus in nonhuman primates. PLOS Pathog. 2011, 7, e1002381. [Google Scholar]
- Valkenburg, S.A.; Rutigliano, J.A.; Ellebedy, A.H.; Doherty, P.C.; Thomas, P.G.; Kedzierska, K. Immunity to seasonal and pandemic influenza A viruses. Microbes. Infect. 2011, 13, 489–501. [Google Scholar] [CrossRef] [PubMed]
- Amsen, D.; Backer, R.A.; Helbig, C. Decisions on the road to memory. Adv. Exp. Med. Biol. 2013, 785, 107–120. [Google Scholar] [PubMed]
- Kreijtz, J.H.; Fouchier, R.A.; Rimmelzwaan, G.F. Immune responses to influenza virus infection. Virus Res. 2011, 162, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Zanker, D.; Valkenburg, S.; Tan, B.; Kedzierska, K.; Zou, Q.M.; Doherty, P.C.; Chen, W. Systematic identification of immunodominant CD8+ T-cell responses to influenza A virus in HLA-A2 individuals. Proc. Natl. Acad. Sci. USA 2011, 108, 9178–9183. [Google Scholar] [CrossRef] [PubMed]
- Shaw, M.L.; Stone, K.L.; Colangelo, C.M.; Gulcicek, E.E.; Palese, P. Cellular proteins in influenza virus particles. PLOS Pathog. 2008, 4, e1000085. [Google Scholar] [CrossRef] [PubMed]
- Topham, D.J.; Tripp, R.A.; Doherty, P.C. CD8+ T cells clear influenza virus by perforin or Fas-dependent processes. J. Immunol. 1997, 159, 5197–5200. [Google Scholar] [PubMed]
- Hufford, M.M.; Kim, T.S.; Sun, J.; Braciale, T.J. Antiviral CD8+ T cell effector activities in situ are regulated by target cell type. J. Exp. Med. 2011, 208, 167–180. [Google Scholar] [CrossRef] [PubMed]
- Altenburg, A.F.; Rimmelzwaan, G.F.; de Vries, R.D. Virus-specific T cells as correlate of (cross-)protective immunity against influenza. Vaccine 2015, 33, 500–506. [Google Scholar] [CrossRef] [PubMed]
- Cerwenka, A.; Morgan, T.M.; Harmsen, A.G.; Dutton, R.W. Migration kinetics and final destination of type 1 and type 2 CD8 effector cells predict protection against pulmonary virus infection. J. Exp. Med. 1999, 189, 423–434. [Google Scholar] [CrossRef] [PubMed]
- Hamada, H.; Garcia-Hernandez Mde, L.; Reome, J.B.; Misra, S.K.; Strutt, T.M.; McKinstry, K.K.; Cooper, A.M.; Swain, S.L.; Dutton, R.W. Tc17, a unique subset of CD8 T cells that can protect against lethal influenza challenge. J. Immunol. 2009, 182, 3469–3481. [Google Scholar] [CrossRef] [PubMed]
- Belz, G.T.; Wodarz, D.; Diaz, G.; Nowak, M.A.; Doherty, P.C. Compromised influenza virus-specific CD8+-T-cell memory in CD4+-T-cell-deficient mice. J. Virol. 2002, 76, 12388–12393. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.C.; Bevan, M.J. Defective CD8 T cell memory following acute infection without CD4 T cell help. Science 2003, 300, 339–342. [Google Scholar] [CrossRef] [PubMed]
- Boyden, A.W.; Legge, K.L.; Waldschmidt, T.J. Pulmonary infection with influenza A virus induces site-specific germinal center and T follicular helper cell responses. PLOS ONE 2012, 7, e40733. [Google Scholar] [CrossRef] [PubMed]
- Hillaire, M.L.; Rimmelzwaan, G.F.; Kreijtz, J.H. Clearance of influenza virus infections by T cells: Risk of collateral damage? Curr. Opin. Virol. 2013, 3, 430–437. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.T.; Wherry, E.J.; Goldrath, A.W. Molecular regulation of effector and memory T cell differentiation. Nat. Immunol. 2014, 15, 1104–1115. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.; Iwasaki, A. Tissue-resident memory T cells. Immunol. Rev. 2013, 255, 165–181. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.H.; Mozdzanowska, K.; Palladino, G.; Gerhard, W. Heterosubtypic immunity to influenza type-A virus in mice—Effector mechanisms and their longevity. J. Immunol. 1994, 152, 1653–1661. [Google Scholar] [PubMed]
- Purwar, R.; Campbell, J.; Murphy, G.; Richards, W.G.; Clark, R.A.; . Kupper, T.S. Resident memory T cells (T(RM)) are abundant in human lung: Diversity, function, and antigen specificity. PLOS ONE 2011, 6, e16245. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Braciale, T.J. Role of T cell immunity in recovery from influenza virus infection. Curr. Opin. Virol. 2013, 3, 425–429. [Google Scholar] [CrossRef] [PubMed]
- Laurie, K.L.; Carolan, L.A.; Middleton, D.; Lowther, S.; Kelso, A.; Barr, I.G. Multiple infections with seasonal influenza A virus induce cross-protective immunity against A(H1N1) pandemic influenza virus in a ferret model. J. Infect. Dis. 2010, 202, 1011–1020. [Google Scholar] [CrossRef] [PubMed]
- Epstein, S.L. Prior H1N1 influenza infection and susceptibility of Cleveland Family Study participants during the H2N2 pandemic of 1957: An experiment of nature. J. Infect. Dis. 2006, 193, 49–53. [Google Scholar] [CrossRef] [PubMed]
- Hillaire, M.L.; Vogelzang-van Trierum, S.E.; Kreijtz, J.H.; de Mutsert, G.; Fouchier, R.A.; Osterhaus, A.D.; Rimmelzwaan, G.F. Human T-cells directed to seasonal influenza A virus cross-react with 2009 pandemic influenza A (H1N1) and swine-origin triple-reassortant H3N2 influenza viruses. J. Gen. Virol. 2013, 94, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Kreijtz, J.H.; de Mutsert, G.; van Baalen, C.A.; Fouchier, R.A.; Osterhaus, A.D.; Rimmelzwaan, G.F. Cross-recognition of avian H5N1 influenza virus by human cytotoxic T-lymphocyte populations directed to human influenza A virus. J. Virol. 2008, 82, 5161–5166. [Google Scholar] [CrossRef] [PubMed]
- Tu, W.; Mao, H.; Zheng, J.; Liu, Y.; Chiu, S.S.; Qin, G.; Chan, P.L.; Lam, K.T.; Guan, J.; Zhang, L.; et al. Cytotoxic T lymphocytes established by seasonal human influenza cross-react against 2009 pandemic H1N1 influenza virus. J. Virol. 2010, 84, 6527–6535. [Google Scholar] [CrossRef] [PubMed]
- Greenbaum, J.A.; Kotturi, M.F.; Kim, Y.; Oseroff, C.; Vaughan, K.; Salimi, N.; Vita, R.; Ponomarenko, J.; Scheuermann, R.H.; Sette, A.; et al. Pre-existing immunity against swine-origin H1N1 influenza viruses in the general human population. Proc. Natl. Acad. Sci. USA 2009, 106, 20365–20370. [Google Scholar] [CrossRef] [PubMed]
- Ge, X.; Tan, V.; Bollyky, P.L.; Standifer, N.E.; James, E.A.; Kwok, W.W. Assessment of seasonal influenza A virus-specific CD4 T-cell responses to 2009 pandemic H1N1 swine-origin influenza A virus. J. Virol. 2010, 84, 3312–3319. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, T.M.; Li, C.K.; Chui, C.S.; Huang, A.K.; Perkins, M.; Liebner, J.C.; Lambkin-Williams, R.; Gilbert, A.; Oxford, J.; Nicholas, B.; et al. Preexisting influenza-specific CD4+ T cells correlate with disease protection against influenza challenge in humans. Nat. Med. 2012, 18, 274–280. [Google Scholar] [CrossRef] [PubMed]
- Sridhar, S.; Begom, S.; Bermingham, A.; Hoschler, K.; Adamson, W.; Carman, W.; Bean, T.; Barclay, W.; Deeks, J.J.; Lalvani, A. Cellular immune correlates of protection against symptomatic pandemic influenza. Nat. Med. 2013, 19, 1305–1312. [Google Scholar] [CrossRef] [PubMed]
- McElhaney, J.E.; Xie, D.; Hager, W.D.; Barry, M.B.; Wang, Y.; Kleppinger, A.; Ewen, C.; Kane, K.P.; Bleackley, R.C. T cell responses are better correlates of vaccine protection in the elderly. J. Immunol. 2006, 176, 6333–6339. [Google Scholar] [CrossRef] [PubMed]
- Forrest, B.D.; Pride, M.W.; Dunning, A.J.; Capeding, M.R.; Chotpitayasunondh, T.; Tam, J.S.; Rappaport, R.; Eldridge, J.H.; Gruber, W.C. Correlation of cellular immune responses with protection against culture-confirmed influenza virus in young children. Clin. Vaccine Immunol. 2008, 15, 1042–1053. [Google Scholar] [CrossRef] [PubMed]
- Beran, J.; Vesikari, T.; Wertzova, V.; Karvonen, A.; Honegr, K.; Lindblad, N.; van Belle, P.; Peeters, M.; Innis, B.L.; Devaster, J.M. Efficacy of inactivated split-virus influenza vaccine against culture-confirmed influenza in healthy adults: a prospective, randomized, placebo-controlled trial. J. Infect. Dis. 2009, 200, 1861–1869. [Google Scholar] [CrossRef] [PubMed]
- Jefferson, T.O.; Rivetti, D.; di Pietrantonj, C.; Rivetti, A.; Demicheli, V. Vaccines for preventing influenza in healthy adults. Cochrane Database Syst. Rev. 2007. [Google Scholar] [CrossRef]
- De Jong, J.C.; Beyer, W.E.; Palache, A.M.; Rimmelzwaan, G.F.; Osterhaus, A.D. Mismatch between the 1997/1998 influenza vaccine and the major epidemic A(H3N2) virus strain as the cause of an inadequate vaccine-induced antibody response to this strain in the elderly. J. Med. Virol. 2000, 61, 94–99. [Google Scholar]
- Jefferson, T.; Rivetti, A.; Harnden, A.; di Pietrantonj, C.; Demicheli, V. Vaccines for preventing influenza in healthy children. Cochrane Database Syst. Rev. 2008. [Google Scholar] [CrossRef]
- Jain, V.K.; Rivera, L.; Zaman, K.; Espos, R.A., Jr.; Sirivichayakul, C.; Quiambao, B.P.; Rivera-Medina, D.M.; Kerdpanich, P.; Ceyhan, M.; Dinleyici, E.C.; et al. Vaccine for prevention of mild and moderate-to-severe influenza in children. N. Engl. J. Med. 2013, 369, 2481–2491. [Google Scholar] [CrossRef] [PubMed]
- Heiny, A.T.; Miotto, O.; Srinivasan, K.N.; Khan, A.M.; Zhang, G.L.; Brusic, V.; Tan, T.W.; August, J.T. Evolutionarily conserved protein sequences of influenza a viruses, avian and human, as vaccine targets. PLOS ONE 2007, 2, e1190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tekamp, P.A.; Penhoet, E.E. Quantification of influenza virus messenger RNAs. J. Gen. Virol. 1980, 47, 449–459. [Google Scholar] [CrossRef] [PubMed]
- Osterholm, M.T.; Kelley, N.S.; Sommer, A.; Belongia, E.A. Efficacy and effectiveness of influenza vaccines: a systematic review and meta-analysis. Lancet Infect. Dis. 2012, 12, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Lambe, T. Novel viral vectored vaccines for the prevention of influenza. Mol. Med. 2012, 18, 1153–1160. [Google Scholar] [CrossRef] [PubMed]
- Barnes, E.; Folgori, A.; Capone, S.; Swadling, L.; Aston, S.; Kurioka, A.; Meyer, J.; Huddart, R.; Smith, K.; Townsend, R.; et al. Novel adenovirus-based vaccines induce broad and sustained T cell responses to HCV in man. Sci. Transl. Med. 2012. [Google Scholar] [CrossRef]
- Keefer, M.C.; Gilmour, J.; Hayes, P.; Gill, D.; Kopycinski, J.; Cheeseman, H.; Cashin-Cox, M.; Naarding, M.; Clark, L.; Fernandez, N.; et al. A phase I double blind, placebo-controlled, randomized study of a multigenic HIV-1 adenovirus subtype 35 vector vaccine in healthy uninfected adults. PLOS ONE 2012, 7, e41936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ewer, K.J.; O’Hara, G.A.; Duncan, C.J.; Collins, K.A.; Sheehy, S.H.; Reyes-Sandoval, A.; Goodman, A.L.; Edwards, N.J.; Elias, S.C.; Halstead, F.D.; et al. Protective CD8+ T-cell immunity to human malaria induced by chimpanzee adenovirus-MVA immunisation. Nat. Commun. 2013. [Google Scholar] [CrossRef]
- Sheehy, S.H.; Duncan, C.J.; Elias, S.C.; Choudhary, P.; Biswas, S.; Halstead, F.D.; Collins, K.A.; Edwards, N.J.; Douglas, A.D.; Anagnostou, N.A.; et al. ChAd63-MVA-vectored blood-stage malaria vaccines targeting MSP1 and AMA1: Assessment of efficacy against mosquito bite challenge in humans. Mol. Ther. 2012, 20, 2355–2368. [Google Scholar] [CrossRef] [PubMed]
- Antrobus, R.D.; Berthoud, T.K.; Mullarkey, C.E.; Hoschler, K.; Coughlan, L.; Zambon, M.; Hill, A.V.; Gilbert, S.C. Coadministration of seasonal influenza vaccine and MVA-NP+M1 simultaneously achieves potent humoral and cell-mediated responses. Mol. Ther. 2014, 22, 233–238. [Google Scholar] [CrossRef] [PubMed]
- Antrobus, R.D.; Coughlan, L.; Berthoud, T.K.; Dicks, M.D.; Hill, A.V.; Lambe, T.; Gilbert, S.C. Clinical assessment of a novel recombinant simian adenovirus ChAdOx1 as a vectored vaccine expressing conserved influenza A antigens. Mol. Ther. 2014, 22, 668–674. [Google Scholar] [CrossRef] [PubMed]
- Antrobus, R.D.; Lillie, P.J.; Berthoud, T.K.; Spencer, A.J.; McLaren, J.E.; Ladell, K.; Lambe, T.; Milicic, A.; Price, D.A.; Hill, A.V.; et al. A T cell-inducing influenza vaccine for the elderly: Safety and immunogenicity of MVA-NP+M1 in adults aged over 50 years. PLOS ONE 2012, 7, e48322. [Google Scholar] [CrossRef] [PubMed]
- Lillie, P.J.; Berthoud, T.K.; Powell, T.J.; Lambe, T.; Mullarkey, C.; Spencer, A.J.; Hamill, M.; Peng, Y.; Blais, M.E.; Duncan, C.J.; et al. Preliminary assessment of the efficacy of a T-cell-based influenza vaccine, MVA-NP+M1, in humans. Clin. Infect. Dis. 2012, 55, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Berthoud, T.K.; Hamill, M.; Lillie, P.J.; Hwenda, L.; Collins, K.A.; Ewer, K.J.; Milicic, A.; Poyntz, H.C.; Lambe, T.; Fletcher, H.A.; et al. Potent CD8+ T-cell immunogenicity in humans of a novel heterosubtypic influenza A vaccine, MVA-NP+M1. Clin. Infect. Dis. 2011, 52, 1–7. [Google Scholar]
- Powell, T.J.; Peng, Y.; Berthoud, T.K.; Blais, M.E.; Lillie, P.J.; Hill, A.V.; Rowland-Jones, S.L.; McMichael, A.J.; Gilbert, S.C.; Dong, T. Examination of influenza specific T cell responses after influenza virus challenge in individuals vaccinated with MVA-NP+M1 vaccine. PLOS ONE 2013, 8, e62778. [Google Scholar] [CrossRef] [PubMed]
- Eichelberger, M.; Golding, H.; Hess, M.; Weir, J.; Subbarao, K.; Luke, C.J. FDA/NIH/WHO public workshop on immune correlates of protection against influenza A viruses in support of pandemic vaccine development, Bethesda, Maryland, US, December 10–11, 2007. Vaccine 2008, 26, 4299–4303. [Google Scholar] [CrossRef] [PubMed]
- Grohskopf, L.A.; Shay, D.K.; Shimabukuro, T.T.; Sokolow, L.Z.; Keitel, W.A.; Bresee, J.S.; Bresee, N.J. Prevention and Control of Seasonal Influenza with Vaccines: Recommendations of the Advisory Committee on Immunization Practices—United States, 2013–2014. Available online: http://www.cdc.gov/mmwr/preview/mmwrhtml/rr6207a1.htm (accessed on 10 February 2015).
- Nichol, K.L.; Mallon, K.P.; Mendelman, P.M. Cost benefit of influenza vaccination in healthy, working adults: An economic analysis based on the results of a clinical trial of trivalent live attenuated influenza virus vaccine. Vaccine 2003, 21, 2207–2217. [Google Scholar] [CrossRef] [PubMed]
- Remick, D.G.; Newcomb, D.E.; Friedland, J.S. Whole-blood assays for cytokine production. Methods Mol. Med. 2000, 36, 101–112. [Google Scholar] [PubMed]
- Calarota, S.A.; Baldanti, F. Enumeration and characterization of human memory T cells by enzyme-linked immunospot assays. Clin. Dev. Immunol. 2013. [Google Scholar] [CrossRef]
- Seder, R.A.; Darrah, P.A.; Roederer, M. T-cell quality in memory and protection: Implications for vaccine design. Nat. Rev. Immunol. 2008, 8, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Leng, S.X.; McElhaney, J.E.; Walston, J.D.; Xie, D.; Fedarko, N.S.; Kuchel, G.A. ELISA and multiplex technologies for cytokine measurement in inflammation and aging research. J. Gerontol. A Biol. Sci. Med. Sci. 2008, 63, 879–884. [Google Scholar] [CrossRef] [PubMed]
- Czerkinsky, C.C.; Nilsson, L.A.; Nygren, H.; Ouchterlony, O.; Tarkowski, A. A solid-phase enzyme-linked immunospot (ELISPOT) assay for enumeration of specific antibody-secreting cells. J. Immunol. Methods 1983, 65, 109–121. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Caspell, R.; Karulin, A.Y.; Ahmad, M.; Haicheur, N.; Abdelsalam, A.; Johannesen, K.; Vignard, V.; Dudzik, P.; Georgakopoulou, K.; et al. ELISPOT assays provide reproducible esults among different laboratories for T-cell immune monitoring—Even in hands of ELISPOT-inexperienced investigators. J. Immunotoxicol. 2009, 6, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, A.C.; Martin, J.N.; Younger, S.R.; Bredt, B.M.; Epling, L.; Ronquillo, R.; Varma, A.; Deeks, S.G.; McCune, J.M.; Nixon, D.F.; et al. Comparison of the ELISPOT and cytokine flow cytometry assays for the enumeration of antigen-specific T cells. J. Immunol. Methods 2003, 283, 141–153. [Google Scholar] [CrossRef] [PubMed]
- Currier, J.R.; Kuta, E.G.; Turk, E.; Earhart, L.B.; Loomis-Price, L.; Janetzki, S.; Ferrari, G.; Birx, D.L.; Cox, J.H. A panel of MHC class I restricted viral peptides for use as a quality control for vaccine trial ELISPOT assays. J. Immunol. Methods 2002, 260, 157–172. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.G.; Liu, X.; Kaufhold, R.M.; Clair, J.; Caulfield, M.J. Development and validation of a gamma interferon ELISPOT assay for quantitation of cellular immune responses to varicella-zoster virus. Clin. Diagn. Lab. Immunol. 2001, 8, 871–879. [Google Scholar] [PubMed]
- Palzer, S.; Bailey, T.; Hartnett, C.; Grant, A.; Tsang, M.; Kalyuzhny, A.E. Simultaneous detection of multiple cytokines in ELISPOT assays. Methods Mol. Biol. 2005, 302, 273–288. [Google Scholar] [PubMed]
- Gazagne, A.; Claret, E.; Wijdenes, J.; Yssel, H.; Bousquet, F.; Levy, E.; Vielh, P.; Scotte, F.; Goupil, T.L.; Fridman, W.H.; et al. A Fluorospot assay to detect single T lymphocytes simultaneously producing multiple cytokines. J. Immunol. Methods 2003, 283, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Todryk, S.M.; Pathan, A.A.; Keating, S.; Porter, D.W.; Berthoud, T.; Thompson, F.; Klenerman, P.; Hill, A.V. The relationship between human effector and memory T cells measured by ex vivo and cultured ELISPOT following recent and distal priming. Immunology 2009, 128, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Todryk, S.M.; Bejon, P.; Mwangi, T.; Plebanski, M.; Urban, B.; Marsh, K.; Hill, A.V.; Flanagan, K.L. Correlation of memory T cell responses against TRAP with protection from clinical malaria, and CD4 CD25 high T cells with susceptibility in Kenyans. PLOS ONE 2008, 3, e2027. [Google Scholar] [CrossRef] [PubMed]
- Keating, S.M.; Bejon, P.; Berthoud, T.; Vuola, J.M.; Todryk, S.; Webster, D.P.; Dunachie, S.J.; Moorthy, V.S.; McConkey, S.J.; Gilbert, S.C.; et al. Durable human memory T cells quantifiable by cultured enzyme-linked immunospot assays are induced by heterologous prime boost immunization and correlate with protection against malaria. J. Immunol. 2005, 175, 5675–5680. [Google Scholar] [CrossRef] [PubMed]
- Goletti, D.; Butera, O.; Bizzoni, F.; Casetti, R.; Girardi, E.; Poccia, F. Region of difference 1 antigen-specific CD4+ memory T cells correlate with a favorable outcome of tuberculosis. J. Infect. Dis. 2006, 194, 984–992. [Google Scholar] [CrossRef] [PubMed]
- Calarota, S.A.; Foli, A.; Maserati, R.; Baldanti, F.; Paolucci, S.; Young, M.A.; Tsoukas, C.M.; Lisziewicz, J.; Lori, F. HIV-1-specific T cell precursors with high proliferative capacity correlate with low viremia and high CD4 counts in untreated individuals. J. Immunol. 2008, 180, 5907–5915. [Google Scholar] [CrossRef] [PubMed]
- Klenerman, P.; Cerundolo, V.; Dunbar, P.R. Tracking T cells with tetramers: new tales from new tools. Nat. Rev. Immunol. 2002, 2, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Vollers, S.S.; Stern, L.J. Class II major histocompatibility complex tetramer staining: Progress, problems, and prospects. Immunology 2008, 123, 305–313. [Google Scholar] [CrossRef]
- Dunbar, P.R.; Ogg, G.S.; Chen, J.; Rust, N.; van der Bruggen, P.; Cerundolo, V. Direct isolation, phenotyping and cloning of low-frequency antigen-specific cytotoxic T lymphocytes from peripheral blood. Curr. Biol. 1998, 8, 413–416. [Google Scholar] [CrossRef] [PubMed]
- Gotch, F.; Rothbard, J.; Howland, K.; Townsend, A.; McMichael, A. Cytotoxic T lymphocytes recognize a fragment of influenza virus matrix protein in association with HLA-A2. Nature 1987, 326, 881–882. [Google Scholar] [CrossRef] [PubMed]
- Maecker, H.T.; Hassler, J.; Payne, J.K.; Summers, A.; Comatas, K.; Ghanayem, M.; Morse, M.A.; Clay, T.M.; Lyerly, H.K.; Bhatia, S.; et al. Precision and linearity targets for validation of an IFNgamma ELISPOT, cytokine flow cytometry, and tetramer assay using CMV peptides. BMC Immunol. 2008. [Google Scholar] [CrossRef]
- Tan, P.T.; Heiny, A.T.; Miotto, O.; Salmon, J.; Marques, E.T.; Lemonnier, F.; August, J.T. Conservation and diversity of influenza A H1N1 HLA-restricted T cell epitope candidates for epitope-based vaccines. PLOS ONE 2010, 5, e8754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, P.T.; Khan, A.M.; August, J.T. Highly conserved influenza A sequences as T cell epitopes-based vaccine targets to address the viral variability. Hum. Vaccin. 2011, 7, 402–409. [Google Scholar] [CrossRef] [PubMed]
- Moise, L.; Terry, F.; Ardito, M.; Tassone, R.; Latimer, H.; Boyle, C.; Martin, W.D.; de Groot, A.S. Universal H1N1 influenza vaccine development: identification of consensus class II hemagglutinin and neuraminidase epitopes derived from strains circulating between 1980 and 2011. Hum. Vaccin. Immunother. 2013, 9, 1598–1607. [Google Scholar] [CrossRef] [PubMed]
- Nepom, G.T. MHC class II tetramers. J. Immunol. 2012, 188, 2477–2482. [Google Scholar] [CrossRef]
- Suni, M.A.; Picker, L.J.; Maino, V.C. Detection of antigen-specific T cell cytokine expression in whole blood by flow cytometry. J. Immunol. Methods 1998, 212, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Nomura, L.E.; Walker, J.M.; Maecker, H.T. Optimization of whole blood antigen-specific cytokine assays for CD4+ T cells. Cytometry 2000, 40, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Mittrucker, H.W.; Visekruna, A.; Huber, M. Heterogeneity in the differentiation and function of CD8+ T cells. Arch. Immunol. Ther. Exp. (Warsz.) 2014, 62, 449–458. [Google Scholar] [CrossRef]
- Zhu, J.; Yamane, H.; Paul, W.E. Differentiation of effector CD4 T cell populations (*). Annu. Rev. Immunol. 2010, 28, 445–489. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Chong, M.M.; Littman, D.R. Plasticity of CD4+ T cell lineage differentiation. Immunity 2009, 30, 646–655. [Google Scholar] [CrossRef] [PubMed]
- Boyton, R.J.; Altmann, D.M. Is selection for TCR affinity a factor in cytokine polarization? Trends Immunol. 2002, 23, 526–529. [Google Scholar] [CrossRef] [PubMed]
- Schroder, K.; Hertzog, P.J.; Ravasi, T.; Hume, D.A. Interferon-gamma: An overview of signals, mechanisms and functions. J. Leukoc. Biol. 2004, 75, 163–189. [Google Scholar] [CrossRef] [PubMed]
- Ciuffreda, D.; Comte, D.; Cavassini, M.; Giostra, E.; Buhler, L.; Perruchoud, M.; Heim, M.H.; Battegay, M.; Genne, D.; Mulhaupt, B.; et al. Polyfunctional HCV-specific T-cell responses are associated with effective control of HCV replication. Eur. J. Immunol. 2008, 38, 2665–2677. [Google Scholar] [CrossRef] [PubMed]
- Kannanganat, S.; Kapogiannis, B.G.; Ibegbu, C.; Chennareddi, L.; Goepfert, P.; Robinson, H.L.; Lennox, J.; Amara, R.R. Human immunodeficiency virus type 1 controllers but not noncontrollers maintain CD4 T cells coexpressing three cytokines. J. Virol. 2007, 81, 12071–12076. [Google Scholar] [CrossRef] [PubMed]
- Darrah, P.A.; Patel, D.T.; de Luca, P.M.; Lindsay, R.W.; Davey, D.F.; Flynn, B.J.; Hoff, S.T.; Andersen, P.; Reed, S.G.; Morris, S.L.; et al. Multifunctional TH1 cells define a correlate of vaccine-mediated protection against Leishmania major. Nat. Med. 2007, 13, 843–850. [Google Scholar] [CrossRef] [PubMed]
- Precopio, M.L.; Betts, M.R.; Parrino, J.; Price, D.A.; Gostick, E.; Ambrozak, D.R.; Asher, T.E.; Douek, D.C.; Harari, A.; Pantaleo, G.; et al. Immunization with vaccinia virus induces polyfunctional and phenotypically distinctive CD8+ T cell responses. J. Exp. Med. 2007, 204, 1405–1416. [Google Scholar] [CrossRef] [PubMed]
- Beveridge, N.E.; Price, D.A.; Casazza, J.P.; Pathan, A.A.; Sander, C.R.; Asher, T.E.; Ambrozak, D.R.; Precopio, M.L.; Scheinberg, P.; Alder, N.C.; et al. Immunisation with BCG and recombinant MVA85A induces long-lasting, polyfunctional Mycobacterium tuberculosis-specific CD4+ memory T lymphocyte populations. Eur. J. Immunol. 2007, 37, 3089–3100. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Z.; Zhang, M.; Zhu, Y.; Zheng, F.; Lu, P.; Liu, H.; Graner, M.W.; Zhou, B.; Chen, X. Multifunctional CD4 T cell responses in patients with active tuberculosis. Sci Rep 2012. [Google Scholar] [CrossRef]
- Shey, M.S.; Hughes, E.J.; de Kock, M.; Barnard, C.; Stone, L.; Kollmann, T.R.; Hanekom, W.A.; Scriba, T.J. Optimization of a whole blood intracellular cytokine assay for measuring innate cell responses to mycobacteria. J. Immunol. Methods 2012, 376, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Hanekom, W.A.; Hughes, J.; Mavinkurve, M.; Mendillo, M.; Watkins, M.; Gamieldien, H.; Gelderbloem, S.J.; Sidibana, M.; Mansoor, N.; Davids, V.; et al. Novel application of a whole blood intracellular cytokine detection assay to quantitate specific T-cell frequency in field studies. J. Immunol. Methods 2004, 291, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Hornef, M.W.; Wagner, H.J.; Kruse, A.; Kirchner, H. Cytokine production in a whole-blood assay after Epstein-Barr virus infection in vivo. Clin. Diagn. Lab. Immunol. 1995, 2, 209–213. [Google Scholar] [PubMed]
- Meddows-Taylor, S.; Shalekoff, S.; Kuhn, L.; Gray, G.E.; Tiemessen, C.T. Development of a whole blood intracellular cytokine staining assay for mapping CD4+ and CD8+ T-cell responses across the HIV-1 genome. J. Virol. Methods 2007, 144, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Stoddard, M.B.; Pinto, V.; Keiser, P.B.; Zollinger, W. Evaluation of a whole-blood cytokine release assay for use in measuring endotoxin activity of group B Neisseria meningitidis vaccines made from lipid A acylation mutants. Clin. Vaccine Immunol. 2010, 17, 98–107. [Google Scholar] [CrossRef] [PubMed]
- Waldrop, S.L.; Davis, K.A.; Maino, V.C.; Picker, L.J. Normal human CD4+ memory T cells display broad heterogeneity in their activation threshold for cytokine synthesis. J. Immunol. 1998, 161, 5284–5295. [Google Scholar] [PubMed]
- Salic, A.; Mitchison, T.J. A chemical method for fast and sensitive detection of DNA synthesis in vivo. Proc. Natl. Acad. Sci. USA 2008, 105, 2415–2420. [Google Scholar] [CrossRef] [PubMed]
- Buck, S.B.; Bradford, J.; Gee, K.R.; Agnew, B.J.; Clarke, S.T.; Salic, A. Detection of S-phase cell cycle progression using 5-ethynyl-2'-deoxyuridine incorporation with click chemistry, an alternative to using 5-bromo-2'-deoxyuridine antibodies. Biotechniques 2008, 44, 927–929. [Google Scholar] [CrossRef] [PubMed]
- Bradford, J.A.; Clarke, S.T. Dual-pulse labeling using 5-ethynyl-2'-deoxyuridine (EdU) and 5-bromo-2'-deoxyuridine (BrdU) in flow cytometry. Curr. Protoc. Cytom. 2011. [Google Scholar] [CrossRef]
- Brunner, K.T.; Mauel, J.; Cerottini, J.C.; Chapuis, B. Quantitative assay of the lytic action of immune lymphoid cells on 51-Cr-labelled allogeneic target cells in vitro; inhibition by isoantibody and by drugs. Immunology 1968, 14, 181–196. [Google Scholar] [PubMed]
- Zaritskaya, L.; Shurin, M.R.; Sayers, T.J.; Malyguine, A.M. New flow cytometric assays for monitoring cell-mediated cytotoxicity. Expert Rev. Vaccines 2010, 9, 601–616. [Google Scholar] [CrossRef] [PubMed]
- Sepp, A.; Binns, R.M.; Lechler, R.I. Improved protocol for colorimetric detection of complement-mediated cytotoxicity based on the measurement of cytoplasmic lactate dehydrogenase activity. J. Immunol. Methods 1996, 196, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Szekeres, J.; Pacsa, A.S.; Pejtsik, B. Measurement of lymphocyte cytotoxicity by assessing endogenous alkaline phosphatase activity of the target cells. J. Immunol. Methods 1981, 40, 151–154. [Google Scholar] [CrossRef] [PubMed]
- Roden, M.M.; Lee, K.H.; Panelli, M.C.; Marincola, F.M. A novel cytolysis assay using fluorescent labeling and quantitative fluorescent scanning technology. J. Immunol. Methods 1999, 226, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Van Baalen, C.A.; Gruters, R.A.; Berkhoff, E.G.; Osterhaus, A.D.; Rimmelzwaan, G.F. FATT-CTL assay for detection of antigen-specific cell-mediated cytotoxicity. Cytometry A 2008, 73, 1058–1065. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, J.E.; Sherwood, S.W.; Clayberger, C. A novel method for measuring CTL and NK cell-mediated cytotoxicity using annexin V and two-color flow cytometry. J. Immunol. Methods 1999, 224, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Jerome, K.R.; Sloan, D.D.; Aubert, M. Measurement of CTL-induced cytotoxicity: The caspase 3 assay. Apoptosis 2003, 8, 563–571. [Google Scholar] [CrossRef]
- Lecoeur, H.; Fevrier, M.; Garcia, S.; Riviere, Y.; Gougeon, M.L. A novel flow cytometric assay for quantitation and multiparametric characterization of cell-mediated cytotoxicity. J. Immunol. Methods 2001, 253, 177–187. [Google Scholar] [CrossRef]
- Sanchez-Ruiz, Y.; Valitutti, S.; Dupre, L. Stepwise maturation of lytic granules during differentiation and activation of human CD8+ T lymphocytes. PLOS ONE 2011, 6, e27057. [Google Scholar] [CrossRef] [PubMed]
- Peters, P.J.; Borst, J.; Oorschot, V.; Fukuda, M.; Krahenbuhl, O.; Tschopp, J.; Slot, J.W.; Geuze, H.J. Cytotoxic T lymphocyte granules are secretory lysosomes, containing both perforin and granzymes. J. Exp. Med. 1991, 173, 1099–1109. [Google Scholar] [CrossRef] [PubMed]
- Fruh, K.; Simmen, K.; Luukkonen, B.G.; Bell, Y.C.; Ghazal, P. Virogenomics: A novel approach to antiviral drug discovery. Drug Discov. Today 2001, 6, 621–627. [Google Scholar] [CrossRef]
- Bucasas, K.L.; Franco, L.M.; Shaw, C.A.; Bray, M.S.; Wells, J.M.; Nino, D.; Arden, N.; Quarles, J.M.; Couch, R.B.; Belmont, J.W. Early patterns of gene expression correlate with the humoral immune response to influenza vaccination in humans. J. Infect. Dis. 2011, 203, 921–929. [Google Scholar] [CrossRef] [PubMed]
- Woods, C.W.; McClain, M.T.; Chen, M.; Zaas, A.K.; Nicholson, B.P.; Varkey, J.; Veldman, T.; Kingsmore, S.F.; Huang, Y.; Lambkin-Williams, R.; et al. A host transcriptional signature for presymptomatic detection of infection in humans exposed to influenza H1N1 or H3N2. PLOS ONE 2013, 8, e52198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davenport, E.E.; Antrobus, R.D.; Lillie, P.J.; Gilbert, S.; Knight, J.C. Transcriptomic profiling facilitates classification of response to influenza challenge. J. Mol. Med. (Berl.) 2015, 93, 105–114. [Google Scholar] [CrossRef]
- Huang, Y.; Zaas, A.K.; Rao, A.; Dobigeon, N.; Woolf, P.J.; Veldman, T.; Oien, N.C.; McClain, M.T.; Varkey, J.B.; Nicholson, B.; et al. Temporal dynamics of host molecular responses differentiate symptomatic and asymptomatic influenza a infection. PLOS Genet. 2011, 7, e1002234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pritchard, C.C.; Cheng, H.H.; Tewari, M. MicroRNA profiling: Approaches and considerations. Nat. Rev. Genet. 2012, 13, 358–369. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.P.; Wu, H.; Zhu, J.; Miao, H. Systematic identification of transcriptional and post-transcriptional regulations in human respiratory epithelial cells during influenza A virus infection. BMC Bioinform. 2014. [Google Scholar] [CrossRef]
- Loveday, E.K.; Svinti, V.; Diederich, S.; Pasick, J.; Jean, F. Temporal- and strain-specific host microRNA molecular signatures associated with swine-origin H1N1 and avian-origin H7N7 influenza A virus infection. J. Virol. 2012, 86, 6109–6122. [Google Scholar] [CrossRef] [PubMed]
- Skovgaard, K.; Cirera, S.; Vasby, D.; Podolska, A.; Breum, S.O.; Durrwald, R.; Schlegel, M.; Heegaard, P.M. Expression of innate immune genes, proteins and microRNAs in lung tissue of pigs infected experimentally with influenza virus (H1N2). Innate Immun. 2013, 19, 531–544. [Google Scholar] [CrossRef] [PubMed]
- Tambyah, P.A.; Sepramaniam, S.; Mohamed Ali, J.; Chai, S.C.; Swaminathan, P.; Armugam, A.; Jeyaseelan, K. microRNAs in circulation are altered in response to influenza A virus infection in humans. PLOS ONE 2013, 8, e76811. [Google Scholar] [CrossRef] [PubMed]
- Satti, I.; Meyer, J.; Harris, S.A.; Manjaly Thomas, Z.R.; Griffiths, K.; Antrobus, R.D.; Rowland, R.; Ramon, R.L.; Smith, M.; Sheehan, S.; et al. Safety and immunogenicity of a candidate tuberculosis vaccine MVA85A delivered by aerosol in BCG-vaccinated healthy adults: A phase 1, double-blind, randomised controlled trial. Lancet Infect. Dis. 2014, 14, 939–946. [Google Scholar] [CrossRef] [PubMed]
- Sarzotti-Kelsoe, M.; Needham, L.K.; Rountree, W.; Bainbridge, J.; Gray, C.M.; Fiscus, S.A.; Ferrari, G.; Stevens, W.S.; Stager, S.L.; Binz, W.; et al. The Center for HIV/AIDS Vaccine Immunology (CHAVI) multi-site quality assurance program for cryopreserved human peripheral blood mononuclear cells. J. Immunol. Methods 2014, 409, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Ducar, C.; Smith, D.; Pinzon, C.; Stirewalt, M.; Cooper, C.; McElrath, M.J.; Hural, J.; N.H.V.T. Network. Benefits of a comprehensive quality program for cryopreserved PBMC covering 28 clinical trials sites utilizing an integrated, analytical web-based portal. J. Immunol. Methods 2014, 409, 9–20. [Google Scholar]
- Sambor, A.; Garcia, A.; Berrong, M.; Pickeral, J.; Brown, S.; Rountree, W.; Sanchez, A.; Pollara, J.; Frahm, N.; Keinonen, S.; et al. Establishment and maintenance of a PBMC repository for functional cellular studies in support of clinical vaccine trials. J. Immunol. Methods 2014, 409, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Boaz, M.J.; Hayes, P.; Tarragona, T.; Seamons, L.; Cooper, A.; Birungi, J.; Kitandwe, P.; Semaganda, A.; Kaleebu, P.; Stevens, G.; et al. Concordant proficiency in measurement of T-cell immunity in human immunodeficiency virus vaccine clinical trials by peripheral blood mononuclear cell and enzyme-linked immunospot assays in laboratories from three continents. Clin. Vaccine Immunol. 2009, 16, 147–155. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Coughlan, L.; Lambe, T. Measuring Cellular Immunity to Influenza: Methods of Detection, Applications and Challenges. Vaccines 2015, 3, 293-319. https://doi.org/10.3390/vaccines3020293
Coughlan L, Lambe T. Measuring Cellular Immunity to Influenza: Methods of Detection, Applications and Challenges. Vaccines. 2015; 3(2):293-319. https://doi.org/10.3390/vaccines3020293
Chicago/Turabian StyleCoughlan, Lynda, and Teresa Lambe. 2015. "Measuring Cellular Immunity to Influenza: Methods of Detection, Applications and Challenges" Vaccines 3, no. 2: 293-319. https://doi.org/10.3390/vaccines3020293
APA StyleCoughlan, L., & Lambe, T. (2015). Measuring Cellular Immunity to Influenza: Methods of Detection, Applications and Challenges. Vaccines, 3(2), 293-319. https://doi.org/10.3390/vaccines3020293