
Cancer Dormancy: A Regulatory Role for Endogenous Immunity in Establishing and Maintaining the Tumor Dormant State

Abstract
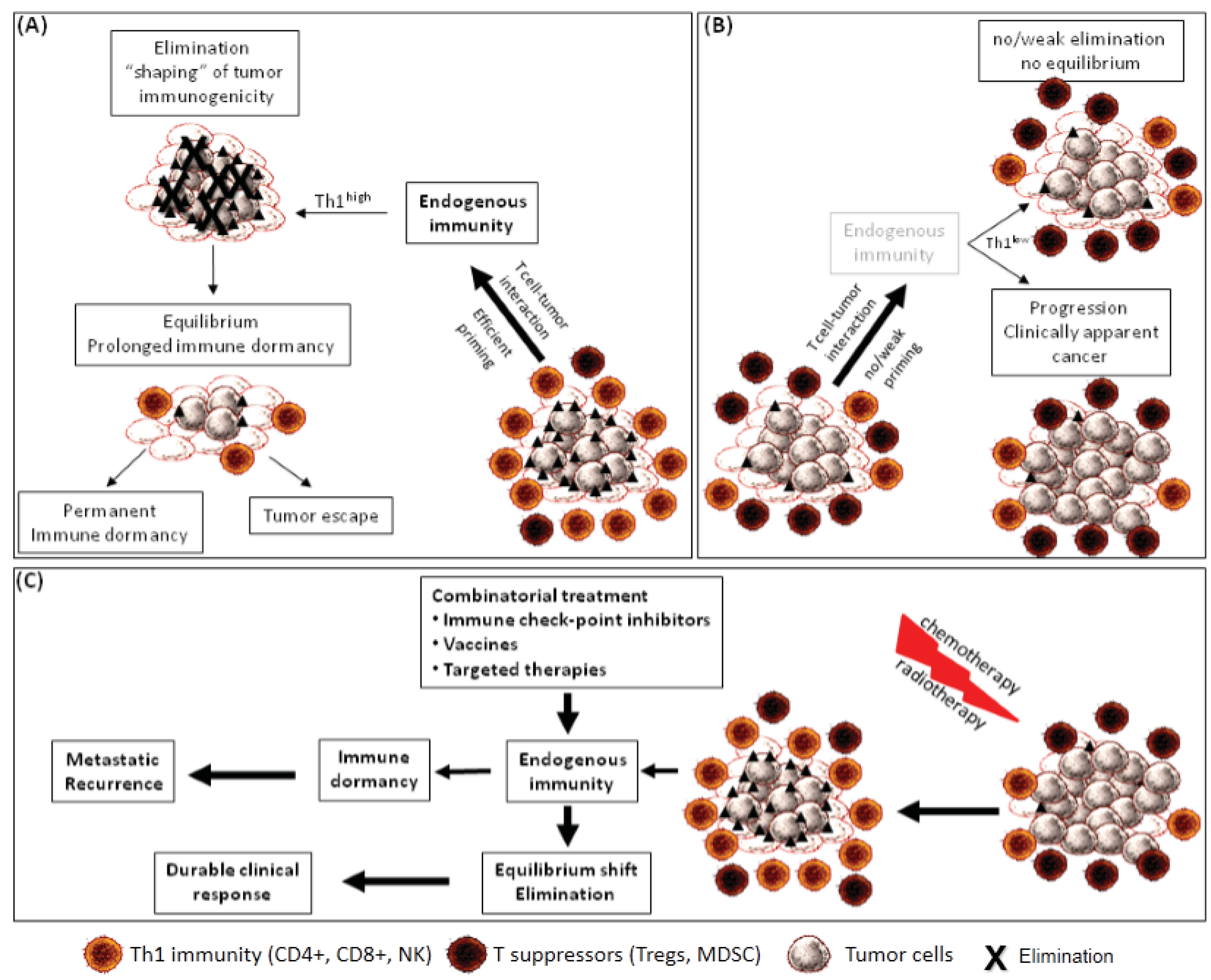
:1. Introduction
2. Modulation of the Endogenous Antitumor Immunity

{kind=link}
{kind=link}
| Study Name | Targeted Antigen | Vaccine | Cancer Type | Refs. |
|---|---|---|---|---|
| IMPACT | PAP | Mo/DCs PAP-GM-CSF | mCRPC | [12] |
| PROSPECT | PSA | rV-PSA-TRICOM rF-PSA-TRICOM | mCRPC | [13] |
| PANVAC | MUC1/CEA | DCs rV-PSA-TRICOM rF-PSA-TRICOM | Colorectal | [14] |
| GVAX | Multiple on allogeneic tumor lines | T47D and SKBR3 lines secreting GM-CSF | Breast | [15] |
| DERMA | MAGE-A3 | Recombinant protein | Melanoma | [16] |
| MAGRIT | MAGE-A3 | Recombinant protein | NSCLC | [17] |
| IMPRINT | Mixture of 11 naturally presented renal Ca tumor peptides | soluble | Renal | [18] |
| BIOVAXID | Id-IgG on B-cell lymphoma | Id-IgG-KLH | Follicular lymphoma | [19] |
| GV1001 | Telomerase | Soluble 16-mer | Melanoma | [20] |
| TG4010 | MUC1 | MVA MUC-1-IL-2 | Prostate NSCLC | [21,22,23] |
| CDX110 | EGFRvIII | Soluble 13-mer | Glioblastoma | [24] |
| Stimuvax | MUC1 | 25-mer MUC1 liposome (BLP25) | NSCLC | [25] |
3. Tumor Dormancy as a Result of Endogenous Immune Surveillance
| Treatment | Mechanisms for Synergistic Effects |
|---|---|
| Chemotherapy | “Immunogenic” cell death Alterations in tumor cell phenotype Inhibition of regulatory cells Homing to tumors |
| Radiation | Increased tumor antigen presentation Release of pro-inflammatory cytokines Promotion of tumor antigen cross-presentation |
| Kinase inhibitors | Promotion of DC maturation T cell priming, differentiation in memory cells Sensitization of tumor cells to immune-mediated killing Impairment of immunosuppression |
| Hormonal therapy | Increase of T cell levels in lymphoid tissues T cell sensitivity to antigen-specific stimulation T cell infiltration into the prostate |
4. Endogenous Immunity and Tumor Cells as Major Players for Tumor Dormancy

5. Local and Peripheral Endogenous Immunity in Immune Equilibrium
6. Immune Editing, Dormancy and Escape from Immune Surveillance

7. Immune Adaptation
8. Cancer Stem Cells in Clinical Tumor Dormancy
9. Conclusions and Future Directions
Acknowledgements
Author Contributions
Conflicts of Interest
References
- Shankaran, V.; Ikeda, H.; Bruce, A.T.; White, J.M.; Swanson, P.E.; Old, L.J.; Schreiber, R.D. IFNgamma and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature 2001, 410, 1107–1111. [Google Scholar] [CrossRef] [PubMed]
- Manjili, M.H. The inherent premise of immunotherapy for cancer dormancy. Cancer Res. 2014, 74, 6745–6749. [Google Scholar] [CrossRef] [PubMed]
- Quezada, S.A.; Peggs, K.S.; Simpson, T.R.; Allison, J.P. Shifting the equilibrium in cancer immunoediting: From tumor tolerance to eradication. Immunol. Rev. 2011, 241, 104–118. [Google Scholar] [CrossRef] [PubMed]
- Bindea, G.; Mlecnik, B.; Fridman, W.H.; Pages, F.; Galon, J. Natural immunity to cancer in humans. Curr. Opin. Immunol. 2010, 22, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Fridman, W.H.; Pages, F.; Sautes-Fridman, C.; Galon, J. The immune contexture in human tumours: Impact on clinical outcome. Nature Rev. Cancer 2012, 12, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Schlom, J.; Hodge, J.W.; Palena, C.; Tsang, K.Y.; Jochems, C.; Greiner, J.W.; Farsaci, B.; Madan, R.A.; Heery, C.R.; Gulley, J.L. Therapeutic cancer vaccines. Adv. Cancer Res. 2014, 121, 67–124. [Google Scholar] [PubMed]
- Melero, I.; Gaudernack, G.; Gerritsen, W.; Huber, C.; Parmiani, G.; Scholl, S.; Thatcher, N.; Wagstaff, J.; Zielinski, C.; Faulkner, I.; et al. Therapeutic vaccines for cancer: An overview of clinical trials. Nature Rev. Clin. Oncol. 2014, 11, 509–524. [Google Scholar] [CrossRef] [PubMed]
- Vanneman, M.; Dranoff, G. Combining immunotherapy and targeted therapies in cancer treatment. Nat. Rev. Cancer 2012, 12, 237–251. [Google Scholar] [CrossRef] [PubMed]
- Gajewski, T.F.; Woo, S.R.; Zha, Y.; Spaapen, R.; Zheng, Y.; Corrales, L.; Spranger, S. Cancer immunotherapy strategies based on overcoming barriers within the tumor microenvironment. Curr. Opin. Immunol. 2013, 25, 268–276. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vacchelli, E.; Eggermont, A.; Fridman, W.H.; Galon, J.; Sautes-Fridman, C.; Tartour, E.; Zitvogel, L.; Kroemer, G. Trial watch: Adoptive cell transfer immunotherapy. Oncoimmunology 2012, 1, 306–315. [Google Scholar] [CrossRef] [PubMed]
- Yamada, A.; Sasada, T.; Noguchi, M.; Itoh, K. Next-generation peptide vaccines for advanced cancer. Cancer Sci. 2013, 104, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Kantoff, P.W.; Higano, C.S.; Shore, N.D.; Berger, E.R.; Small, E.J.; Penson, D.F.; Redfern, C.H.; Ferrari, A.C.; Dreicer, R.; Sims, R.B.; et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N. Engl. J. Med. 2010, 363, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Gulley, J.L.; Giacchino, J.L.; Breitmeyer, J.B.; Franzusoff, A.J.; Panicali, D.; Schlom, J.; Kantoff, P.W. Prospect: A randomized double-blind phase 3 efficacy study of PROSTVAC-VF immunotherapy in men with asymptomatic/minimally symptomatic metastatic castration-resistant prostate cancer. J. Clin. Oncol. 2015, 33. Abstract Number: TPS5081. [Google Scholar]
- Morse, M.A.; Niedzwiecki, D.; Marshall, J.L.; Garrett, C.; Chang, D.Z.; Aklilu, M.; Crocenzi, T.S.; Cole, D.J.; Dessureault, S.; Hobeika, A.C.; et al. A randomized phase II study of immunization with dendritic cells modified with poxvectors encoding CEA and MUC1 compared with the same poxvectors plus GM-CSF for resected metastatic colorectal cancer. Ann. Surg. 2013, 258, 879–886. [Google Scholar] [CrossRef] [PubMed]
- Emens, L.A.; Asquith, J.M.; Leatherman, J.M.; Kobrin, B.J.; Petrik, S.; Laiko, M.; Levi, J.; Daphtary, M.M.; Biedrzycki, B.; Wolff, A.C.; et al. Timed sequential treatment with cyclophosphamide, doxorubicin, and an allogeneic granulocyte-macrophage colony-stimulating factor-secreting breast tumor vaccine: A chemotherapy dose-ranging factorial study of safety and immune activation. J. Clin. Oncol. 2009, 27, 5911–5918. [Google Scholar] [CrossRef] [PubMed]
- Kruit, W.H.; Suciu, S.; Dreno, B.; Mortier, L.; Robert, C.; Chiarion-Sileni, V.; Maio, M.; Testori, A.; Dorval, T.; Grob, J.J.; et al. Selection of immunostimulant AS15 for active immunization with MAGE-A3 protein: Results of a randomized phase II study of the European Organisation for Research and Treatment of Cancer Melanoma Group in Metastatic Melanoma. J. Clin. Oncol. 2013, 31, 2413–2420. [Google Scholar] [CrossRef] [PubMed]
- Vansteenkiste, J.; Zielinski, M.; Linder, A.; Dahabreh, J.; Gonzalez, E.E.; Malinowski, W.; Lopez-Brea, M.; Vanakesa, T.; Jassem, J.; Kalofonos, H.; et al. Adjuvant MAGE-A3 immunotherapy in resected non-small-cell lung cancer: Phase II randomized study results. J. Clin. Oncol. 2013, 31, 2396–2403. [Google Scholar] [CrossRef] [PubMed]
- Rini, B.I.; Eisen, T.; Stenzl, A.; Brugger, W.; Weinschenk, T.; Mahr, A.; Fritsche, J.; Hilf, N.; Mendrzyk, R.; Lindner, J.; et al. IMA901 multipeptide vaccine randomized international PHASE III trial (IMPRINT): A randomized, controlled study investigating IMA901 multipeptide cancer vaccine in patients receiving sunitinib as first-line therapy for advanced/metastatic RCC. J. Clin. Oncol. 2015, 33. Abtract Number: TPS183. [Google Scholar]
- Schuster, S.J.; Neelapu, S.S.; Gause, B.L.; Janik, J.E.; Muggia, F.M.; Gockerman, J.P.; Winter, J.N.; Flowers, C.R.; Nikcevich, D.A.; Sotomayor, E.M.; et al. Vaccination with patient-specific tumor-derived antigen in first remission improves disease-free survival in follicular lymphoma. J. Clin. Oncol. 2011, 29, 2787–2794. [Google Scholar] [CrossRef] [PubMed]
- Kyte, J.A.; Gaudernack, G.; Dueland, S.; Trachsel, S.; Julsrud, L.; Aamdal, S. Telomerase peptide vaccination combined with temozolomide: A clinical trial in stage iv melanoma patients. Clin. Cancer Res. 2011, 17, 4568–4580. [Google Scholar] [CrossRef] [PubMed]
- Dreicer, R.; Stadler, W.M.; Ahmann, F.R.; Whiteside, T.; Bizouarne, N.; Acres, B.; Limacher, J.M.; Squiban, P.; Pantuck, A. MVA-MUC1-IL2 vaccine immunotherapy (TG4010) improves PSA doubling time in patients with prostate cancer with biochemical failure. Investig. New Drugs 2009, 27, 379–386. [Google Scholar] [CrossRef] [PubMed]
- Ramlau, R.; Quoix, E.; Rolski, J.; Pless, M.; Lena, H.; Levy, E.; Krzakowski, M.; Hess, D.; Tartour, E.; Chenard, M.P.; et al. A phase II study OF TG4010 (MVA-MUC1-IL2) in association with chemotherapy in patients with stage III/IV non-small cell lung cancer. J. Thorac. Oncol. 2008, 3, 735–744. [Google Scholar] [CrossRef] [PubMed]
- Quoix, E.; Ramlau, R.; Westeel, V.; Papai, Z.; Madroszyk, A.; Riviere, A.; Koralewski, P.; Breton, J.L.; Stoelben, E.; Braun, D.; et al. Therapeutic vaccination with TG4010 and first-line chemotherapy in advanced non-small-cell lung cancer: A controlled phase 2B trial. Lancet. Oncol. 2011, 12, 1125–1133. [Google Scholar] [CrossRef]
- Sampson, J.H.; Heimberger, A.B.; Archer, G.E.; Aldape, K.D.; Friedman, A.H.; Friedman, H.S.; Gilbert, M.R.; Herndon, J.E., 2nd; McLendon, R.E.; Mitchell, D.A.; et al. Immunologic escape after prolonged progression-free survival with epidermal growth factor receptor variant III peptide vaccination in patients with newly diagnosed glioblastoma. J. Clin. Oncol. 2010, 28, 4722–4729. [Google Scholar] [CrossRef] [PubMed]
- Butts, C.; Murray, N.; Maksymiuk, A.; Goss, G.; Marshall, E.; Soulieres, D.; Cormier, Y.; Ellis, P.; Price, A.; Sawhney, R.; et al. Randomized phase IIB trial of BLP25 liposome vaccine in stage IIIB and IV non-small-cell lung cancer. J. Clin. Oncol. 2005, 23, 6674–6681. [Google Scholar] [CrossRef] [PubMed]
- Melero, I.; Grimaldi, A.M.; Perez-Gracia, J.L.; Ascierto, P.A. Clinical development of immunostimulatory monoclonal antibodies and opportunities for combination. Clin. Cancer Res. 2013, 19, 997–1008. [Google Scholar] [CrossRef] [PubMed]
- Jochems, C.; Tucker, J.A.; Tsang, K.Y.; Madan, R.A.; Dahut, W.L.; Liewehr, D.J.; Steinberg, S.M.; Gulley, J.L.; Schlom, J. A combination trial of vaccine plus ipilimumab in metastatic castration-resistant prostate cancer patients: Immune correlates. Cancer Immunol. Immunother. 2014, 63, 407–418. [Google Scholar] [CrossRef] [PubMed]
- Madan, R.A.; Mohebtash, M.; Arlen, P.M.; Vergati, M.; Rauckhorst, M.; Steinberg, S.M.; Tsang, K.Y.; Poole, D.J.; Parnes, H.L.; Wright, J.J.; et al. Ipilimumab and a poxviral vaccine targeting prostate-specific antigen in metastatic castration-resistant prostate cancer: A phase 1 dose-escalation trial. Lancet. Oncol. 2012, 13, 501–508. [Google Scholar] [CrossRef]
- Dranoff, G. Immunotherapy at large: Balancing tumor immunity and inflammatory pathology. Nat. Med. 2013, 19, 1100–1101. [Google Scholar] [CrossRef] [PubMed]
- Kwek, S.S.; Cha, E.; Fong, L. Unmasking the immune recognition of prostate cancer with CTLA4 blockade. Nat. Rev. Cancer 2012, 12, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Luke, J.J.; Ott, P.A. Kinase inhibitors and immune check-point blockade for the treatment of metastatic melanoma and advanced cancer: Synergistic or antagonistic? Expert Opin. Pharmacother. 2013, 14, 2457–2462. [Google Scholar] [CrossRef] [PubMed]
- Peng, W.; Lizee, G.; Hwu, P. Blockade of the PD-1 pathway enhances the efficacy of adoptive cell therapy against cancer. Oncoimmunology 2013, 2, e22691. [Google Scholar] [CrossRef] [PubMed]
- Galon, J.; Angell, H.K.; Bedognetti, D.; Marincola, F.M. The continuum of cancer immunosurveillance: Prognostic, predictive, and mechanistic signatures. Immunity 2013, 39, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Galon, J.; Mlecnik, B.; Bindea, G.; Angell, H.K.; Berger, A.; Lagorce, C.; Lugli, A.; Zlobec, I.; Hartmann, A.; Bifulco, C.; et al. Towards the introduction of the “immunoscore” in the classification of malignant tumours. J. Pathol. 2014, 232, 199–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baxevanis, C.N.; Papamichail, M.; Perez, S.A. Immune classification of colorectal cancer patients: Impressive but how complete? Expert Opin. Pharmacother. 2013, 13, 517–526. [Google Scholar] [CrossRef] [PubMed]
- Denkert, C.; von Minckwitz, G.; Brase, J.C.; Sinn, B.V.; Gade, S.; Kronenwett, R.; Pfitzner, B.M.; Salat, C.; Loi, S.; Schmitt, W.D.; et al. Tumor-infiltrating lymphocytes and response to neoadjuvant chemotherapy with or without carboplatin in human epidermal growth factor receptor 2-positive and triple-negative primary breast cancers. J. Clin. Oncol. 2015, 33, 983–991. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, H.; Koo, S.L.; Dent, R.; Tan, P.H.; Iqbal, J. Role of inflammatory infiltrates in triple negative breast cancer. J. Clin. Pathol. 2015, 68, 506–510. [Google Scholar] [CrossRef] [PubMed]
- Zitvogel, L.; Kepp, O.; Kroemer, G. Immune parameters affecting the efficacy of chemotherapeutic regimens. Nat. Rev. Clin. Oncol. 2011, 8, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Zitvogel, L.; Galluzzi, L.; Smyth, M.J.; Kroemer, G. Mechanism of action of conventional and targeted anticancer therapies: Reinstating immunosurveillance. Immunity 2013, 39, 74–88. [Google Scholar] [CrossRef] [PubMed]
- Antonarakis, E.S.; Drake, C.G. Combining immunological and androgen-directed approaches: An emerging concept in prostate cancer immunotherapy. Curr. Opin. Oncol. 2012, 24, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Formenti, S.C.; Demaria, S. Combining radiotherapy and cancer immunotherapy: A paradigm shift. J. Nal. Cancer Inst. 2013, 105, 256–265. [Google Scholar] [CrossRef] [PubMed]
- Kalbasi, A.; June, C.H.; Haas, N.; Vapiwala, N. Radiation and immunotherapy: A synergistic combination. J. Clin. Investig. 2013, 123, 2756–2763. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A.; Wolchok, J.D. Combining cancer immunotherapy and targeted therapy. Curr. Opin. Immunol. 2013, 25, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Drake, C.G.; Pardoll, D.M. Immune checkpoint blockade: A common denominator approach to cancer therapy. Cancer Cell 2015, 27, 450–461. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015, 331, 124–128. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Vesely, M.D.; Kershaw, M.H.; Schreiber, R.D.; Smyth, M.J. Natural innate and adaptive immunity to cancer. Ann. Rev. Immunol. 2011, 29, 235–271. [Google Scholar] [CrossRef] [PubMed]
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R.D. Cancer immunoediting: From immunosurveillance to tumor escape. Nat. Immunol. 2002, 3, 991–998. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef] [PubMed]
- Garrido, F.; Cabrera, T.; Lopez-Nevot, M.A.; Ruiz-Cabello, F. HLA class I antigens in human tumors. Adv. Cancer Res. 1995, 67, 155–195. [Google Scholar] [PubMed]
- Seliger, B.; Ritz, U.; Abele, R.; Bock, M.; Tampe, R.; Sutter, G.; Drexler, I.; Huber, C.; Ferrone, S. Immune escape of melanoma: First evidence of structural alterations in two distinct components of the mhc class I antigen processing pathway. Cancer Res. 2001, 61, 8647–8650. [Google Scholar] [PubMed]
- Matsushita, H.; Vesely, M.D.; Koboldt, D.C.; Rickert, C.G.; Uppaluri, R.; Magrini, V.J.; Arthur, C.D.; White, J.M.; Chen, Y.S.; Shea, L.K.; et al. Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting. Nature 2012, 482, 400–404. [Google Scholar] [CrossRef] [PubMed]
- Romero, I.; Garrido, C.; Algarra, I.; Collado, A.; Garrido, F.; Garcia-Lora, A.M. T lymphocytes restrain spontaneous metastases in permanent dormancy. Cancer Res. 2014, 74, 1958–1968. [Google Scholar] [CrossRef] [PubMed]
- Garrido, C.; Paco, L.; Romero, I.; Berruguilla, E.; Stefansky, J.; Collado, A.; Algarra, I.; Garrido, F.; Garcia-Lora, A.M. MHC class I molecules act as tumor suppressor genes regulating the cell cycle gene expression, invasion and intrinsic tumorigenicity of melanoma cells. Carcinogenesis 2012, 33, 687–693. [Google Scholar] [CrossRef] [PubMed]
- Romero, I.; Garrido, F.; Garcia-Lora, A.M. Metastases in immune-mediated dormancy: A new opportunity for targeting cancer. Cancer Res. 2014, 74, 6750–6757. [Google Scholar] [CrossRef] [PubMed]
- Dewan, M.Z.; Galloway, A.E.; Kawashima, N.; Dewyngaert, J.K.; Babb, J.S.; Formenti, S.C.; Demaria, S. Fractionated but not single-dose radiotherapy induces an immune-mediated abscopal effect when combined with anti-CTLA-4 antibody. Clin. Cancer Res. 2009, 15, 5379–5388. [Google Scholar] [CrossRef] [PubMed]
- Vacchelli, E.; Aranda, F.; Eggermont, A.; Galon, J.; Sautes-Fridman, C.; Cremer, I.; Zitvogel, L.; Kroemer, G.; Galluzzi, L. Trial watch: Chemotherapy with immunogenic cell death inducers. Oncoimmunology 2014, 3, e27878. [Google Scholar] [CrossRef] [PubMed]
- Hensel, J.A.; Flaig, T.W.; Theodorescu, D. Clinical opportunities and challenges in targeting tumour dormancy. Nat. Rev. Clin. Oncol. 2013, 10, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Koebel, C.M.; Vermi, W.; Swann, J.B.; Zerafa, N.; Rodig, S.J.; Old, L.J.; Smyth, M.J.; Schreiber, R.D. Adaptive immunity maintains occult cancer in an equilibrium state. Nature 2007, 450, 903–907. [Google Scholar] [CrossRef] [PubMed]
- Quesnel, B. Tumor dormancy and immunoescape. APMIS 2008, 116, 685–694. [Google Scholar] [CrossRef] [PubMed]
- Teng, M.W.; Vesely, M.D.; Duret, H.; McLaughlin, N.; Towne, J.E.; Schreiber, R.D.; Smyth, M.J. Opposing roles for IL-23 and IL-12 in maintaining occult cancer in an equilibrium state. Cancer Res. 2012, 72, 3987–3996. [Google Scholar] [CrossRef] [PubMed]
- Binder, D.C.; Engels, B.; Arina, A.; Yu, P.; Slauch, J.M.; Fu, Y.X.; Karrison, T.; Burnette, B.; Idel, C.; Zhao, M.; et al. Antigen-specific bacterial vaccine combined with anti-PD-l1 rescues dysfunctional endogenous t cells to reject long-established cancer. Cancer Immunol. Res. 2013, 1, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Disis, M.L. Immunologic biomarkers as correlates of clinical response to cancer immunotherapy. Cancer Immunol. Immunother. 2011, 60, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Bernards, R.; Weinberg, R.A. A progression puzzle. Nature 2002. [Google Scholar] [CrossRef]
- Eyles, J.; Puaux, A.L.; Wang, X.; Toh, B.; Prakash, C.; Hong, M.; Tan, T.G.; Zheng, L.; Ong, L.C.; Jin, Y.; et al. Tumor cells disseminate early, but immunosurveillance limits metastatic outgrowth, in a mouse model of melanoma. J. Clin. Investig. 2010, 120, 2030–2039. [Google Scholar] [CrossRef] [PubMed]
- Pages, F.; Kirilovsky, A.; Mlecnik, B.; Asslaber, M.; Tosolini, M.; Bindea, G.; Lagorce, C.; Wind, P.; Marliot, F.; Bruneval, P.; et al. In situ cytotoxic and memory T cells predict outcome in patients with early-stage colorectal cancer. J. Clinical Oncol. 2009, 27, 5944–5951. [Google Scholar] [CrossRef] [PubMed]
- Whiteside, T.L. Regulatory T cell subsets in human cancer: Are they regulating for or against tumor progression? Cancer Immunol. Immunother. 2014, 63, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Peng, M.; Huang, B.; Zhang, H.; Wang, H.; Xue, Z.; Zhang, L.; Da, Y.; Yang, D.; Yao, Z.; et al. Immune microenvironment profiles of tumor immune equilibrium and immune escape states of mouse sarcoma. Cancer Lett. 2013, 340, 124–133. [Google Scholar] [CrossRef] [PubMed]
- Piras, F.; Colombari, R.; Minerba, L.; Murtas, D.; Floris, C.; Maxia, C.; Corbu, A.; Perra, M.T.; Sirigu, P. The predictive value of CD8, CD4, CD68, and human leukocyte antigen-D-related cells in the prognosis of cutaneous malignant melanoma with vertical growth phase. Cancer 2005, 104, 1246–1254. [Google Scholar] [CrossRef] [PubMed]
- Harlin, H.; Meng, Y.; Peterson, A.C.; Zha, Y.; Tretiakova, M.; Slingluff, C.; McKee, M.; Gajewski, T.F. Chemokine expression in melanoma metastases associated with CD8+ T-cell recruitment. Cancer Res. 2009, 69, 3077–3085. [Google Scholar] [CrossRef] [PubMed]
- Hillen, F.; Baeten, C.I.; van de Winkel, A.; Creytens, D.; van der Schaft, D.W.; Winnepenninckx, V.; Griffioen, A.W. Leukocyte infiltration and tumor cell plasticity are parameters of aggressiveness in primary cutaneous melanoma. Cancer Immunol. Immunother. CII 2008, 57, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Perez, S.A.; Anastasopoulou, E.A.; Papamichail, M.; Baxevanis, C.N. AE37 peptide vaccination in prostate cancer: Identification of biomarkers in the context of prognosis and prediction. Cancer Immunol. Immunother. 2014, 63, 1141–1150. [Google Scholar] [CrossRef] [PubMed]
- Perez, S.A.; Anastasopoulou, E.A.; Tzonis, P.; Gouttefangeas, C.; Kalbacher, H.; Papamichail, M.; Baxevanis, C.N. AE37 peptide vaccination in prostate cancer: A 4-year immunological assessment updates on a phase I trial. Cancer Immunol. Immunother. 2013, 62, 1599–1608. [Google Scholar] [CrossRef] [PubMed]
- Palena, C.; Schlom, J. Vaccines against human carcinomas: Strategies to improve antitumor immune responses. J. Biomed. Biotechnol. 2010. [Google Scholar] [CrossRef] [PubMed]
- Aguirre-Ghiso, J.A. Models, mechanisms and clinical evidence for cancer dormancy. Nat. Rev. Cancer 2007, 7, 834–846. [Google Scholar] [CrossRef] [PubMed]
- Whiteside, T.L. Immune responses to cancer: Are they potential biomarkers of prognosis? Front. Oncol. 2013. [Google Scholar] [CrossRef] [PubMed]
- Russo, V.; Pilla, L.; Lunghi, F.; Crocchiolo, R.; Greco, R.; Ciceri, F.; Maggioni, D.; Fontana, R.; Mukenge, S.; Rivoltini, L.; et al. Clinical and immunologic responses in melanoma patients vaccinated with MAGE-A3-genetically modified lymphocytes. Int. J. Cancer 2013, 132, 2557–2566. [Google Scholar] [CrossRef] [PubMed]
- Andres, A. Cancer incidence after immunosuppressive treatment following kidney transplantation. Crit. Rev. Oncol. Hematol. 2005, 56, 71–85. [Google Scholar] [CrossRef] [PubMed]
- Patel, P.; Chen, E.I. Cancer stem cells, tumor dormancy, and metastasis. Front. Endocrinol. 2012. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, T.; Qian, B.Z.; Pollard, J.W. Immune cell promotion of metastasis. Nat. Rev. Immunol. 2015, 15, 73–86. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, H.; Lethe, B.; Lehmann, F.; van Baren, N.; Baurain, J.F.; de Smet, C.; Chambost, H.; Vitale, M.; Moretta, A.; Boon, T.; et al. Characterization of an antigen that is recognized on a melanoma showing partial HLA loss by CTL expressing an NK inhibitory receptor. Immunity 1997, 6, 199–208. [Google Scholar] [CrossRef]
- Yamshchikov, G.V.; Mullins, D.W.; Chang, C.C.; Ogino, T.; Thompson, L.; Presley, J.; Galavotti, H.; Aquila, W.; Deacon, D.; Ross, W.; et al. Sequential immune escape and shifting of T cell responses in a long-term survivor of melanoma. J. Immunol. 2005, 174, 6863–6871. [Google Scholar] [CrossRef] [PubMed]
- Bruttel, V.S.; Wischhusen, J. Cancer stem cell immunology: Key to understanding tumorigenesis and tumor immune escape? Front. Immunol. 2014. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.S.; Avivar-Valderas, A.; Estrada, Y.; Bragado, P.; Sosa, M.S.; Aguirre-Ghiso, J.A.; Segall, J.E. Dormancy signatures and metastasis in estrogen receptor positive and negative breast cancer. PLoS ONE 2012, 7, e35569. [Google Scholar] [CrossRef] [PubMed]
- Mahnke, Y.D.; Schwendemann, J.; Beckhove, P.; Schirrmacher, V. Maintenance of long-term tumour-specific T-cell memory by residual dormant tumour cells. Immunology 2005, 115, 325–336. [Google Scholar] [CrossRef] [PubMed]
- Beatty, G.L.; Paterson, Y. IFN-gamma can promote tumor evasion of the immune system in vivo by down-regulating cellular levels of an endogenous tumor antigen. J. Immunol. 2000, 165, 5502–5508. [Google Scholar] [CrossRef] [PubMed]
- Beatty, G.L.; Paterson, Y. Regulation of tumor growth by IFN-gamma in cancer immunotherapy. Immunol. Res. 2001, 24, 201–210. [Google Scholar] [CrossRef]
- Kmieciak, M.; Knutson, K.L.; Dumur, C.I.; Manjili, M.H. Her-2/neu antigen loss and relapse of mammary carcinoma are actively induced by T cell-mediated anti-tumor immune responses. Eur. J. Immunol. 2007, 37, 675–685. [Google Scholar] [CrossRef] [PubMed]
- Kmieciak, M.; Payne, K.K.; Wang, X.Y.; Manjili, M.H. IFN-gamma ralpha is a key determinant of CD8+ T cell-mediated tumor elimination or tumor escape and relapse in FVB mouse. PLoS ONE 2013, 8, e82544. [Google Scholar] [CrossRef] [PubMed]
- Farrar, J.D.; Katz, K.H.; Windsor, J.; Thrush, G.; Scheuermann, R.H.; Uhr, J.W.; Street, N.E. Cancer dormancy. VII. A regulatory role for CD8+ T cells and IFN-gamma in establishing and maintaining the tumor-dormant state. J. Immunol. 1999, 162, 2842–2849. [Google Scholar] [PubMed]
- Campbell, C.; Risueno, R.M.; Salati, S.; Guezguez, B.; Bhatia, M. Signal control of hematopoietic stem cell fate: Wnt, notch, and hedgehog as the usual suspects. Curr. Opin. Hematol. 2008, 15, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Bohin, N.; Wen, T.; Ng, V.; Magee, J.; Chen, S.C.; Shannon, K.; Morrison, S.J. Oncogenic nras has bimodal effects on stem cells that sustainably increase competitiveness. Nature 2013, 504, 143–147. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Ward, P.S.; Kapoor, G.S.; Rohle, D.; Turcan, S.; Abdel-Wahab, O.; Edwards, C.R.; Khanin, R.; Figueroa, M.E.; Melnick, A.; et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 2012, 483, 474–478. [Google Scholar] [CrossRef] [PubMed]
- Taipale, J.; Beachy, P.A. The hedgehog and wnt signalling pathways in cancer. Nature 2001, 411, 349–354. [Google Scholar] [CrossRef] [PubMed]
- Takebe, N.; Harris, P.J.; Warren, R.Q.; Ivy, S.P. Targeting cancer stem cells by inhibiting Wnt, Notch, and Hedgehog pathways. Nat. Rev. Clin. Oncol. 2011, 8, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Zardawi, S.J.; O’Toole, S.A.; Sutherland, R.L.; Musgrove, E.A. Dysregulation of Hedgehog, Wnt and Notch signalling pathways in breast cancer. Histol. Histopathol. 2009, 24, 385–398. [Google Scholar] [PubMed]
- Kaplan, R.N.; Riba, R.D.; Zacharoulis, S.; Bramley, A.H.; Vincent, L.; Costa, C.; MacDonald, D.D.; Jin, D.K.; Shido, K.; Kerns, S.A.; et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature 2005, 438, 820–827. [Google Scholar] [CrossRef] [PubMed]
- Sceneay, J.; Smyth, M.J.; Moller, A. The pre-metastatic niche: Finding common ground. Cancer Metastasis Rev. 2013, 32, 449–464. [Google Scholar] [CrossRef] [PubMed]
- Zitvogel, L.; Tesniere, A.; Kroemer, G. Cancer despite immunosurveillance: Immunoselection and immunosubversion. Nat. Rev. Immunol. 2006, 6, 715–727. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baxevanis, C.N.; Perez, S.A. Cancer Dormancy: A Regulatory Role for Endogenous Immunity in Establishing and Maintaining the Tumor Dormant State. Vaccines 2015, 3, 597-619. https://doi.org/10.3390/vaccines3030597
Baxevanis CN, Perez SA. Cancer Dormancy: A Regulatory Role for Endogenous Immunity in Establishing and Maintaining the Tumor Dormant State. Vaccines. 2015; 3(3):597-619. https://doi.org/10.3390/vaccines3030597
Chicago/Turabian StyleBaxevanis, Constantin N., and Sonia A. Perez. 2015. "Cancer Dormancy: A Regulatory Role for Endogenous Immunity in Establishing and Maintaining the Tumor Dormant State" Vaccines 3, no. 3: 597-619. https://doi.org/10.3390/vaccines3030597