
Highly-Immunogenic Virally-Vectored T-cell Vaccines Cannot Overcome Subversion of the T-cell Response by HCV during Chronic Infection

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patient Enrolment and Study Arms
2.2. Vaccine Constructs
2.3. Peptides and Antigens
2.4. IFNγ-ELISpot Assays
2.5. Intracellular Cytokine Staining
2.6. Thymidine Incorporation Proliferation Assay
2.7. HLA Class I Pentamer Staining
2.8. HCV RNA Quantification
2.9. HCV Viral Sequencing
2.10. Sequence Variability at T-cell Epitopes at a Population Level
2.11. nAbs to ChAd3 Vector
2.12. Statistical Analysis
3. Results
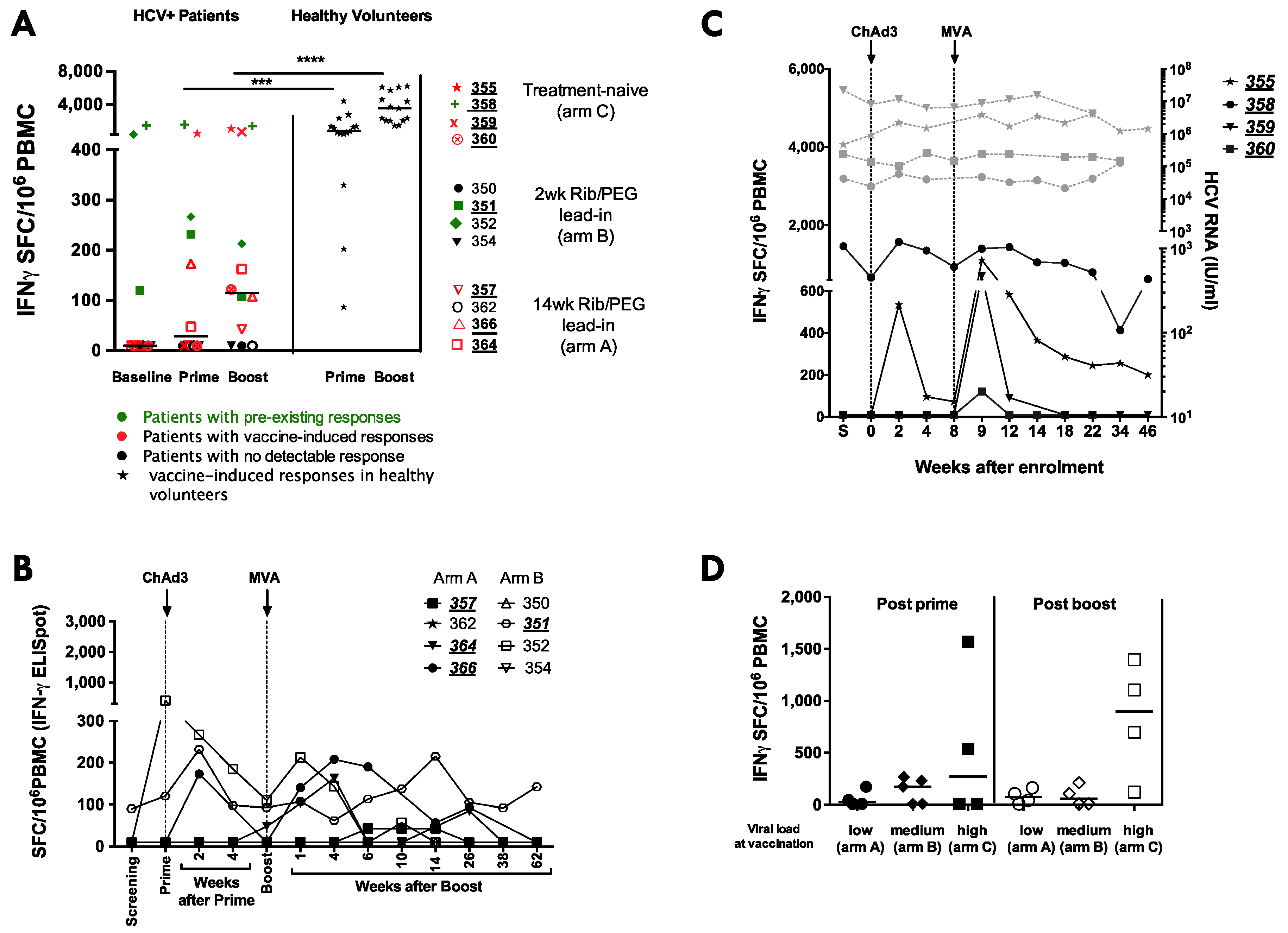
3.1. Patient Characteristics and Virological Outcome
3.2. Vaccination with ChAd3-NSmut and MVA-NSmut Is Well Tolerated
3.3. Pre-Existing HCV-Specific T-Cell Responses
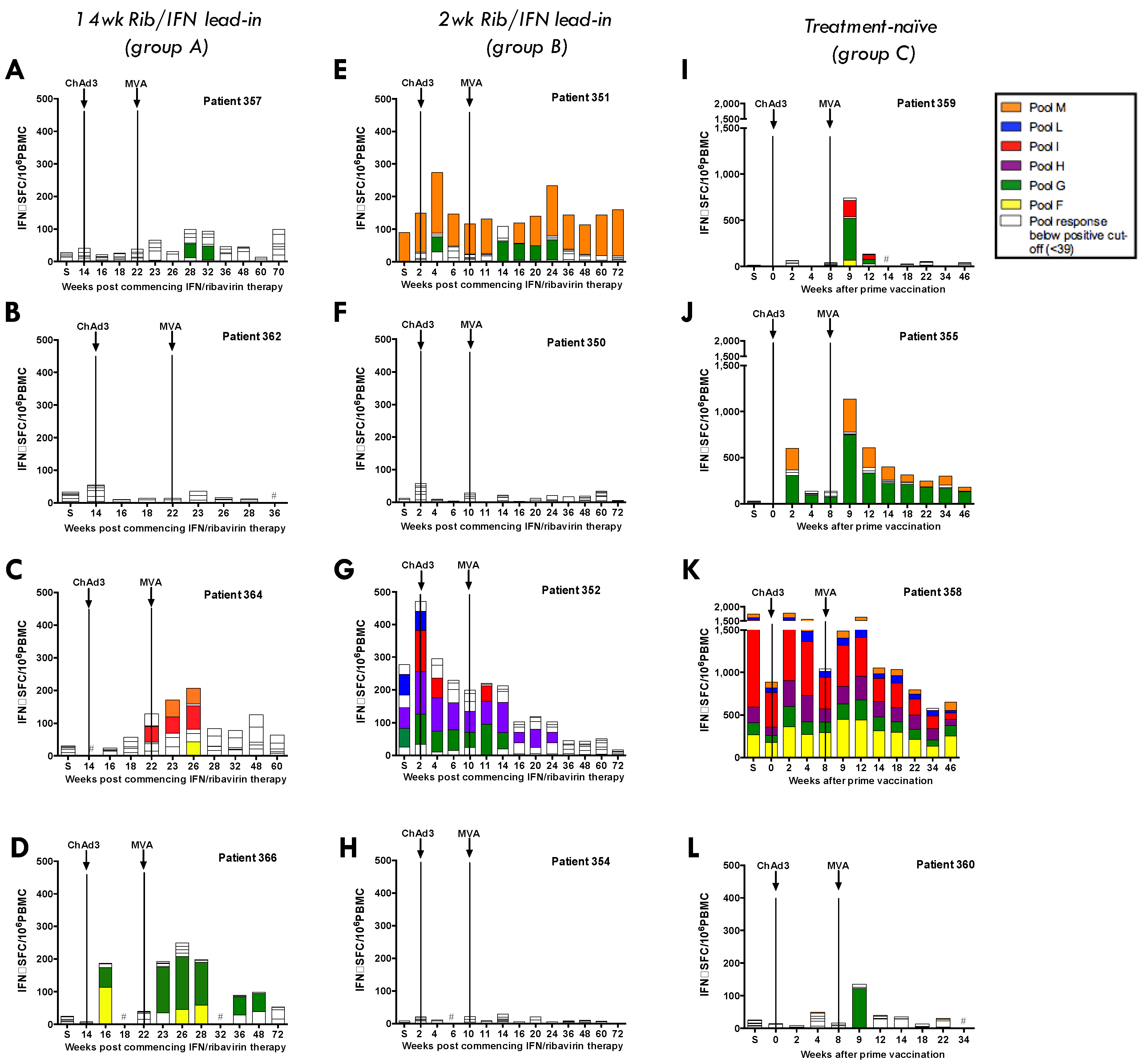
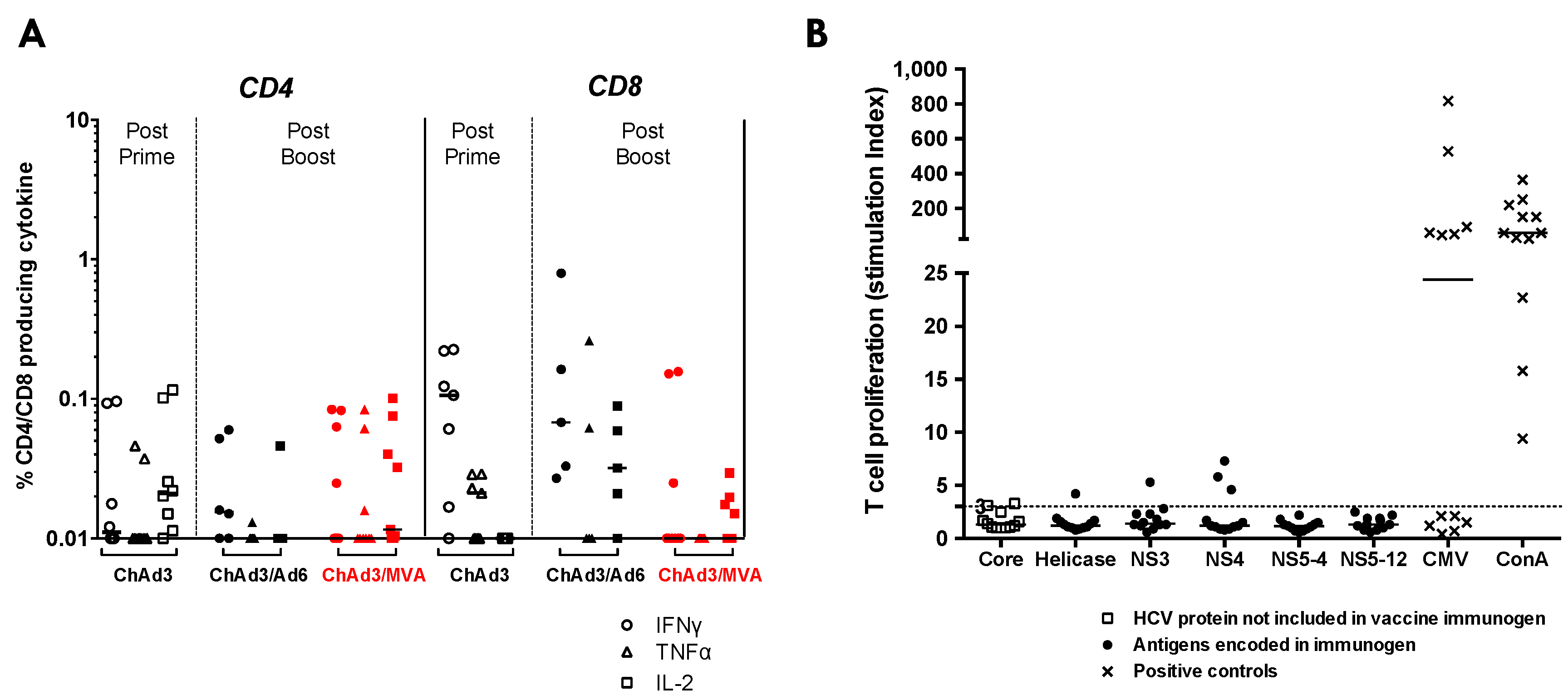
3.4. Heterologous ChAd3-NSmut Prime/MVA-NSmut Boost Vaccination Can Induce HCV-specific T-cell Responses in Patients Chronically Infected with HCV, But at a Reduced Magnitude When Compared to Healthy Volunteers
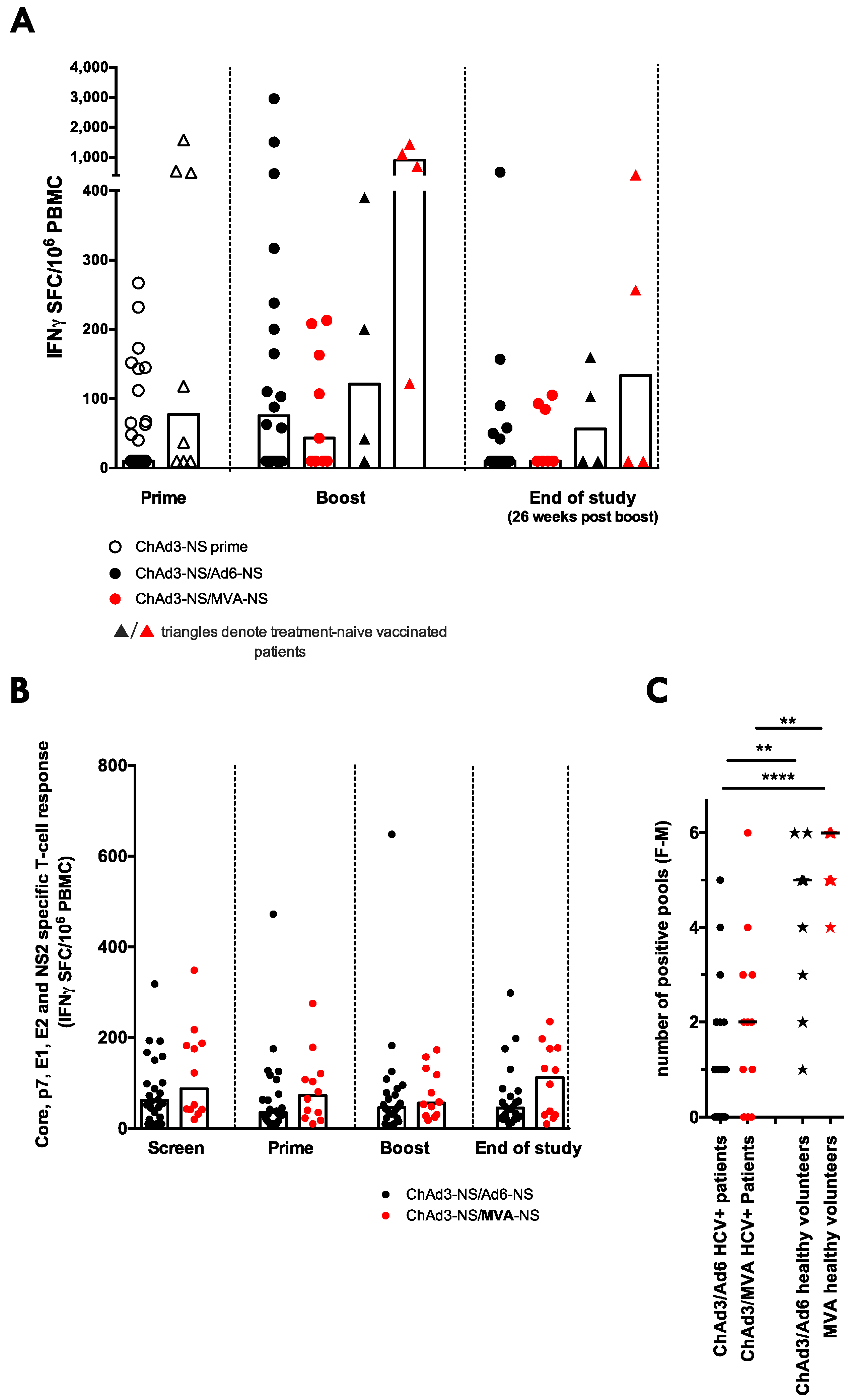
3.5. ChAd3/Ad6 vs. ChAd3/MVA Vaccine Regimens in HCV Infected Patients
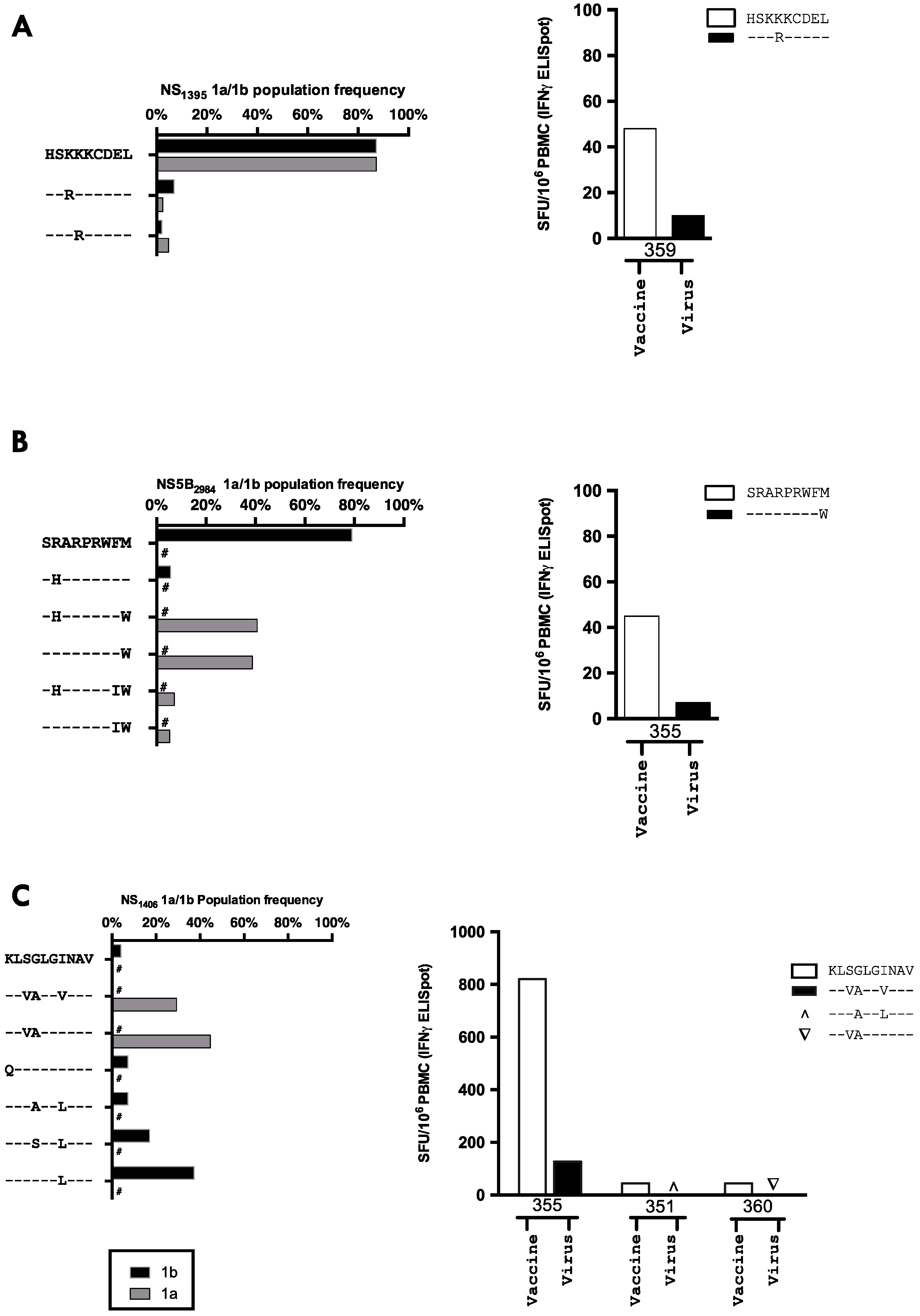
3.6. The Relationship between Vaccine Induced T-cell Specificity and Endogenous Viral Sequence
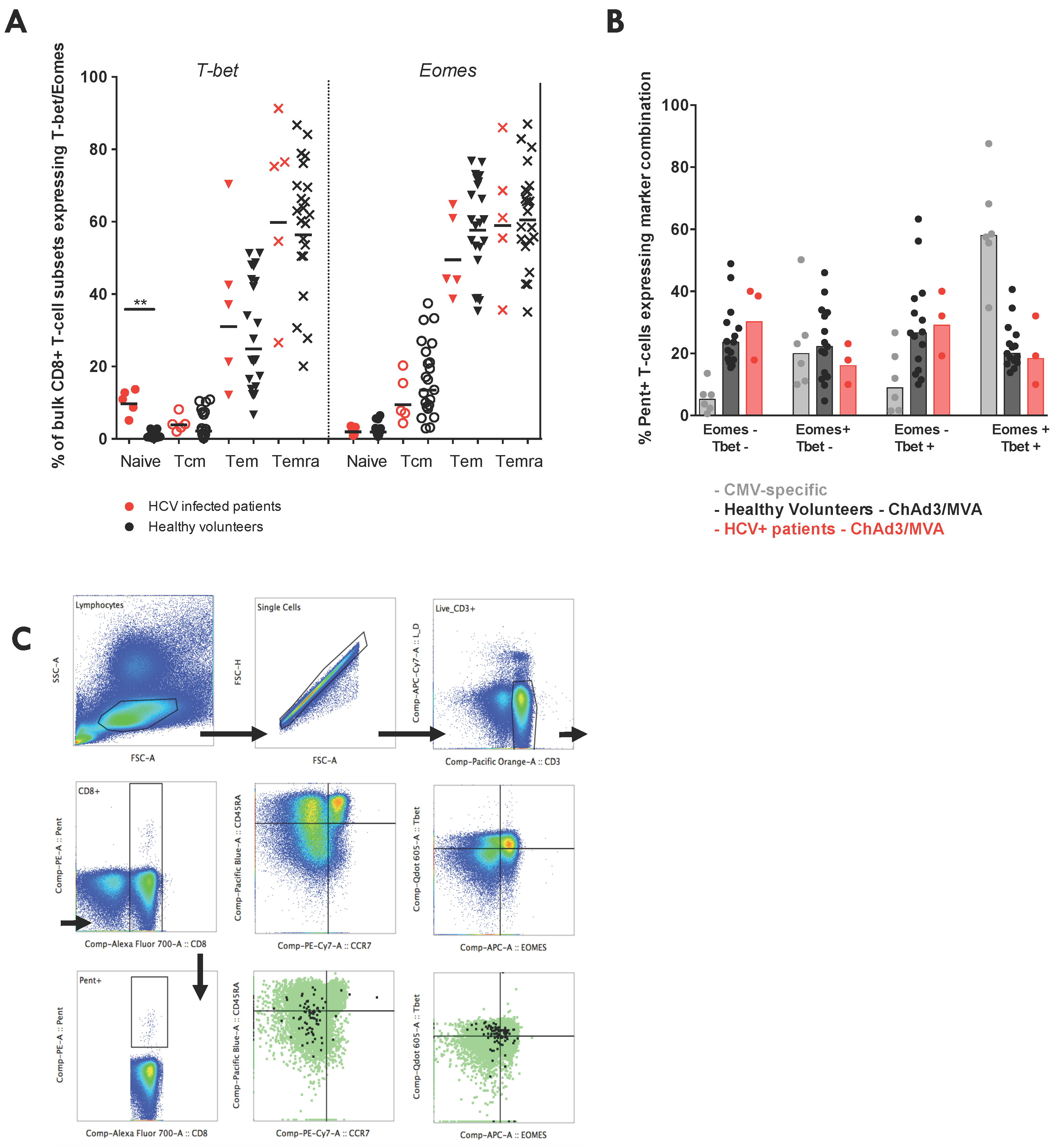
3.7. Vaccine-Induced T-Cell Function and Phenotype in Patients with Chronic HCV
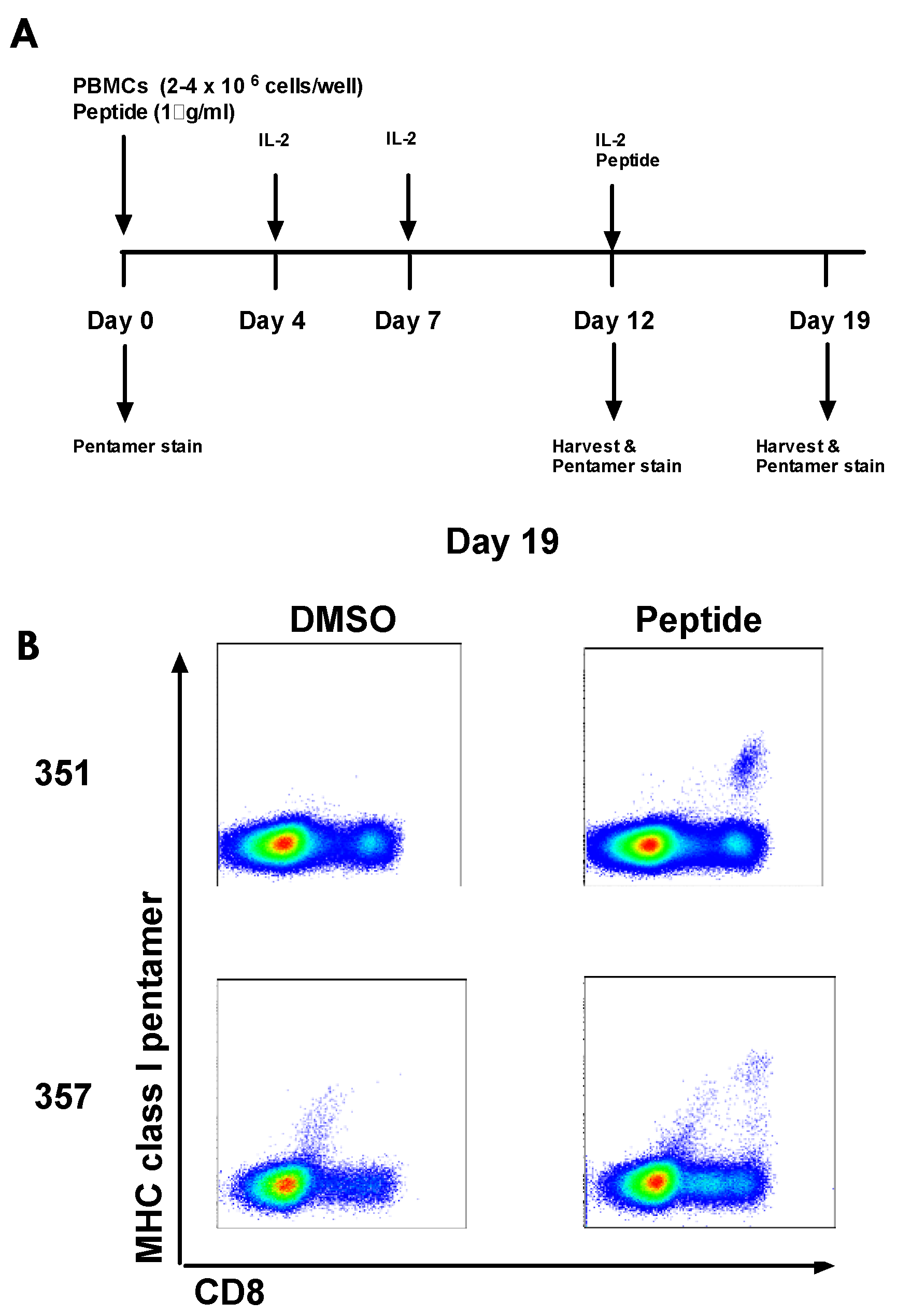
3.8. Culturing Pre-Vaccination HCV-Specific T-cells
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Messina, J.P.; Messina, I.X.; Flaxman, A.; Brown, A.; Cooke, G.S.; Pybus, O.G.; Barnes, E. Global distribution and prevalence of hepatitis C virus genotypes. Hepatology 2015, 61, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Kohli, A.; Shaffer, A.; Sherman, A.; Kottilil, S. Treatment of hepatitis C: A systematic review. JAMA 2014, 312, 631–640. [Google Scholar] [CrossRef] [PubMed]
- Walker, C.M.; Grakoui, A. Hepatitis C virus: Why do we need a vaccine to prevent a curable persistent infection? Curr. Opin. Immunol. 2015, 35, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Martin, N.K.; Foster, G.R.; Vilar, J.; Ryder, S.; Cramp, M.E.; Gordon, F.; Dillon, J.F.; Craine, N.; Busse, H.; Clements, A.; et al. HCV treatment rates and sustained viral response among people who inject drugs in seven UK sites: Real world results and modelling of treatment impact. J. Viral Hepat. 2015, 22, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Dutta, A.; Barker, C.; Kallarakal, A. The HIV treatment gap: Estimates of the financial resources needed versus available for scale-up of antiretroviral therapy in 97 countries from 2015 to 2020. PLoS Med. 2015, 12, e1001907. [Google Scholar] [CrossRef] [PubMed]
- Grady, B.P.; Schinkel, J.; Thomas, X.V.; Dalgard, O. Hepatitis C virus reinfection following treatment among people who use drugs. Clin. Infect. Dis. 2013, 57, S105–S110. [Google Scholar] [CrossRef] [PubMed]
- Aspinall, E.J.; Corson, S.; Doyle, J.S.; Grebely, J.; Hutchinson, S.J.; Dore, G.J.; Goldberg, D.J.; Hellard, M.E. Treatment of hepatitis C virus infection among people who are actively injecting drugs: A systematic review and meta-analysis. Clin. Infect. Dis. 2013, 57, S80–S89. [Google Scholar] [CrossRef] [PubMed]
- Osburn, W.O.; Fisher, B.E.; Dowd, K.A.; Urban, G.; Liu, L.; Ray, S.C.; Thomas, D.L.; Cox, A.L. Spontaneous control of primary hepatitis C virus infection and immunity against persistent reinfection. Gastroenterology 2010, 138, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Dahari, H.; Feinstone, S.M.; Major, M.E. Meta-analysis of hepatitis C virus vaccine efficacy in chimpanzees indicates an importance for structural proteins. Gastroenterology 2010, 139, 965–974. [Google Scholar] [CrossRef] [PubMed]
- Mehta, S.H.; Cox, A.; Hoover, D.R.; Wang, X.-H.; Mao, Q.; Ray, S.; Strathdee, S.A.; Vlahov, D.; Thomas, D.L. Protection against persistence of hepatitis C. Lancet 2002, 359, 1478–1483. [Google Scholar] [CrossRef]
- Ishii, S.; Koziel, M.J. Immune responses during acute and chronic infection with hepatitis C virus. Clin. Immunol. 2008, 128, 133–147. [Google Scholar] [CrossRef] [PubMed]
- Rehermann, B. Hepatitis C virus versus innate and adaptive immune responses: A tale of coevolution and coexistence. J. Clin. Investig. 2009, 119, 1745–1754. [Google Scholar] [CrossRef] [PubMed]
- Bowen, D.G.; Walker, C.M. Adaptive immune responses in acute and chronic hepatitis C virus infection. Nature 2005, 436, 946–952. [Google Scholar] [CrossRef] [PubMed]
- Klenerman, P.; Thimme, R. T cell responses in hepatitis C: The good, the bad and the unconventional. Gut 2012, 61, 1226–1234. [Google Scholar] [CrossRef] [PubMed]
- Swadling, L.; Klenerman, P.; Barnes, E. Ever closer to a prophylactic vaccine for HCV. Expert Opin. Biol. Ther. 2013, 13, 1109–1124. [Google Scholar] [CrossRef] [PubMed]
- Halliday, J.; Klenerman, P.; Barnes, E. Vaccination for hepatitis C virus: Closing in on an evasive target. Expert Rev. Vaccines 2011, 10, 659–672. [Google Scholar] [CrossRef] [PubMed]
- Klade, C.S.; Schuller, E.; Boehm, T.; von Gabain, A.; Manns, M.P. Sustained viral load reduction in treatment-naive HCV genotype 1 infected patients after therapeutic peptide vaccination. Vaccine 2012, 30, 2943–2950. [Google Scholar] [CrossRef] [PubMed]
- Habersetzer, F.; Baumert, T.F.; Stoll-Keller, F. GI-5005, a yeast vector vaccine expressing an NS3-core fusion protein for chronic HCV infection. Curr. Opin. Mol. Ther. 2009, 11, 456–462. [Google Scholar] [PubMed]
- Gowans, E.J.; Roberts, S.; Jones, K.; Dinatale, I.; Latour, P.A.; Chua, B.; Eriksson, E.M.Y.; Chin, R.; Li, S.; Wall, D.M.; et al. A phase I clinical trial of dendritic cell immunotherapy in HCV-infected individuals. J. Hepatol. 2010, 53, 599–607. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Lajonchere, L.; Shoukry, N.H.; Gra, B.; Amador-Cañizares, Y.; Helle, F.; Bédard, N.; Guerra, I.; Drouin, C.; Dubuisson, J.; González-Horta, E.E.; et al. Immunogenicity of CIGB-230, a therapeutic DNA vaccine preparation, in HCV-chronically infected individuals in a Phase I clinical trial. J. Viral Hepat. 2009, 16, 156–167. [Google Scholar] [CrossRef] [PubMed]
- Di Bisceglie, A.M.; Janczweska-Kazek, E.; Habersetzer, F.; Mazur, W.; Stanciu, C.; Carreno, V.; Tanasescu, C.; Flisiak, R.; Romero-Gómez, M.; Fich, A.; et al. Efficacy of immunotherapy with TG4040, peg-interferon, and ribavirin in a phase 2 study of patients with chronic HCV infection. Gastroenterology 2014, 147, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Habersetzer, F.; Honnet, G.; Bain, C.; Maynard Muet, M.; Leroy, V.; Zarski, J.P.; Feray, C.; Baumert, T.F.; Bronowicki, J.P.; Doffoël, M.; et al. A poxvirus vaccine is safe, induces T-cell responses, and decreases viral load in patients with chronic hepatitis C. Gastroenterology 2011, 141, 890–899. [Google Scholar] [CrossRef] [PubMed]
- Barnes, E.; Folgori, A.; Capone, S.; Swadling, L.; Aston, S.; Kurioka, A.; Meyer, J.; Huddart, R.; Smith, K.; Townsend, R.; et al. Novel adenovirus-based vaccines induce broad and sustained T-cell responses to HCV in man. Sci. Transl. Med. 2012. [Google Scholar] [CrossRef] [PubMed]
- Kelly, C.; Swadling, L.; Capone, S.; Brown, A.; Richardson, R.; Halliday, J.; von Delft, A.; Oo, Y.; Mutimer, D.; Kurioka, A.; et al. Chronic Hepatitis C Virus infection subverts vaccine induced T-cell immunity in humans. Hepatology 2015, 63, 1455–1470. [Google Scholar] [CrossRef] [PubMed]
- Swadling, L.; Capone, S.; Antrobus, R.D.; Brown, A.; Richardson, R.; Newell, E.W.; Halliday, J.; Kelly, C.; Bowen, D.; Fergusson, J.; et al. A human vaccine strategy based on chimpanzee adenoviral and MVA vectors that primes, boosts, and sustains functional HCV-specific T-cell memory. Sci. Transl. Med. 2014. [Google Scholar] [CrossRef] [PubMed]
- Wherry, E.J.; Blattman, J.N.; Ahmed, R. Low CD8 T-cell proliferative potential and high viral load limit the effectiveness of therapeutic vaccination. J. Virol. 2005, 79, 8960–8968. [Google Scholar] [CrossRef] [PubMed]
- Klenerman, P.; Hill, A. T cells and viral persistence: Lessons from diverse infections. Nat. Immunol. 2005, 6, 873–879. [Google Scholar] [CrossRef] [PubMed]
- Sheehy, S.H.; Duncan, C.J.A.; Elias, S.C.; Choudhary, P.; Biswas, S.; Halstead, F.D.; Collins, K.A.; Edwards, N.J.; Douglas, A.D.; Anagnostou, N.A.; et al. ChAd63-MVA-vectored blood-stage malaria vaccines targeting MSP1 and AMA1: Assessment of efficacy against mosquito bite challenge in humans. Mol. Ther. 2012, 20, 2355–2368. [Google Scholar] [CrossRef] [PubMed]
- Porter, D.W.; Thompson, F.M.; Berthoud, T.K.; Hutchings, C.L.; Andrews, L.; Biswas, S.; Poulton, I.; Prieur, E.; Correa, S.; Rowland, R.; et al. A human Phase I/IIa malaria challenge trial of a polyprotein malaria vaccine. Vaccine 2011, 29, 7514–7522. [Google Scholar] [CrossRef] [PubMed]
- Folgori, A.; Capone, S.; Ruggeri, L.; Meola, A.; Sporeno, E.; Ercole, B.B.; Pezzanera, M.; Tafi, R.; Arcuri, M.; Fattori, E.; et al. A T-cell HCV vaccine eliciting effective immunity against heterologous virus challenge in chimpanzees. Nat. Med. 2006, 12, 190–197. [Google Scholar] [CrossRef] [PubMed]
- Colloca, S.; Barnes, E.; Folgori, A.; Ammendola, V.; Capone, S.; Cirillo, A.; Siani, L.; Naddeo, M.; Grazioli, F.; Esposito, M.L.; et al. Vaccine vectors derived from a large collection of simian adenoviruses induce potent cellular immunity across multiple species. Sci. Transl. Med. 2012. [Google Scholar] [CrossRef] [PubMed]
- Batty, E.M.; Wong, T.H.N.; Trebes, A.; Argoud, K.; Attar, M.; Buck, D.; Ip, C.L.C.; Golubchik, T.; Cule, M.; Bowden, R.; et al. A modified RNA-Seq approach for whole genome sequencing of RNA viruses from faecal and blood samples. PLoS ONE 2013. [Google Scholar] [CrossRef] [PubMed]
- Lamble, S.; Batty, E.; Attar, M.; Buck, D.; Bowden, R.; Lunter, G.; Crook, D.; El-Fahmawi, B.; Piazza, P. Improved workflows for high throughput library preparation using the transposome-based Nextera system. BMC Biotechnol. 2013. [Google Scholar] [CrossRef] [PubMed]
- Gaidatzis, D.; Lerch, A.; Hahne, F.; Stadler, M.B. QuasR: Quantification and annotation of short reads in R. Bioinformatics 2015, 31, 1130–1132. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.B.; Bukh, J.; Kuiken, C.; Muerhoff, A.S.; Rice, C.M.; Stapleton, J.T.; Simmonds, P. Expanded classification of hepatitis C virus into 7 genotypes and 67 subtypes: Updated criteria and genotype assignment web resource. Hepatology 2014, 59, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Charlebois, P.; Gnerre, S.; Coole, M.G.; Lennon, N.J.; Levin, J.Z.; Qu, J.; Ryan, E.M.; Zody, M.C.; Henn, M.R. De novo assembly of highly diverse viral populations. BMC Genom. 2012. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.P.; Stromberg, M.P.; Ward, A.; Stewart, C.; Garrison, E.P.; Marth, G.T. MOSAIK: A hash-based algorithm for accurate next-generation sequencing short-read mapping. PLoS ONE 2014. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Charlebois, P.; Macalalad, A.; Henn, M.R.; Zody, M.C. V-Phaser 2: Variant inference for viral populations. BMC Genom. 2013. [Google Scholar] [CrossRef] [PubMed]
- Henn, M.R.; Boutwell, C.L.; Charlebois, P.; Lennon, N.J.; Power, K.A.; Macalalad, A.R.; Berlin, A.M.; Malboeuf, C.M.; Ryan, E.M.; Gnerre, S.; et al. Whole genome deep sequencing of HIV-1 reveals the impact of early minor variants upon immune recognition during acute infection. PLoS Pathog. 2012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aste-Amézaga, M.; Bett, A.J.; Wang, F.; Casimiro, D.R.; Antonello, J.M.; Patel, D.K.; Dell, E.C.; Franlin, L.L.; Dougherty, N.M.; Bennett, P.S.; et al. Quantitative adenovirus neutralization assays based on the secreted alkaline phosphatase reporter gene: Application in epidemiologic studies and in the design of adenovector vaccines. Hum. Gene Ther. 2004, 15, 293–304. [Google Scholar] [CrossRef] [PubMed]
- Hakamada, T.; Funatsuki, K.; Morita, H.; Ugajin, T.; Nakamura, I.; Ishiko, H.; Matsuzaki, Y.; Tanaka, N.; Imawari, M. Identification of novel hepatitis C virus-specific cytotoxic T lymphocyte epitopes by ELISpot assay using peptides with human leukocyte antigen-A*2402-binding motifs. J. Gen. Virol. 2004, 85, 1521–1531. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Zhang, H.; Rao, H.; Jiang, D.; Cong, X.; Feng, B.; Wang, J.; Wei, L.; Chen, H. DCs pulsed with novel HLA-A2-restricted CTL epitopes against hepatitis C virus induced a broadly reactive anti-HCV-specific T lymphocyte response. PLoS ONE 2012. [Google Scholar] [CrossRef]
- Barnes, E.; Gelderblom, H.C.; Humphreys, I.; Semmo, N.; Reesink, H.W.; Beld, M.G.; van Lier, R.A.; Klenerman, P. Cellular Immune Responses during High-Dose Interferon-α Induction Therapy for Hepatitis C Virus Infection. J. Infect. Dis. 2009, 199, 819–828. [Google Scholar] [CrossRef] [PubMed]
- Kuntzen, T.; Timm, J.; Berical, A.; Lewis-Ximenez, L.L.; Jones, A.; Nolan, B.; Schulze zur Wiesch, J.; Li, B.; Schneidewind, A.; Kim, A.Y.; et al. Viral sequence evolution in acute hepatitis C virus infection. J. Virol. 2007, 81, 11658–11668. [Google Scholar] [CrossRef] [PubMed]
- Timm, J.; Lauer, G.M.; Kavanagh, D.G.; Sheridan, I.; Kim, A.Y.; Lucas, M.; Pillay, T.; Ouchi, K.; Reyor, L.L.; zur Wiesch, J.S.; et al. CD8 epitope escape and reversion in acute HCV infection. J. Exp. Med. 2004, 200, 1593–1604. [Google Scholar] [CrossRef] [PubMed]
- Bengsch, B.; Seigel, B.; Ruhl, M.; Timm, J.; Kuntz, M.; Blum, H.E.; Pircher, H.; Thimme, R. Coexpression of PD-1, 2B4, CD160 and KLRG1 on exhausted HCV-specific CD8+ T-cells is linked to antigen recognition and T-cell differentiation. PLoS Pathog. 2010, 6, e1000947. [Google Scholar] [CrossRef] [PubMed]
- Bolinger, B.; Sims, S.; Swadling, L.; O’Hara, G.; de Lara, C.; Baban, D.; Saghal, N.; Lee, L.N.; Marchi, E.; Davis, M.; et al. Adenoviral vector vaccination induces a conserved program of CD8(+) T-cell memory differentiation in mouse and man. Cell Rep. 2015, 13, 1578–1588. [Google Scholar] [CrossRef] [PubMed]
- Alanio, C.; Nicoli, F.; Sultanik, P.; Flecken, T.; Perot, B.; Duffy, D.; Bianchi, E.; Lim, A.; Clave, E.; van Buuren, M.M.; et al. Bystander hyperactivation of preimmune CD8(+) T-cells in chronic HCV patients. Elife 2015. [Google Scholar] [CrossRef] [PubMed]
- Swadling, L.; Nuffield Department of Medicine, University of Oxford, Oxford, UK. Unpublished data. 2016.
- Callendret, B.; Eccleston, H.B.; Satterfield, W.; Capone, S.; Folgori, A.; Cortese, R.; Nicosia, A.; Walker, C.M. Persistent HCV replication despite priming of functional CD8+ T-cells by combined therapy with a vaccine and direct acting antiviral. Hepatology 2015. [Google Scholar] [CrossRef]
- Rizza, P.; Capone, I.; Moretti, F.; Proietti, E.; Belardelli, F. IFN-α as a vaccine adjuvant: Recent insights into the mechanisms and perspectives for its clinical use. Expert Rev. Vaccines 2011, 10, 487–498. [Google Scholar] [CrossRef] [PubMed]
- Penna, A.; Pilli, M.; Zerbini, A.; Orlandini, A.; Mezzadri, S.; Sacchelli, L.; Missale, G.; Ferrari, C. Dysfunction and functional restoration of HCV-specific CD8 responses in chronic hepatitis C virus infection. Hepatology 2007, 45, 588–601. [Google Scholar] [CrossRef] [PubMed]
- Wherry, E.J. T cell exhaustion. Nat. Immunol. 2011, 12, 492–499. [Google Scholar] [CrossRef] [PubMed]
- Doering, T.A.; Crawford, A.; Angelosanto, J.M.; Paley, M.A.; Ziegler, C.G.; Wherry, E.J. Network analysis reveals centrally connected genes and pathways involved in CD8(+) T-cell exhaustion versus memory. Immunity 2012, 37, 1130–1144. [Google Scholar] [CrossRef] [PubMed]
- Rao, R.R.; Li, Q.; Odunsi, K.; Shrikant, P.A. The mTOR kinase determines effector versus memory CD8+ T-cell fate by regulating the expression of transcription factors T-bet and eomesodermin. Immunity 2010, 32, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; Gordon, S.M.; Intlekofer, A.M.; Paley, M.A.; Mooney, E.C.; Lindsten, T.; Wherry, E.J.; Reiner, S.L. Cutting edge: The transcription factor eomesodermin enables CD8+ T-cells to compete for the memory cell niche. J. Immunol. 2010, 185, 4988–4992. [Google Scholar] [CrossRef] [PubMed]
- Paley, M.A.; Kroy, D.C.; Odorizzi, P.M.; Johnnidis, J.B.; Dolfi, D.V.; Barnett, B.E.; Bikoff, E.K.; Robertson, E.J.; Lauer, G.M.; Reiner, S.L.; Wherry, E.J. Progenitor and terminal subsets of CD8+ T-cells cooperate to contain chronic viral infection. Science 2012, 338, 1220–1225. [Google Scholar] [CrossRef] [PubMed]
- Kurktschiev, P.D.; Raziorrouh, B.; Schraut, W.; Backmund, M.; Wächtler, M.; Wendtner, C.-M.; Bengsch, B.; Thimme, R.; Denk, G.; Zachoval, R.; et al. Dysfunctional CD8+ T-cells in hepatitis B and C are characterized by a lack of antigen-specific T-bet induction. J. Exp. Med. 2014, 211, 2047–2059. [Google Scholar] [CrossRef] [PubMed]
- Fuller, M.J.; Shoukry, N.H.; Gushima, T.; Bowen, D.G.; Callendret, B.; Campbell, K.J.; Hasselschwert, D.L.; Hughes, A.L.; Walker, C.M. Selection-driven immune escape is not a significant factor in the failure of CD4 T-cell responses in persistent hepatitis C virus infection. Hepatology 2010, 51, 378–387. [Google Scholar] [CrossRef] [PubMed]
- Puig, M.; Mihalik, K.; Tilton, J.C.; Williams, O.; Merchlinsky, M.; Connors, M.; Feinstone, S.M.; Major, M.E. CD4+ immune escape and subsequent T-cell failure following chimpanzee immunization against hepatitis C virus. Hepatology 2006, 44, 736–745. [Google Scholar] [CrossRef] [PubMed]
- Kanagavelu, S.; Termini, J.M.; Gupta, S.; Raffa, F.N.; Fuller, K.A.; Rivas, Y.; Philip, S.; Kornbluth, R.S.; Stone, G.W. HIV-1 adenoviral vector vaccines expressing multi-trimeric BAFF and 4-1BBL enhance T-cell mediated anti-viral immunity. PLoS ONE 2014, 9, e90100. [Google Scholar] [CrossRef] [PubMed]
- Ragonnaud, E.; Andersson, A.-M.C.; Pedersen, A.E.; Laursen, H.; Holst, P.J. An adenoviral cancer vaccine co-encoding a tumor associated antigen together with secreted 4-1BBL leads to delayed tumor progression. Vaccine 2016, 34, 2147–2156. [Google Scholar] [CrossRef] [PubMed]
- Gardiner, D.; Lalezari, J.; Lawitz, E.; DiMicco, M.; Ghalib, R.; Reddy, K.R.; Chang, K.M.; Sulkowski, M.; Marro, S.O.; Anderson, J.; et al. A randomized, double-blind, placebo-controlled assessment of BMS-936558, a fully human monoclonal antibody to programmed death-1 (PD-1), in patients with chronic hepatitis C virus infection. PLoS ONE 2013, 8, e63818. [Google Scholar] [CrossRef] [PubMed]
- Fuller, M.J.; Callendret, B.; Zhu, B.; Freeman, G.J.; Hasselschwert, D.L.; Satterfield, W.; Sharpe, A.H.; Dustin, L.B.; Rice, C.M.; Grakoui, A.; et al. Immunotherapy of chronic hepatitis C virus infection with antibodies against programmed cell death-1 (PD-1). Proc. Natl. Acad. Sci. USA 2013, 110, 15001–15006. [Google Scholar] [CrossRef] [PubMed]
- Nakamoto, N.; Kaplan, D.E.; Coleclough, J.; Li, Y.; Valiga, M.E.; Kaminski, M.; Shaked, A.; Olthoff, K.; Gostick, E.; Price, D.A.; et al. Functional restoration of HCV-specific CD8 T cells by PD-1 blockade is defined by PD-1 expression and compartmentalization. Gastroenterology 2008, 134, 1927–1937. [Google Scholar] [CrossRef] [PubMed]
- Penaloza-MacMaster, P.; Provine, N.M.; Blass, E.; Barouch, D.H. CD4 T cell depletion substantially augments the rescue potential of PD-L1 blockade for deeply exhausted CD8 T cells. J. Immunol. 2015, 195, 1054–1063. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Regimen | Patient | Arm | Age (years) | Sex | HCV Genotype | Baseline Viral Load (IU/mL) | HLA-A | HLA-B | HLA-C | HLA-DR | HLA-DQ | RVR | EVR | SVR |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 14wk IFNa/Rib lead-in pre-vaccination | 357 | A | 58 | M | 1a | 33.7 × 106 | 02/30 | 13/62 | 06/10 | 04/07 | 02/08 | No | No | No |
| 362 | A | 43 | M | 1a | 32,000 | 01/29 | 08/44 | 07/16 | 01/07/53 | 02/05 | Yes | Yes | No | |
| 364 | A | 46 | F | 1a | 109,382 | 03/24 | 7 | 07/16 | 01/103 | 05 | Yes | Yes | No | |
| 366 | A | 36 | F | 1a | 2.0 × 106 | 02/33 | 07/65 | 07/08 | 11/103 | 05/07 | Yes | Yes | Yes | |
| 2 weeks IFNa/Rib lead-in pre-vaccination | 350 | B | 56 | M | 1a | 1.8 × 106 | 24/26 | 35/51 | 04/15 | 04/09 | 08/09 | Yes | Yes | No |
| 351 | B | 38 | M | 1b | 2.7 × 106 | 02 | 36/50 | 06/10 | - | - | Yes | Yes | Yes | |
| 352 | B | 52 | M | 1b | 9.4 × 106 | 03/24 | 07 | 07 | 01/15/51 | 05/06 | No | Yes | No | |
| 354 | B | 51 | M | 1a | 3.2 × 106 | 01/02 | 08/62 | 07/09 | 52/53 | 02/08 | No | No | No | |
| 356 | B | 38 | M | 1b | 173,764 | 11/66 | 35/41 | 04/17 | 13 | 06 | Yes | Yes | w/d | |
| Vaccinated treatment-naïve | 355 | C | 59 | M | 1a | 467,000 | 02/03 | 27/65 | 02/08 | 52/53 | 06/07 | n/a | n/a | n/a |
| 358 | C | 49 | F | 1a | 42,000 | 03/68 | 44/60 | 10/16 | 07/13/52/53 | 02/06 | n/a | n/a | n/a | |
| 359 | C | 35 | M | 1a | 237,000 | 01/66 | 08/41 | 07/17 | 52/53 | 02 | n/a | n/a | n/a | |
| 360 | C | 51 | F | 1a | 21.7 × 106 | 02/24 | 35/44 | 07/09 | 04/11/52/53 | 07 | n/a | n/a | n/a |
| 15mer Peptide | Amino Acid Location | Responding Patient | Likely Epitope | Predicted HLA-restriction |
|---|---|---|---|---|
| AYAAQGYKVLVLNPS | NS3 1243–1252 | 358 | AYAAQGYKVL^ | Cw03 |
| LTTGSVVIVGRIILS | NS4A 1677–1686 | 358 | TGSVVIVGR* | A68 |
| TEVDGVRLHRYAPAC & PEFFTEVDGVRLHRY | NS5A 2124–2132 | 358 | EVDGVRLHRY* | A3/B44 |
| LEFWESVFTGLTHID | NS3 1555–1564 | 358 | LEFWESVFTG* | B44/B60 |
| PASSQLDLSGWFVAG | NS5B 2963–2971 | 351 | QLDLSGWFV* | A2 |
| Regimen | Group | Patient | HLA-A | HLA-B | HLA-C | pre-vaccination +ve pools | post vaccination+ve pools | Mapped to peptide (parent pool) |
|---|---|---|---|---|---|---|---|---|
| 14wk IFNa/Rib lead-in pre-vaccination | A | 357 | 02/30 | 13/62 | 06/10 | - | G | KLSGLGINAV (G) |
| 362 | 01/29 | 08/44 | 07/16 | - | - | - | ||
| 364 | 03/24 | 07 | 07/16 | - | F, I, M | - | ||
| 366 | 02/33 | 07/65 | 07/08 | - | F, G | KLSGLGINAV (G) | ||
| 2 weeks IFNa/Rib lead-in pre-vaccination | B | 350 | 24/26 | 35/51 | 04/15 | - | - | - |
| 351 | 02 | 36/50 | 06/10 | M | , M | KLSGLGINAV (G), PASSQLDLSGWFVAG (M) | ||
| 352 | 03/24 | 07 | 07 | G, H, I, L | G, H, I | - | ||
| 354 | 01/02 | 08/62 | 07/09 | - | - | - | ||
| Vaccinated treatment-naïve | C | 355 | 02/03 | 27/65 | 02/08 | - | G, M | KLSGLGINAV (G), SRARPRWFM (M) |
| 358 | 03/68 | 44/60 | 10/16 | F, G, H, I, L, M | F, G, H, I, L, M | AYAAQGYKVLVLNPS (F), LEFWESVFTGLTHID (G), LTTGSVVIVGRIILS (H), TEVDGVRLHRYAPAC (I) | ||
| 359 | 01/66 | 08/41 | 07/17 | - | F, G, I | HSKKKCDEL (G) | ||
| 360 | 02/24 | 35/44 | 07/09 | - | G | KLSGLGINAV (G) |
| (A) | Patient | H | S | K | K | K | C | D | E | L | (B) | Patient | K | L | S | G | L | G | I | N | A | V | (C) | Patient | S | R | A | R | P | R | W | F | M |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HLA-B8 + ve pts | 359 | - | - | - | R | - | - | - | - | - | HLA-A2 +ve pts | 357 | - | - | V | A | - | - | V | - | - | - | 357 | - | - | - | - | - | - | - | - | W | |
| 359pv | - | - | - | R | - | - | - | - | - | 351 | - | - | - | A | - | - | L | - | - | - | 351 | - | - | - | - | - | - | - | - | - | |||
| 354 | - | - | - | - | - | - | - | - | - | 354 | - | - | T | A | M | - | - | - | - | - | 354 | - | H | - | - | - | - | - | - | W | |||
| 362 | - | - | - | - | - | - | - | - | - | 355 | - | - | V | A | - | - | V | - | - | - | 355 | - | - | - | - | - | - | - | - | W | |||
| HLA-B8 neg pts | 357 | - | - | - | - | - | - | - | - | - | 355pv | - | - | V | A | - | - | V | - | - | - | 355pv | - | - | - | - | - | - | - | - | W | ||
| 350 | - | - | - | - | - | - | - | - | - | 360 | - | - | V | A | - | - | - | - | - | - | 360 | - | - | - | - | - | - | - | - | W | |||
| 351 | - | - | - | - | - | - | - | - | - | 360pv | - | - | V | A | - | - | - | - | - | - | 360pv | - | - | - | - | - | - | - | - | W | |||
| 352 | - | - | - | - | - | - | - | - | - | 366 | - | - | V | S | - | - | - | - | - | - | 366 | - | H | - | - | - | - | - | - | W | |||
| 356 | - | - | - | - | - | - | - | - | - | HLA-A2 Neg pts | 350 | - | - | V | A | - | - | - | - | - | - | 350 | - | - | - | - | - | - | - | - | W | ||
| 355 | - | - | - | - | - | - | - | - | - | 352 | - | - | V | A | - | - | V | - | - | - | 352 | - | - | - | - | - | - | - | - | W | |||
| 355pv | - | - | - | - | - | - | - | - | - | 356 | - | - | - | - | - | - | L | - | - | - | 356 | - | - | - | - | - | - | - | I | - | |||
| 358 | - | - | - | - | - | - | - | - | - | 358 | - | - | V | A | - | - | V | - | - | - | 358 | - | - | - | - | - | - | - | - | W | |||
| 358 pv | - | - | - | - | - | - | - | - | - | 358pv | - | - | V | A | - | - | V | - | - | - | 358pv | - | - | - | - | - | - | - | - | W | |||
| 360 | - | - | - | - | - | - | - | - | - | 359 | - | - | V | A | - | - | V | - | - | - | 359 | - | - | - | - | - | - | - | - | W | |||
| 360pv | - | - | - | - | - | - | - | - | - | 359 | - | - | V | A | - | - | V | - | - | - | 359pv | - | - | - | - | - | - | - | - | W | |||
| 364 | n.d | 364 | n.d | 364 | n.d | ||||||||||||||||||||||||||||
| 366 | - | - | - | - | - | - | - | - | - | 362 | - | - | V | A | - | - | - | - | - | - | 362 | - | - | - | - | - | - | - | L | W | |||
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Swadling, L.; Halliday, J.; Kelly, C.; Brown, A.; Capone, S.; Ansari, M.A.; Bonsall, D.; Richardson, R.; Hartnell, F.; Collier, J.; et al. Highly-Immunogenic Virally-Vectored T-cell Vaccines Cannot Overcome Subversion of the T-cell Response by HCV during Chronic Infection. Vaccines 2016, 4, 27. https://doi.org/10.3390/vaccines4030027
Swadling L, Halliday J, Kelly C, Brown A, Capone S, Ansari MA, Bonsall D, Richardson R, Hartnell F, Collier J, et al. Highly-Immunogenic Virally-Vectored T-cell Vaccines Cannot Overcome Subversion of the T-cell Response by HCV during Chronic Infection. Vaccines. 2016; 4(3):27. https://doi.org/10.3390/vaccines4030027
Chicago/Turabian StyleSwadling, Leo, John Halliday, Christabel Kelly, Anthony Brown, Stefania Capone, M. Azim Ansari, David Bonsall, Rachel Richardson, Felicity Hartnell, Jane Collier, and et al. 2016. "Highly-Immunogenic Virally-Vectored T-cell Vaccines Cannot Overcome Subversion of the T-cell Response by HCV during Chronic Infection" Vaccines 4, no. 3: 27. https://doi.org/10.3390/vaccines4030027
APA StyleSwadling, L., Halliday, J., Kelly, C., Brown, A., Capone, S., Ansari, M. A., Bonsall, D., Richardson, R., Hartnell, F., Collier, J., Ammendola, V., Del Sorbo, M., Von Delft, A., Traboni, C., Hill, A. V. S., Colloca, S., Nicosia, A., Cortese, R., Klenerman, P., ... Barnes, E. (2016). Highly-Immunogenic Virally-Vectored T-cell Vaccines Cannot Overcome Subversion of the T-cell Response by HCV during Chronic Infection. Vaccines, 4(3), 27. https://doi.org/10.3390/vaccines4030027