Virus-Like-Vaccines against HIV


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
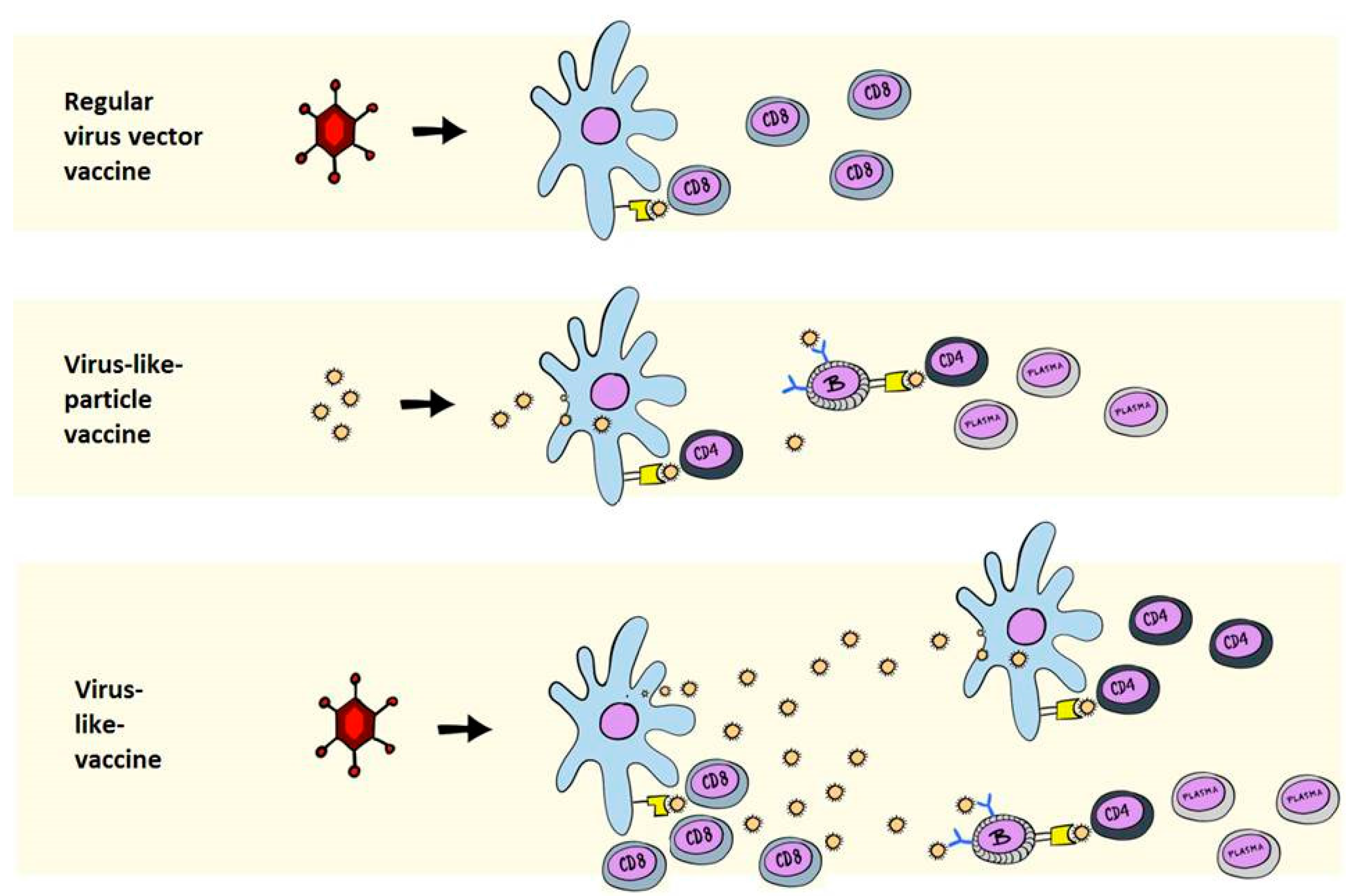
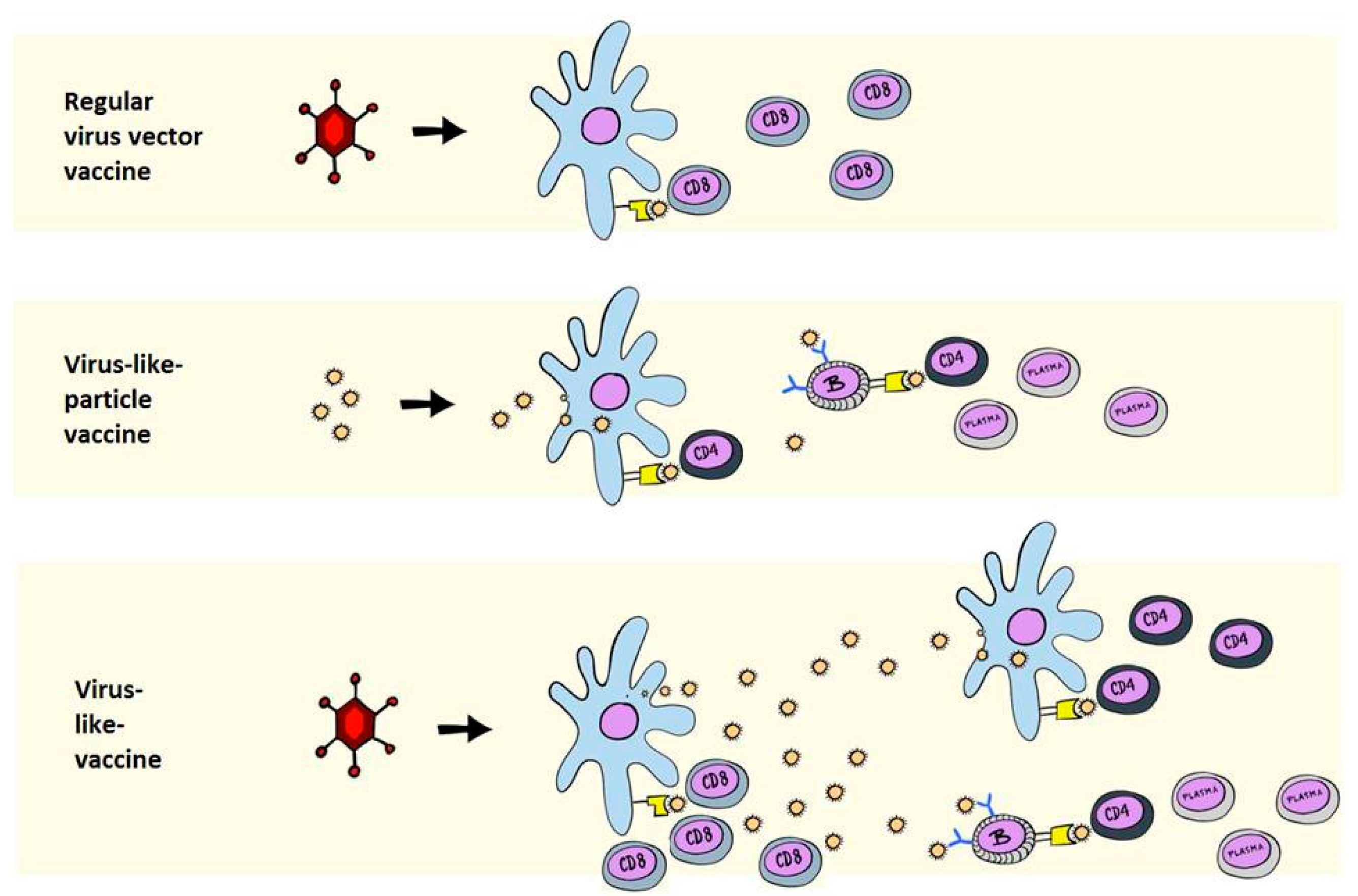
2. Immunological Differences between VLPs and VLVs
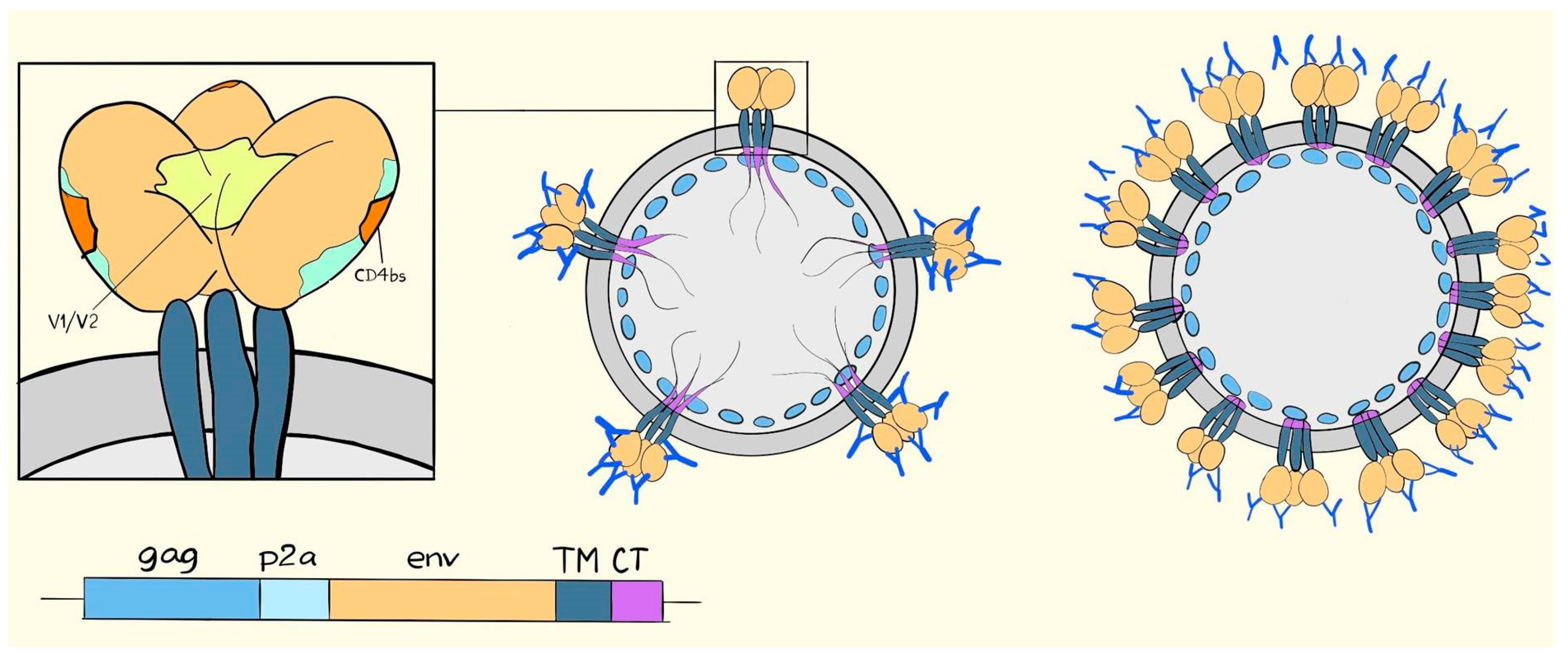
3. Poxviral Vectors Encoding HIV and SIV VLPs
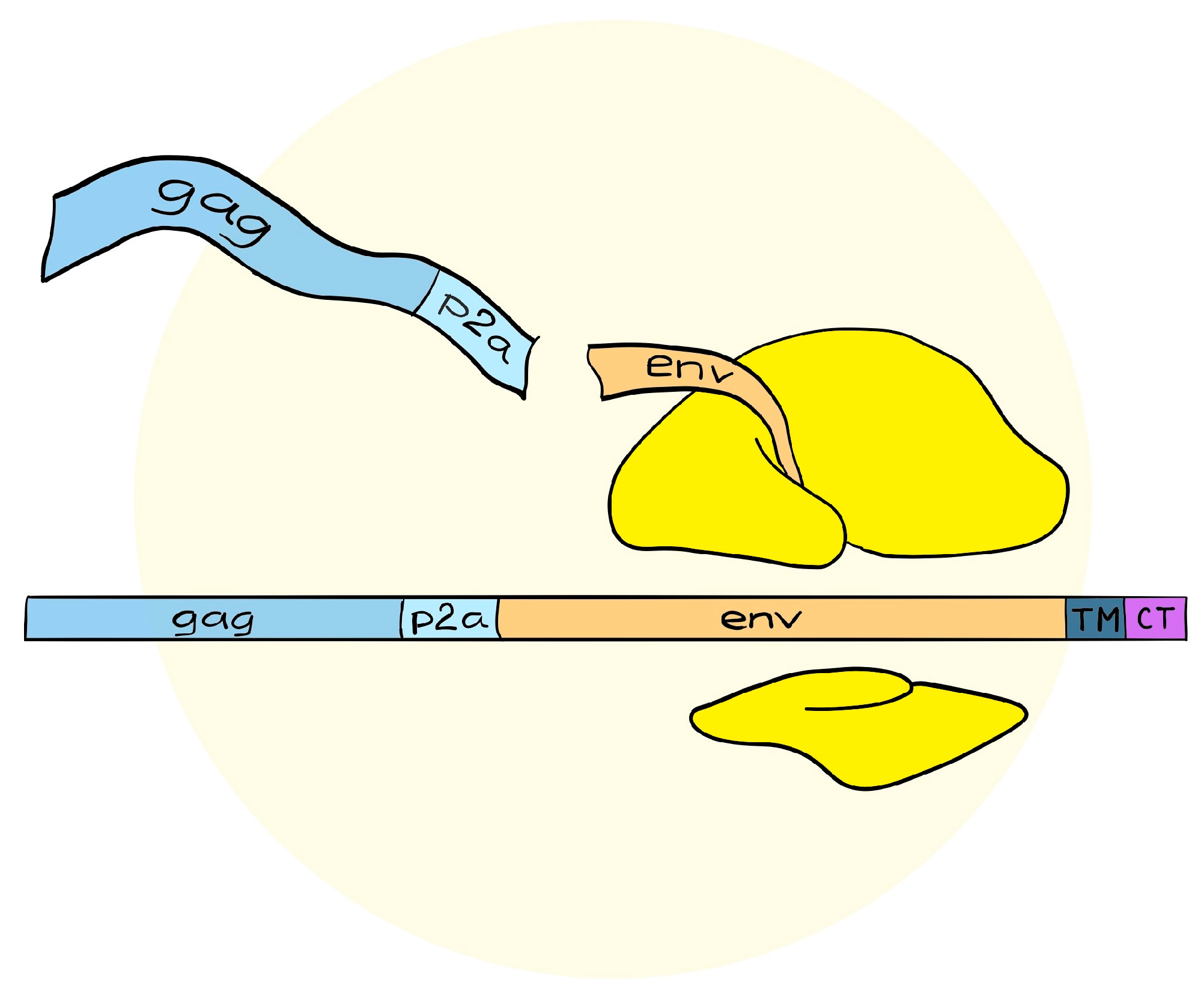
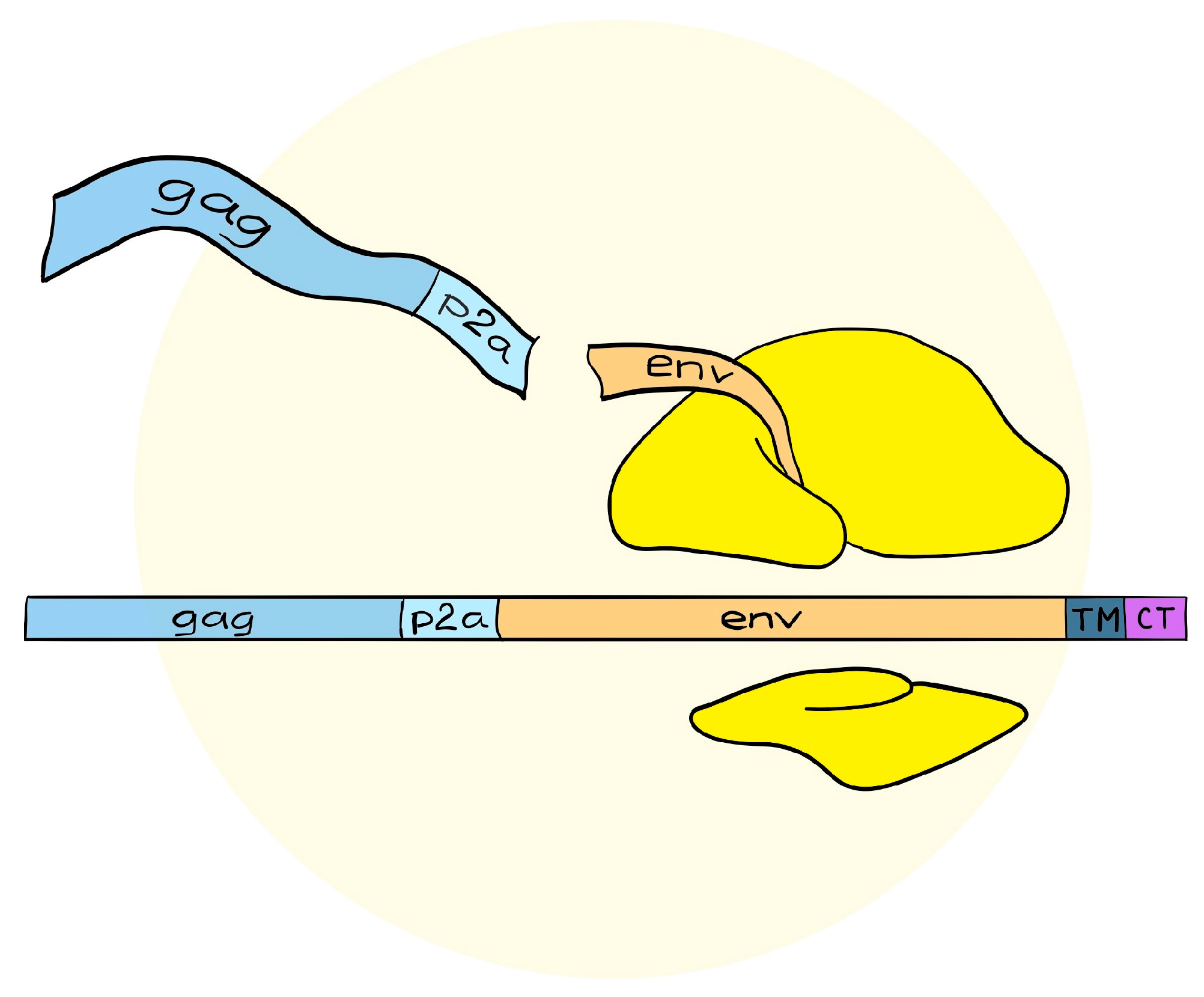
4. A Simplified Approach to the Generation of Adenovirus-based VLVs
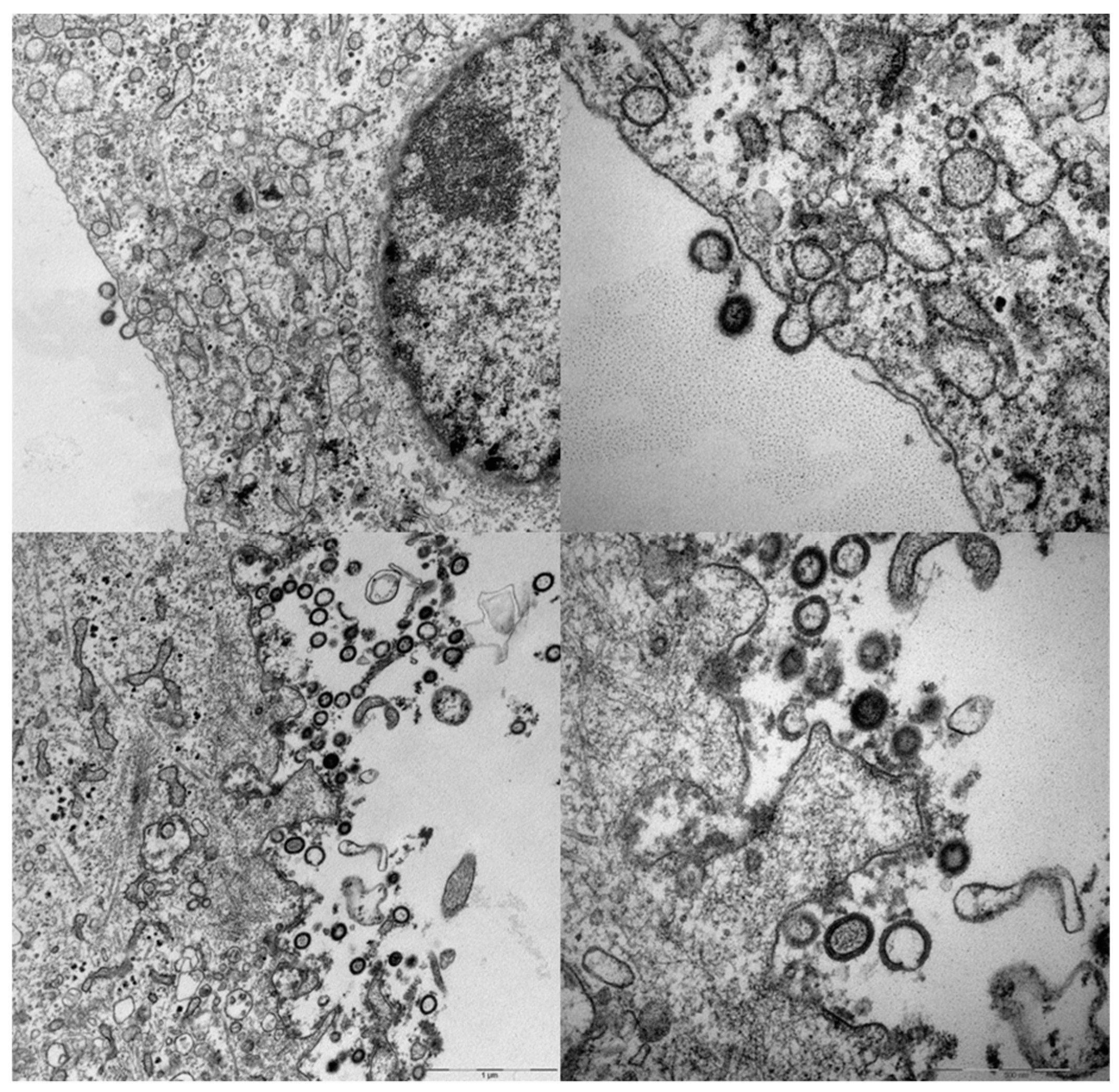
5. Adenovirus Vectored VLVs Targeting HIV, SIV, and P. falciparum
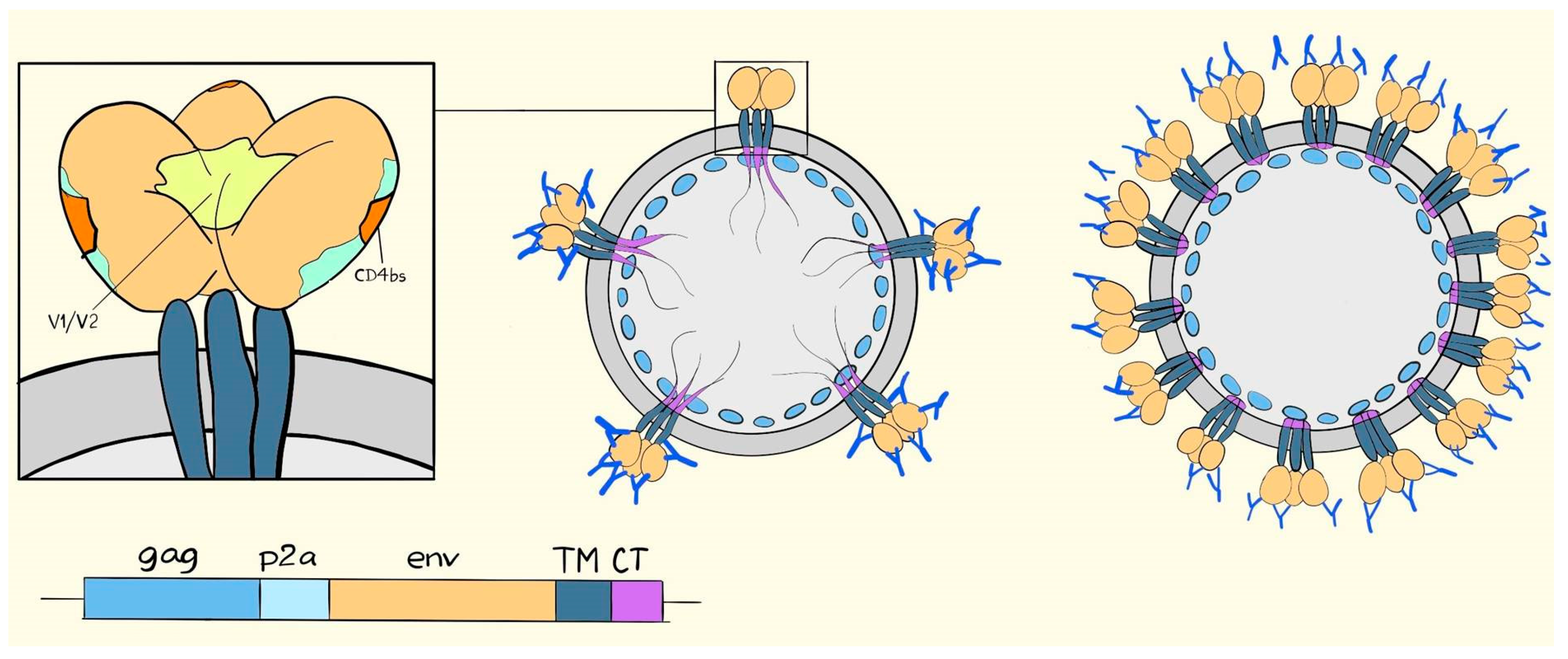
6. Suggested Mechanisms of Antibody-Mediated Protection from SIV, SHIV, or HIV Infection
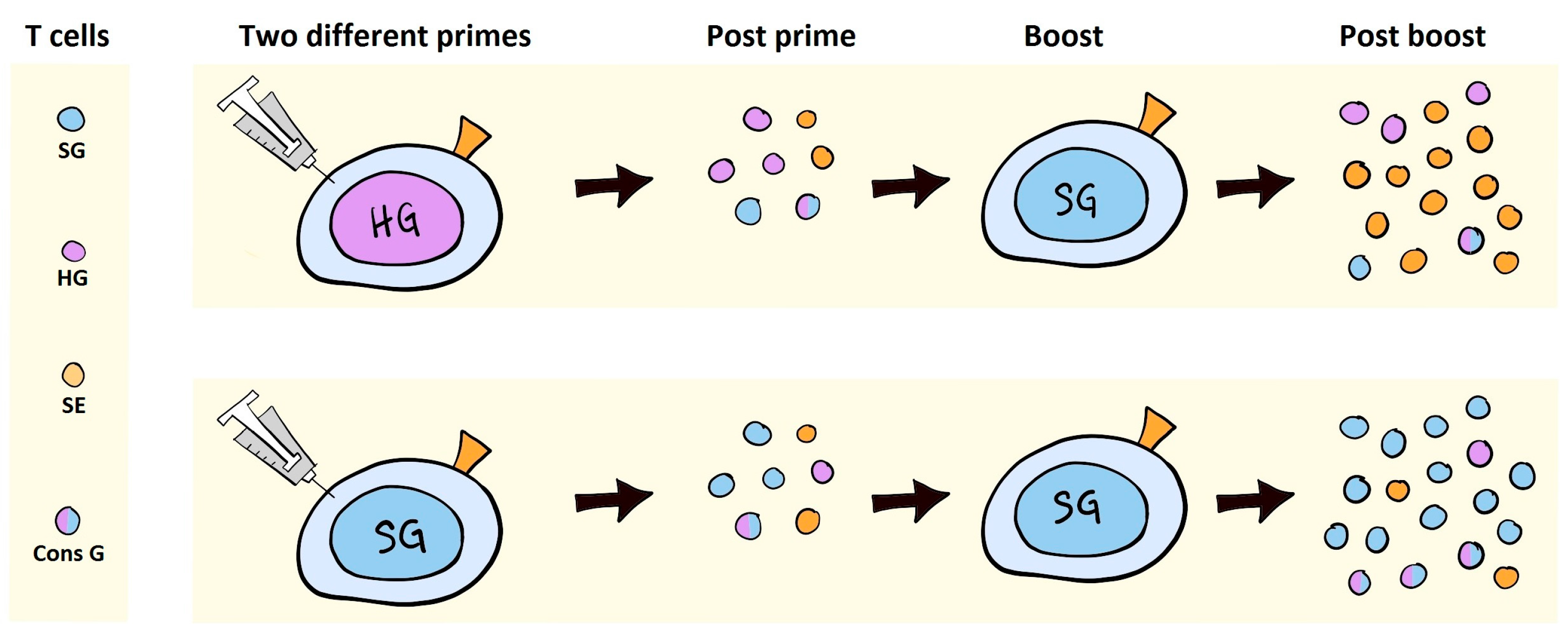
7. VLV Induced Antibodies in Prime-Boost Regimens
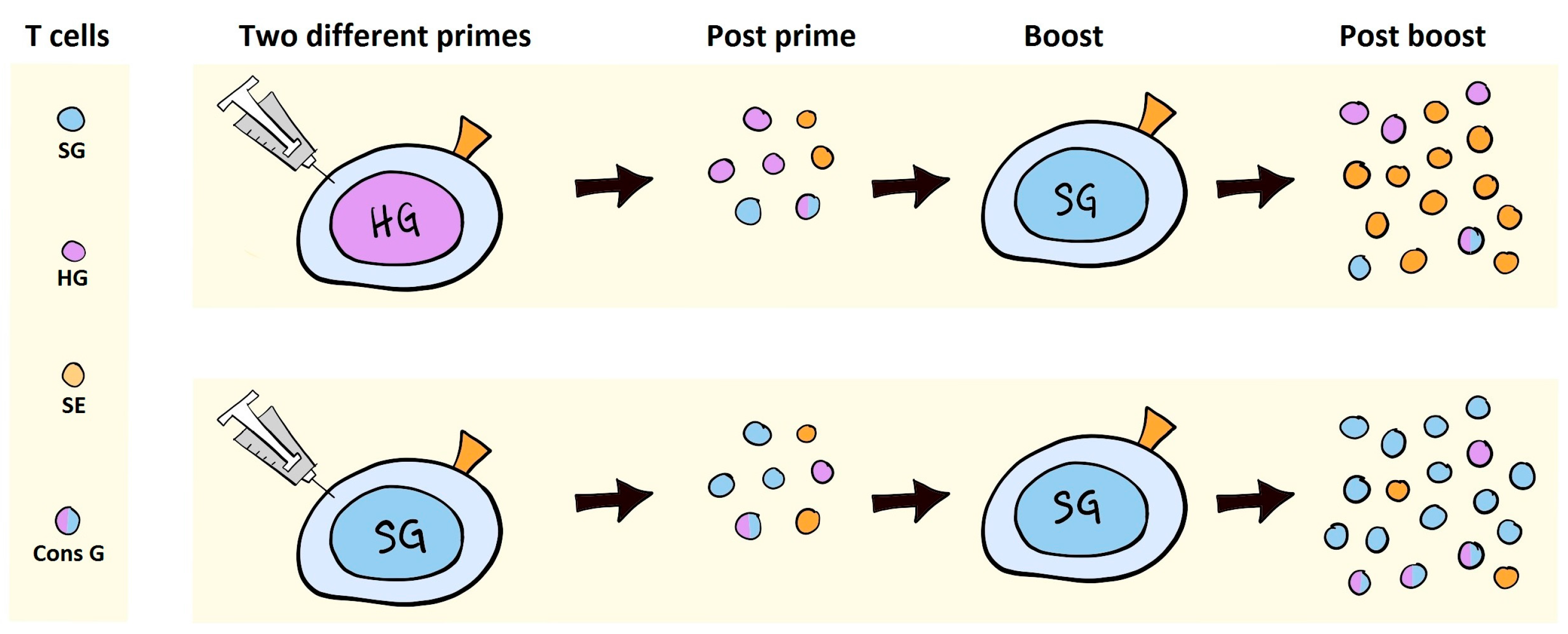
8. VLVs as Inducers of T Cell Responses
9. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Wang, H.; Wolock, T.M.; Carter, A.; Nguyen, G.; Kyu, H.H.; Gakidou, E.; Hay, S.I.; Mills, E.J.; Trickey, A.; Msemburi, W.; et al. Estimates of global, regional, and national incidence, prevalence, and mortality of HIV, 1980–2015: The Global Burden of Disease Study 2015. Lancet HIV 2016, 3, e361–e387. [Google Scholar] [CrossRef]
- Rerks-Ngarm, S.; Pitisuttithum, P.; Nitayaphan, S.; Kaewkungwal, J.; Chiu, J.; Paris, R.; Premsri, N.; Namwat, C.; de Souza, M.; Adams, E.; et al. Vaccination with ALVAC and AIDSVAX to prevent HIV-1 infection in Thailand. N. Engl. J. Med. 2009, 361, 2209–2220. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; O’Brien, K.L.; Lynch, D.M.; Simmons, N.L.; La, P.A.; Riggs, A.M.; Abbink, P.; Coffey, R.T.; Grandpre, L.E.; Seaman, M.S.; et al. Immune control of an SIV challenge by a T-cell-based vaccine in rhesus monkeys. Nature 2009, 457, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Daniel, M.D.; Kirchhoff, F.; Czajak, S.C.; Sehgal, P.K.; Desrosiers, R.C. Protective effects of a live attenuated SIV vaccine with a deletion in the nef gene. Science 1992, 258, 1938–1941. [Google Scholar] [CrossRef] [PubMed]
- Lifson, J.D.; Rossio, J.L.; Piatak, M., Jr.; Parks, T.; Li, L.; Kiser, R.; Coalter, V.; Fisher, B.; Flynn, B.M.; Czajak, S.; et al. Role of CD8(+) lymphocytes in control of simian immunodeficiency virus infection and resistance to rechallenge after transient early antiretroviral treatment. J. Virol. 2001, 75, 10187–10199. [Google Scholar] [CrossRef] [PubMed]
- Haynes, B.F.; Shaw, G.M.; Korber, B.; Kelsoe, G.; Sodroski, J.; Hahn, B.H.; Borrow, P.; McMichael, A.J. HIV-Host Interactions: Implications for Vaccine Design. Cell Host Microbe 2016, 19, 292–303. [Google Scholar] [CrossRef] [PubMed]
- Fukazawa, Y.; Lum, R.; Okoye, A.A.; Park, H.; Matsuda, K.; Bae, J.Y.; Hagen, S.I.; Shoemaker, R.; Deleage, C.; Lucero, C.; et al. B cell follicle sanctuary permits persistent productive simian immunodeficiency virus infection in elite controllers. Nat. Med. 2015, 21, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Fukazawa, Y.; Park, H.; Cameron, M.J.; Lefebvre, F.; Lum, R.; Coombes, N.; Mahyari, E.; Hagen, S.I.; Bae, J.Y.; Reyes, M.D., III; et al. Lymph node T cell responses predict the efficacy of live attenuated SIV vaccines. Nat. Med. 2012, 18, 1673–1681. [Google Scholar] [CrossRef] [PubMed]
- Williamson, A.L.; Rybicki, E.P. Justification for the inclusion of Gag in HIV vaccine candidates. Expert Rev. Vaccines 2016, 15, 585–598. [Google Scholar] [CrossRef] [PubMed]
- Griffin, D.E. The Immune Response in Measles: Virus Control, Clearance and Protective Immunity. Viruses 2016, 8, 282. [Google Scholar] [CrossRef] [PubMed]
- Minor, P.D. Live attenuated vaccines: Historical successes and current challenges. Virology 2015, 479–480, 379–392. [Google Scholar] [CrossRef] [PubMed]
- Mohsen, M.O.; Zha, L.; Cabral-Miranda, G.; Bachmann, M.F. Major findings and recent advances in virus-like particle (VLP)-based vaccines. Semin. Immunol. 2017, 34, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Kirnbauer, R.; Taub, J.; Greenstone, H.; Roden, R.; Durst, M.; Gissmann, L.; Lowy, D.R.; Schiller, J.T. Efficient self-assembly of human papillomavirus type 16 L1 and L1-L2 into virus-like particles. J. Virol. 1993, 67, 6929–6936. [Google Scholar] [PubMed]
- Wagner, R.; Teeuwsen, V.J.; Deml, L.; Notka, F.; Haaksma, A.G.; Jhagjhoorsingh, S.S.; Niphuis, H.; Wolf, H.; Heeney, J.L. Cytotoxic T cells and neutralizing antibodies induced in rhesus monkeys by virus-like particle HIV vaccines in the absence of protection from SHIV infection. Virology 1998, 245, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Pereyra, F.; Jia, X.; McLaren, P.J.; Telenti, A.; de Bakker, P.I.; Walker, B.D.; Ripke, S.; Brumme, C.J.; Pulit, S.L.; Carrington, M.; et al. The major genetic determinants of HIV-1 control affect HLA class I peptide presentation. Science 2010, 330, 1551–1557. [Google Scholar] [PubMed]
- Andersson, A.C.; Ragonnaud, E.; Seaton, K.E.; Sawant, S.; Folgori, A.; Colloca, S.; Labranche, C.; Montefiori, D.C.; Tomaras, G.D.; Holst, P.J. Effect of HIV-1 envelope cytoplasmic tail on adenovirus primed virus encoded virus-like particle immunizations. Vaccine 2016, 34, 5344–5351. [Google Scholar] [CrossRef] [PubMed]
- Leneghan, D.B.; Miura, K.; Taylor, I.J.; Li, Y.; Jin, J.; Brune, K.D.; Bachmann, M.F.; Howarth, M.; Long, C.A.; Biswas, S. Nanoassembly routes stimulate conflicting antibody quantity and quality for transmission-blocking malaria vaccines. Sci. Rep. 2017, 7, 3811. [Google Scholar] [CrossRef] [PubMed]
- Thrane, S.; Janitzek, C.M.; Matondo, S.; Resende, M.; Gustavsson, T.; Jongh, W.A.; Clemmensen, S.; Roeffen, W.; Vegte-Bolmer, M.; Gemert, G.J.; et al. Bacterial superglue enables easy development of efficient virus-like particle based vaccines. J. Nanobiotechnol. 2016, 14, 30. [Google Scholar] [CrossRef] [PubMed]
- Huber, B.; Schellenbacher, C.; Shafti-Keramat, S.; Jindra, C.; Christensen, N.; Kirnbauer, R. Chimeric L2-Based Virus-Like Particle (VLP) Vaccines Targeting Cutaneous Human Papillomaviruses (HPV). PLoS ONE 2017, 12, e0169533. [Google Scholar] [CrossRef] [PubMed]
- Calazans, A.; Boggiano, C.; Lindsay, R. A DNA inducing VLP vaccine designed for HIV and tested in mice. PLoS ONE 2017, 12, e0183803. [Google Scholar] [CrossRef] [PubMed]
- Pinto, L.A.; Edwards, J.; Castle, P.E.; Harro, C.D.; Lowy, D.R.; Schiller, J.T.; Wallace, D.; Kopp, W.; Adelsberger, J.W.; Baseler, M.W.; et al. Cellular immune responses to human papillomavirus (HPV)-16 L1 in healthy volunteers immunized with recombinant HPV-16 L1 virus-like particles. J. Infect. Dis. 2003, 188, 327–338. [Google Scholar] [CrossRef] [PubMed]
- Bayer, W.; Tenbusch, M.; Lietz, R.; Johrden, L.; Schimmer, S.; Uberla, K.; Dittmer, U.; Wildner, O. Vaccination with an adenoviral vector that encodes and displays a retroviral antigen induces improved neutralizing antibody and CD4+ T-cell responses and confers enhanced protection. J. Virol. 2010, 84, 1967–1976. [Google Scholar] [CrossRef] [PubMed]
- Andersson, A.C.; Holst, P.J. Increased T cell breadth and antibody response elicited in prime-boost regimen by viral vector encoded homologous SIV Gag/Env in outbred CD1 mice. J. Transl. Med. 2016, 14, 343. [Google Scholar] [CrossRef] [PubMed]
- Cimica, V.; Galarza, J.M. Adjuvant formulations for virus-like particle (VLP) based vaccines. Clin. Immunol. 2017, 183, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Fougeroux, C.; Holst, P.J. Future Prospects for the Development of Cost-Effective Adenovirus Vaccines. Int. J. Mol. Sci. 2017, 18, 686. [Google Scholar] [CrossRef] [PubMed]
- Colloca, S.; Barnes, E.; Folgori, A.; Ammendola, V.; Capone, S.; Cirillo, A.; Siani, L.; Naddeo, M.; Grazioli, F.; Esposito, M.L.; et al. Vaccine vectors derived from a large collection of simian adenoviruses induce potent cellular immunity across multiple species. Sci. Transl. Med. 2012, 4, 115ra2. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.J.; Petrovas, C.; Yamamoto, T.; Lindsay, R.W.; Lore, K.; Gall, J.G.; Gostick, E.; Lefebvre, F.; Cameron, M.J.; Price, D.A.; et al. Type I IFN induced by adenovirus serotypes 28 and 35 has multiple effects on T cell immunogenicity. J. Immunol. 2012, 188, 6109–6118. [Google Scholar] [CrossRef] [PubMed]
- Karasavvas, N.; Billings, E.; Rao, M.; Williams, C.; Zolla-Pazner, S.; Bailer, R.T.; Koup, R.A.; Madnote, S.; Arworn, D.; Shen, X.; et al. The Thai Phase III HIV Type 1 Vaccine trial (RV144) regimen induces antibodies that target conserved regions within the V2 loop of gp120. AIDS Res. Hum. Retrovir. 2012, 28, 1444–1457. [Google Scholar] [CrossRef] [PubMed]
- Amara, R.R.; Villinger, F.; Altman, J.D.; Lydy, S.L.; O’Neil, S.P.; Staprans, S.I.; Montefiori, D.C.; Xu, Y.; Herndon, J.G.; Wyatt, L.S.; et al. Control of a mucosal challenge and prevention of AIDS by a multiprotein DNA/MVA vaccine. Science 2001, 292, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Goepfert, P.A.; Elizaga, M.L.; Seaton, K.; Tomaras, G.D.; Montefiori, D.C.; Sato, A.; Hural, J.; Derosa, S.C.; Kalams, S.A.; McElrath, M.J.; et al. Specificity and 6-Month Durability of Immune Responses Induced by DNA and Recombinant Modified Vaccinia Ankara Vaccines Expressing HIV-1 Virus-Like Particles. J. Infect. Dis. 2014, 210, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Bett, A.J.; Dubey, S.A.; Mehrotra, D.V.; Guan, L.; Long, R.; Anderson, K.; Collins, K.; Gaunt, C.; Fernandez, R.; Cole, S.; et al. Comparison of T cell immune responses induced by vectored HIV vaccines in non-human primates and humans. Vaccine 2010, 28, 7881–7889. [Google Scholar] [CrossRef] [PubMed]
- Barouch, D.H.; Liu, J.; Li, H.; Maxfield, L.F.; Abbink, P.; Lynch, D.M.; Iampietro, M.J.; SanMiguel, A.; Seaman, M.S.; Ferrari, G.; et al. Vaccine protection against acquisition of neutralization-resistant SIV challenges in rhesus monkeys. Nature 2012, 482, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Ewer, K.J.; O’Hara, G.A.; Duncan, C.J.; Collins, K.A.; Sheehy, S.H.; Reyes-Sandoval, A.; Goodman, A.L.; Edwards, N.J.; Elias, S.C.; Halstead, F.D.; et al. Protective CD8+ T-cell immunity to human malaria induced by chimpanzee adenovirus-MVA immunisation. Nat. Commun. 2013, 4, 2836. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, B.K.; Feng, Y.; Sharma, S.K.; McKee, K.; Karlsson Hedestam, G.B.; LaBranche, C.C.; Montefiori, D.C.; Mascola, J.R.; Wyatt, R.T. Robust neutralizing antibodies elicited by HIV-1 JRFL envelope glycoprotein trimers in nonhuman primates. J. Virol. 2013, 87, 13239–13251. [Google Scholar] [CrossRef] [PubMed]
- Wyatt, L.S.; Belyakov, I.M.; Earl, P.L.; Berzofsky, J.A.; Moss, B. Enhanced cell surface expression, immunogenicity and genetic stability resulting from a spontaneous truncation of HIV Env expressed by a recombinant MVA. Virology 2008, 372, 260–272. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Kovacs, J.M.; Peng, H.; Rits-Volloch, S.; Lu, J.; Park, D.; Zablowsky, E.; Seaman, M.S.; Chen, B. HIV-1 ENVELOPE. Effect of the cytoplasmic domain on antigenic characteristics of HIV-1 envelope glycoprotein. Science 2015, 349, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Cottingham, M.G.; Carroll, F.; Morris, S.J.; Turner, A.V.; Vaughan, A.M.; Kapulu, M.C.; Colloca, S.; Siani, L.; Gilbert, S.C.; Hill, A.V. Preventing spontaneous genetic rearrangements in the transgene cassettes of adenovirus vectors. Biotechnol. Bioeng. 2012, 109, 719–728. [Google Scholar] [CrossRef] [PubMed]
- Chackerian, B.; Lenz, P.; Lowy, D.R.; Schiller, J.T. Determinants of autoantibody induction by conjugated papillomavirus virus-like particles. J. Immunol. 2002, 169, 6120–6126. [Google Scholar] [CrossRef] [PubMed]
- Ozorowski, G.; Pallesen, J.; de Val, N.; Lyumkis, D.; Cottrell, C.A.; Torres, J.L.; Copps, J.; Stanfield, R.L.; Cupo, A.; Pugach, P.; et al. Open and closed structures reveal allostery and pliability in the HIV-1 envelope spike. Nature 2017, 547, 360–363. [Google Scholar] [CrossRef] [PubMed]
- Julien, J.P.; Lee, J.H.; Ozorowski, G.; Hua, Y.; de la Peña, A.T.; de Taeye, S.W.; Nieusma, T.; Cupo, A.; Yasmeen, A.; Golabek, M.; et al. Design and structure of two HIV-1 clade C SOSIP.664 trimers that increase the arsenal of native-like Env immunogens. Proc. Natl. Acad. Sci. USA 2015, 112, 11947–11952. [Google Scholar] [CrossRef] [PubMed]
- Pitisuttithum, P.; Rerks-Ngarm, S.; Bussaratid, V.; Dhitavat, J.; Maekanantawat, W.; Pungpak, S.; Suntharasamai, P.; Vanijanonta, S.; Nitayapan, S.; Kaewkungwal, J.; et al. Safety and reactogenicity of canarypox ALVAC-HIV (vCP1521) and HIV-1 gp120 AIDSVAX B/E vaccination in an efficacy trial in Thailand. PLoS ONE 2011, 6, e27837. [Google Scholar] [CrossRef] [PubMed]
- Sanders, R.W.; van Gils, M.J.; Derking, R.; Sok, D.; Ketas, T.J.; Burger, J.A.; Ozorowski, G.; Cupo, A.; Simonich, C.; Goo, L.; et al. HIV-1 VACCINES. HIV-1 neutralizing antibodies induced by native-like envelope trimers. Science 2015, 349, aac4223. [Google Scholar] [CrossRef] [PubMed]
- Crooks, E.T.; Tong, T.; Chakrabarti, B.; Narayan, K.; Georgiev, I.S.; Menis, S.; Huang, X.; Kulp, D.; Osawa, K.; Muranaka, J.; et al. Vaccine-Elicited Tier 2 HIV-1 Neutralizing Antibodies Bind to Quaternary Epitopes Involving Glycan-Deficient Patches Proximal to the CD4 Binding Site. PLoS Pathog. 2015, 11, e1004932. [Google Scholar] [CrossRef] [PubMed]
- McCoy, L.E.; van Gils, M.J.; Ozorowski, G.; Messmer, T.; Briney, B.; Voss, J.E.; Kulp, D.W.; Macauley, M.S.; Sok, D.; Pauthner, M.; et al. Holes in the Glycan Shield of the Native HIV Envelope Are a Target of Trimer-Elicited Neutralizing Antibodies. Cell Rep. 2016, 16, 2327–2338. [Google Scholar] [CrossRef] [PubMed]
- Capucci, S.; Wee, E.G.; Schiffner, T.; LaBranche, C.C.; Borthwick, N.; Cupo, A.; Dodd, J.; Dean, H.; Sattentau, Q.; Montefiori, D.; et al. HIV-1-neutralizing antibody induced by simian adenovirus- and poxvirus MVA-vectored BG505 native-like envelope trimers. PLoS ONE 2017, 12, e0181886. [Google Scholar] [CrossRef] [PubMed]
- Zolla-Pazner, S.; DeCamp, A.; Gilbert, P.B.; Williams, C.; Yates, N.L.; Williams, W.T.; Howington, R.; Fong, Y.; Morris, D.E.; Soderberg, K.A.; et al. Vaccine-induced IgG antibodies to V1V2 regions of multiple HIV-1 subtypes correlate with decreased risk of HIV-1 infection. PLoS ONE 2014, 9, e87572. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Duffy, R.; Howington, R.; Cope, A.; Sadagopal, S.; Park, H.; Pal, R.; Kwa, S.; Ding, S.; Yang, O.O.; et al. Vaccine-Induced Linear Epitope-Specific Antibodies to Simian Immunodeficiency Virus SIVmac239 Envelope Are Distinct from Those Induced to the Human Immunodeficiency Virus Type 1 Envelope in Nonhuman Primates. J. Virol. 2015, 89, 8643–8650. [Google Scholar] [CrossRef] [PubMed]
- Pegu, P.; Vaccari, M.; Gordon, S.; Keele, B.F.; Doster, M.; Guan, Y.; Ferrari, G.; Pal, R.; Ferrari, M.G.; Whitney, S.; et al. Antibodies with high avidity to the gp120 envelope protein in protection from simian immunodeficiency virus SIV(mac251) acquisition in an immunization regimen that mimics the RV-144 Thai trial. J. Virol. 2013, 87, 1708–1719. [Google Scholar] [CrossRef] [PubMed]
- Tomaras, G.D.; Plotkin, S.A. Complex immune correlates of protection in HIV-1 vaccine efficacy trials. Immunol. Rev. 2017, 275, 245–261. [Google Scholar] [CrossRef] [PubMed]
- Golding, H.; Khurana, S.; Zaitseva, M. What Is the Predictive Value of Animal Models for Vaccine Efficacy in Humans? The Importance of Bridging Studies and Species-Independent Correlates of Protection. Cold Spring Harb. Perspect. Biol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wang, S.; Kong, R.; Ding, W.; Lee, F.H.; Parker, Z.; Kim, E.; Learn, G.H.; Hahn, P.; Policicchio, B.; et al. Envelope residue 375 substitutions in simian-human immunodeficiency viruses enhance CD4 binding and replication in rhesus macaques. Proc. Natl. Acad. Sci. USA 2016, 113, E3413–E3422. [Google Scholar] [CrossRef] [PubMed]
- Andersson, A.C.; Resende, M.; Salanti, A.; Nielsen, M.A.; Holst, P.J. Novel adenovirus encoded virus-like particles displaying the placental malaria associated VAR2CSA antigen. Vaccine 2017, 35, 1140–1147. [Google Scholar] [CrossRef] [PubMed]
- Hviid, L.; Salanti, A. VAR2CSA and protective immunity against pregnancy-associated Plasmodium falciparum malaria. Parasitology 2007, 134, 1871–1876. [Google Scholar] [CrossRef] [PubMed]
- Khan, F.; Porter, M.; Schwenk, R.; DeBot, M.; Saudan, P.; Dutta, S. Head-to-Head Comparison of Soluble vs. Qbeta VLP Circumsporozoite Protein Vaccines Reveals Selective Enhancement of NANP Repeat Responses. PLoS ONE 2015, 10, e0142035. [Google Scholar] [CrossRef] [PubMed]
- Storcksdieck genannt, B.M.; Niezold, T.; Temchura, V.; Pissani, F.; Ehrhardt, K.; Brown, E.P.; Osei-Owusu, N.Y.; Hannaman, D.; Hengel, H.; Ackerman, M.E.; et al. Enhancing the Quality of Antibodies to HIV-1 Envelope by GagPol-Specific Th Cells. J. Immunol. 2015, 195, 4861–4872. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.K.; de, V.N.; Bale, S.; Guenaga, J.; Tran, K.; Feng, Y.; Dubrovskaya, V.; Ward, A.B.; Wyatt, R.T. Cleavage-Independent HIV-1 Env Trimers Engineered as Soluble Native Spike Mimetics for Vaccine Design. Cell Rep. 2015, 11, 539–550. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Lai, L.; Amara, R.R.; Montefiori, D.C.; Villinger, F.; Chennareddi, L.; Wyatt, L.S.; Moss, B.; Robinson, H.L. Preclinical studies of human immunodeficiency virus/AIDS vaccines: Inverse correlation between avidity of anti-Env antibodies and peak postchallenge viremia. J. Virol. 2009, 83, 4102–4111. [Google Scholar] [CrossRef] [PubMed]
- Dugast, A.S.; Chan, Y.; Hoffner, M.; Licht, A.; Nkolola, J.; Li, H.; Streeck, H.; Suscovich, T.J.; Ghebremichael, M.; Ackerman, M.E.; et al. Lack of protection following passive transfer of polyclonal highly functional low-dose non-neutralizing antibodies. PLoS ONE 2014, 9, e97229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Page, M.; Stebbings, R.; Berry, N.; Hull, R.; Ferguson, D.; Davis, L.; Duffy, L.; Elsley, W.; Hall, J.; Ham, C.; et al. Heterologous protection elicited by candidate monomeric recombinant HIV-1 gp120 vaccine in the absence of cross neutralising antibodies in a macaque model. Retrovirology 2012, 9, 56. [Google Scholar] [CrossRef] [PubMed]
- Tuero, I.; Mohanram, V.; Musich, T.; Miller, L.; Vargas-Inchaustegui, D.A.; Demberg, T.; Venzon, D.; Kalisz, I.; Kalyanaraman, V.S.; Pal, R.; et al. Mucosal B Cells Are Associated with Delayed SIV Acquisition in Vaccinated Female but Not Male Rhesus Macaques Following SIVmac251 Rectal Challenge. PLoS Pathog. 2015, 11, e1005101. [Google Scholar] [CrossRef] [PubMed]
- Lee, F.H.; Mason, R.; Welles, H.; Learn, G.H.; Keele, B.F.; Roederer, M.; Bar, K.J. Breakthrough Virus Neutralization Resistance as a Correlate of Protection in a Nonhuman Primate Heterologous Simian Immunodeficiency Virus Vaccine Challenge Study. J. Virol. 2015, 89, 12388–12400. [Google Scholar] [CrossRef] [PubMed]
- Veillette, M.; Coutu, M.; Richard, J.; Batraville, L.A.; Dagher, O.; Bernard, N.; Tremblay, C.; Kaufmann, D.E.; Roger, M.; Finzi, A. The HIV-1 gp120 CD4-bound conformation is preferentially targeted by antibody-dependent cellular cytotoxicity-mediating antibodies in sera from HIV-1-infected individuals. J. Virol. 2015, 89, 545–551. [Google Scholar] [CrossRef] [PubMed]
- Kannanganat, S.; Nigam, P.; Velu, V.; Earl, P.L.; Lai, L.; Chennareddi, L.; Lawson, B.; Wilson, R.L.; Montefiori, D.C.; Kozlowski, P.A.; et al. Preexisting vaccinia virus immunity decreases SIV-specific cellular immunity but does not diminish humoral immunity and efficacy of a DNA/MVA vaccine. J. Immunol. 2010, 185, 7262–7273. [Google Scholar] [CrossRef] [PubMed]
- Lai, L.; Kwa, S.F.; Kozlowski, P.A.; Montefiori, D.C.; Nolen, T.L.; Hudgens, M.G.; Johnson, W.E.; Ferrari, G.; Hirsch, V.M.; Felber, B.K.; et al. SIVmac239 MVA vaccine with and without a DNA prime, similar prevention of infection by a repeated dose SIVsmE660 challenge despite different immune responses. Vaccine 2012, 30, 1737–1745. [Google Scholar] [CrossRef] [PubMed]
- Wyatt, L.S.; Earl, P.L.; Vogt, J.; Eller, L.A.; Chandran, D.; Liu, J.; Robinson, H.L.; Moss, B. Correlation of immunogenicities and in vitro expression levels of recombinant modified vaccinia virus Ankara HIV vaccines. Vaccine 2008, 26, 486–493. [Google Scholar] [CrossRef] [PubMed]
- Baur, K.; Brinkmann, K.; Schweneker, M.; Patzold, J.; Meisinger-Henschel, C.; Hermann, J.; Steigerwald, R.; Chaplin, P.; Suter, M.; Hausmann, J. Immediate-early expression of a recombinant antigen by modified vaccinia virus ankara breaks the immunodominance of strong vector-specific B8R antigen in acute and memory CD8 T-cell responses. J. Virol. 2010, 84, 8743–8752. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.A.; Surman, S.L.; Sealy, R.; Jones, B.G.; Slobod, K.S.; Branum, K.; Lockey, T.D.; Howlett, N.; Freiden, P.; Flynn, P.; et al. Heterologous Prime-Boost HIV-1 Vaccination Regimens in Pre-Clinical and Clinical Trials. Viruses 2010, 2, 435–467. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, K.E.; Li, H.; Walker, B.D.; Michael, N.L.; Barouch, D.H. Gag-Specific Cellular Immunity Determines In Vitro Viral Inhibition and In Vivo Virologic Control Following SIV Challenges of Vaccinated Rhesus Monkeys. J. Virol. 2012, 86, 9583–9589. [Google Scholar] [CrossRef] [PubMed]
- Janes, H.; Friedrich, D.P.; Krambrink, A.; Smith, R.J.; Kallas, E.G.; Horton, H.; Casimiro, D.R.; Carrington, M.; Geraghty, D.E.; Gilbert, P.B.; et al. Vaccine-induced gag-specific T cells are associated with reduced viremia after HIV-1 infection. J. Infect. Dis. 2013, 208, 1231–1239. [Google Scholar] [CrossRef] [PubMed]
- Mudd, P.A.; Martins, M.A.; Ericsen, A.J.; Tully, D.C.; Power, K.A.; Bean, A.T.; Piaskowski, S.M.; Duan, L.; Seese, A.; Gladden, A.D.; et al. Vaccine-induced CD8+ T cells control AIDS virus replication. Nature 2012, 491, 129–133. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Rao, U.; McClure, J.; Konopa, P.; Manocheewa, S.; Kim, M.; Chen, L.; Troyer, R.M.; Tebit, D.M.; Holte, S.; et al. Impact of mutations in highly conserved amino acids of the HIV-1 Gag-p24 and Env-gp120 proteins on viral replication in different genetic backgrounds. PLoS ONE 2014, 9, e94240. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, D.R.; Li, F.; Cruz, A.N.; Self, S.G.; Barouch, D.H. Focus and breadth of cellular immune responses elicited by a heterologous insert prime-boost vaccine regimen in rhesus monkeys. Vaccine 2012, 30, 506–509. [Google Scholar] [CrossRef] [PubMed]
- Bauer, A.; Podola, L.; Mann, P.; Missanga, M.; Haule, A.; Sudi, L.; Nilsson, C.; Kaluwa, B.; Lueer, C.; Mwakatima, M.; et al. Preferential Targeting of Conserved Gag Regions after Vaccination with a Heterologous DNA Prime-Modified Vaccinia Virus Ankara Boost HIV-1 Vaccine Regimen. J. Virol. 2017, 91, e00730-17. [Google Scholar] [CrossRef] [PubMed]
- Walsh, S.R.; Moodie, Z.; Fiore-Gartland, A.J.; Morgan, C.; Wilck, M.B.; Hammer, S.M.; Buchbinder, S.P.; Kalams, S.A.; Goepfert, P.A.; Mulligan, M.J.; et al. Vaccination With Heterologous HIV-1 Envelope Sequences and Heterologous Adenovirus Vectors Increases T-Cell Responses to Conserved Regions: HVTN 083. J. Infect. Dis. 2016, 213, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Ragonnaud, E.; Pedersen, A.G.; Holst, P.J. Breadth of T cell responses after immunization with adenovirus vectors encoding ancestral antigens or polyvalent papillomavirus antigens. Scand. J. Immunol. 2017, 85, 182–190. [Google Scholar] [CrossRef] [PubMed]
- Mothe, B.; Hu, X.; Llano, A.; Rosati, M.; Olvera, A.; Kulkarni, V.; Valentin, A.; Alicea, C.; Pilkington, G.R.; Sardesai, N.Y.; et al. A human immune data-informed vaccine concept elicits strong and broad T-cell specificities associated with HIV-1 control in mice and macaques. J. Transl. Med. 2015, 13, 60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Storcksdieck genannt, B.M.; Niezold, T.; Hannaman, D.; Uberla, K.; Tenbusch, M. The improved antibody response against HIV-1 after a vaccination based on intrastructural help is complemented by functional CD8+ T cell responses. Vaccine 2016, 34, 1744–1751. [Google Scholar] [CrossRef] [PubMed]
- Hancock, G.; Yang, H.; Yorke, E.; Wainwright, E.; Bourne, V.; Frisbee, A.; Payne, T.L.; Berrong, M.; Ferrari, G.; Chopera, D.; et al. Identification of effective subdominant anti-HIV-1 CD8+ T cells within entire post-infection and post-vaccination immune responses. PLoS Pathog. 2015, 11, e1004658. [Google Scholar] [CrossRef] [PubMed]
- Monaco, D.C.; Dilernia, D.A.; Fiore-Gartland, A.; Yu, T.; Prince, J.L.; Dennis, K.K.; Qin, K.; Schaefer, M.; Claiborne, D.T.; Kilembe, W.; et al. Balance between transmitted HLA preadapted and nonassociated polymorphisms is a major determinant of HIV-1 disease progression. J. Exp. Med. 2016, 213, 2049–2063. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, V.; Valentin, A.; Rosati, M.; Alicea, C.; Singh, A.K.; Jalah, R.; Broderick, K.E.; Sardesai, N.Y.; Le, G.S.; Mothe, B.; et al. Altered response hierarchy and increased T-cell breadth upon HIV-1 conserved element DNA vaccination in macaques. PLoS ONE 2014, 9, e86254. [Google Scholar] [CrossRef] [PubMed]
- Kron, M.W.; Engler, T.; Schmidt, E.; Schirmbeck, R.; Kochanek, S.; Kreppel, F. High-capacity adenoviral vectors circumvent the limitations of DeltaE1 and DeltaE1/DeltaE3 adenovirus vectors to induce multispecific transgene product-directed CD8 T-cell responses. J. Gene Med. 2011, 13, 648–657. [Google Scholar] [CrossRef] [PubMed]
- Schirmbeck, R.; Reimann, J.; Kochanek, S.; Kreppel, F. The Immunogenicity of Adenovirus Vectors Limits the Multispecificity of CD8 T-cell Responses to Vector-encoded Transgenic Antigens. Mol. Ther. 2008, 16, 1609–1616. [Google Scholar] [CrossRef] [PubMed]
- Betts, M.R.; Yusim, K.; Koup, R.A. Optimal antigens for HIV vaccines based on CD8+ T response, protein length, and sequence variability. DNA Cell Biol. 2002, 21, 665–670. [Google Scholar] [CrossRef] [PubMed]
- Masemola, A.; Mashishi, T.; Khoury, G.; Mohube, P.; Mokgotho, P.; Vardas, E.; Colvin, M.; Zijenah, L.; Katzenstein, D.; Musonda, R.; et al. Hierarchical targeting of subtype C human immunodeficiency virus type 1 proteins by CD8+ T cells: Correlation with viral load. J. Virol. 2004, 78, 3233–3243. [Google Scholar] [CrossRef] [PubMed]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Andersson, A.-M.C.; Schwerdtfeger, M.; Holst, P.J. Virus-Like-Vaccines against HIV. Vaccines 2018, 6, 10. https://doi.org/10.3390/vaccines6010010
Andersson A-MC, Schwerdtfeger M, Holst PJ. Virus-Like-Vaccines against HIV. Vaccines. 2018; 6(1):10. https://doi.org/10.3390/vaccines6010010
Chicago/Turabian StyleAndersson, Anne-Marie C., Melanie Schwerdtfeger, and Peter J. Holst. 2018. "Virus-Like-Vaccines against HIV" Vaccines 6, no. 1: 10. https://doi.org/10.3390/vaccines6010010