Polyacrylate–Peptide Antigen Conjugate as a Single-Dose Oral Vaccine against Group A Streptococcus

 ,
, 

 ,
,  and
and
Abstract
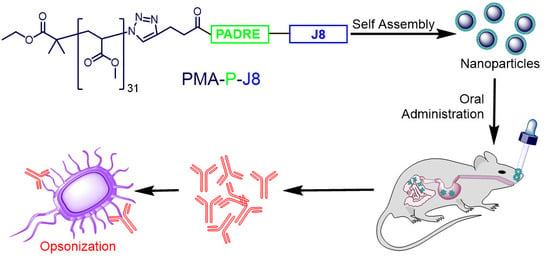
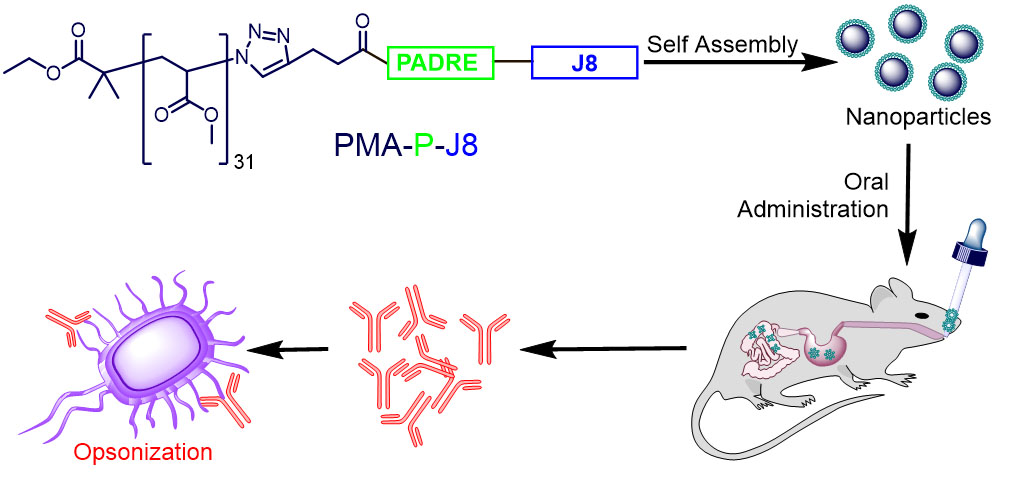
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Equipment
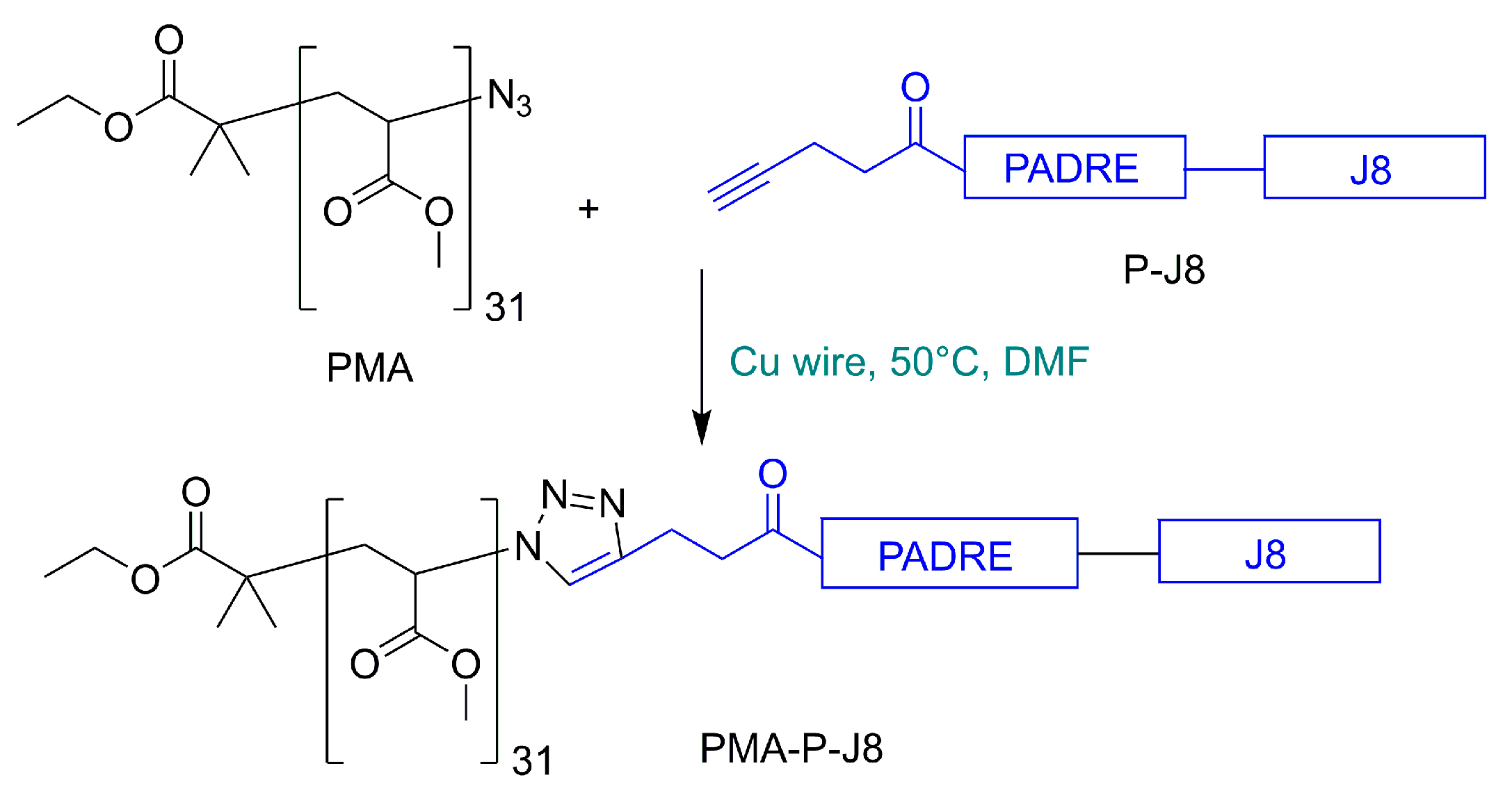
2.3. Synthesis of 4-Pentynoyl Derivative of PADRE-J8 Peptide
2.4. Polymer–Peptide Conjugation
2.5. Immunization Study
2.6. Determination of Antibody Titers (IgG and IgA)
2.7. Opsonization Assay
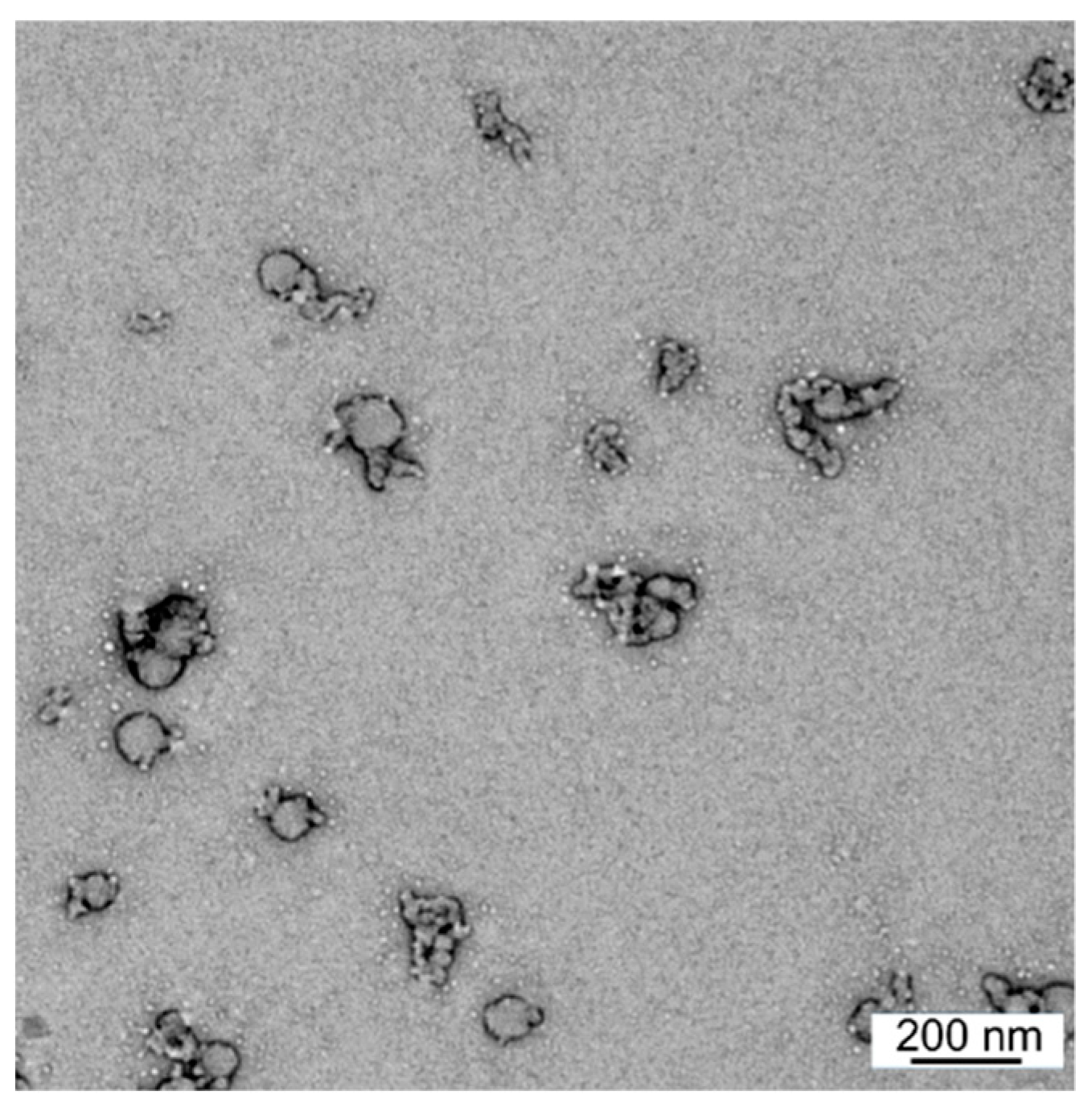
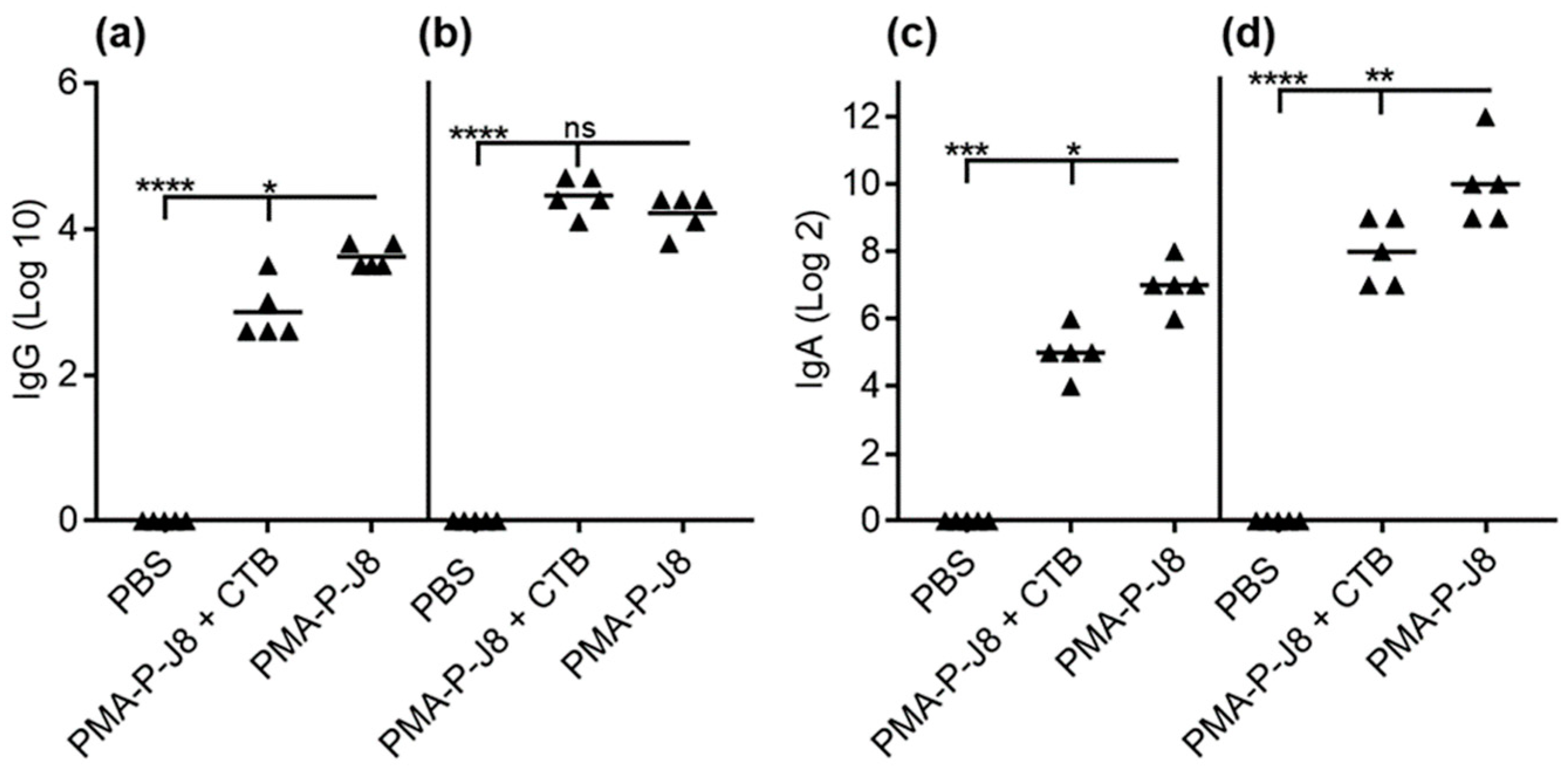
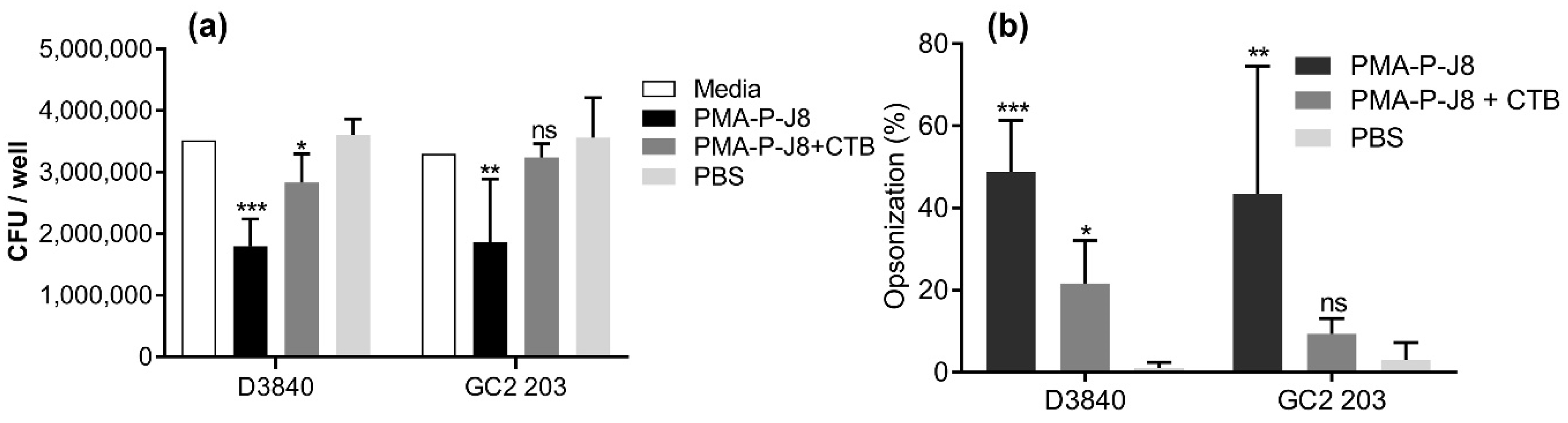
3. Results
3.1. Synthesis and Characterization
3.2. Immunization Study
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Purcell, A.W.; McCluskey, J.; Rossjohn, J. More than one reason to rethink the use of peptides in vaccine design. Nat. Rev. Drug Discov. 2007, 6, 404–414. [Google Scholar] [CrossRef] [PubMed]
- Nevagi, R.J.; Toth, I.; Skwarczynski, M. Peptide-based vaccines. In Peptide Applications in Biomedicine, Biotechnology and Bioengineering; Koutsopoulos, S., Ed.; Woodhead Publishing: Cambridge, UK, 2018; pp. 327–358. [Google Scholar] [CrossRef]
- Flower, D.R. Designing immunogenic peptides. Nat. Chem. Biol. 2013, 9, 749–753. [Google Scholar] [CrossRef] [PubMed]
- Skwarczynski, M.; Toth, I. Peptide-based synthetic vaccines. Chem. Sci. 2016, 7, 842–854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azmi, F.; Ahmad Fuaad, A.A.; Skwarczynski, M.; Toth, I. Recent progress in adjuvant discovery for peptide-based subunit vaccines. Hum. Vaccines Immunother. 2014, 10, 778–796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reed, S.G.; Orr, M.T.; Fox, C.B. Key roles of adjuvants in modern vaccines. Nat. Med. 2013, 19, 1597–1608. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Gao, S.; Cui, X.; Sun, D.; Zhao, K. Adjuvants and delivery systems based on polymeric nanoparticles for mucosal vaccines. Int. J. Pharm. 2019, 118731. [Google Scholar] [CrossRef]
- Yusuf, H.; Kett, V. Current prospects and future challenges for nasal vaccine delivery. Hum. Vaccines Immunother. 2017, 13, 34–45. [Google Scholar] [CrossRef]
- Vela Ramirez, J.E.; Sharpe, L.A.; Peppas, N.A. Current state and challenges in developing oral vaccines. Adv. Drug Deliv. Rev. 2017, 114, 116–131. [Google Scholar] [CrossRef]
- Marasini, N.; Skwarczynski, M.; Toth, I. Oral delivery of nanoparticle-based vaccines. Expert Rev. Vaccines 2014, 13, 1361–1376. [Google Scholar] [CrossRef]
- Lynskey, N.N.; Lawrenson, R.A.; Sriskandan, S. New understandings in Streptococcus pyogenes. Curr. Opin. Infect. Dis. 2011, 24, 196–202. [Google Scholar] [CrossRef]
- Carapetis, J.R.; Steer, A.C.; Mulholland, E.K.; Weber, M. The global burden of group A streptococcal diseases. Lancet Infect. Dis. 2005, 5, 685–694. [Google Scholar] [CrossRef]
- Cunningham, M.W. Pathogenesis of group A streptococcal infections. Clin. Microbiol. Rev. 2000, 13, 470–511. [Google Scholar] [CrossRef] [PubMed]
- Sanyahumbi, A.S.; Colquhoun, S.; Wyber, R.; Carapetis, J.R. Global disease burden of group A Streptococcus. In Streptococcus Pyogenes: Basic Biology to Clinical Manifestations [Internet]; University of Oklahoma Health Sciences Center: Oklahoma City, OK, USA, 2016. Available online: https://www.ncbi.nlm.nih.gov/books/NBK333415/ (accessed on 12 December 2019).
- Paar, J.A.; Berrios, N.M.; Rose, J.D.; Caceres, M.; Pena, R.; Perez, W.; Chen-Mok, M.; Jolles, E.; Dale, J.B. Prevalence of Rheumatic Heart Disease in Children and Young Adults in Nicaragua. Am. J. Cardiol. 2010, 105, 1809–1814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watkins, D.A.; Johnson, C.O.; Colquhoun, S.M.; Karthikeyan, G.; Beaton, A.; Bukhman, G.; Forouzanfar, M.H.; Longenecker, C.T.; Mayosi, B.M.; Mensah, G.A.; et al. Global, Regional, and National Burden of Rheumatic Heart Disease, 1990–2015. N. Engl. J. Med. 2017, 377, 713–722. [Google Scholar] [CrossRef] [PubMed]
- Steer, A.C.; Carapetis, J.R. Prevention and treatment of rheumatic heart disease in the developing world. Nat. Rev. Cardiol. 2009, 6, 689–698. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.M.; Green, M. Group A Streptococcus. Semin. Pediatr. Infect. Dis. 2006, 17, 140–148. [Google Scholar] [CrossRef]
- Steer, A.C.; Carapetis, J.R.; Dale, J.B.; Fraser, J.D.; Good, M.F.; Guilherme, L.; Moreland, N.J.; Mulholland, E.K.; Schodel, F.; Smeesters, P.R. Status of research and development of vaccines for Streptococcus pyogenes. Vaccine 2016, 34, 2953–2958. [Google Scholar] [CrossRef]
- Azuar, A.; Jin, W.; Mukaida, S.; Hussein, W.M.; Toth, I.; Skwarczynski, M. Recent Advances in the Development of Peptide Vaccines and Their Delivery Systems Against Group A Streptococcus. Vaccines 2019, 7, 58. [Google Scholar] [CrossRef] [Green Version]
- Steer, A.C.; Batzloff, M.R.; Mulholland, K.; Carapetis, J.R. Group A streptococcal vaccines: Facts versus fantasy. Curr. Opin. Infect. Dis. 2009, 22, 544–552. [Google Scholar] [CrossRef]
- McArthur, J.D.; Walker, M.J. Domains of group A streptococcal M protein that confer resistance to phagocytosis, opsonization and protection: Implications for vaccine development. Mol. Microbiol. 2006, 59, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Good, M.F.; Xu, H.; Batzloff, M. Adapting immunity with subunit vaccines: Case studies with group A Streptococcus and malaria. Int. J. Parasitol. 2002, 32, 575–580. [Google Scholar] [CrossRef]
- Brandt, E.R.; Sriprakash, K.S.; Hobb, R.I.; Hayman, W.A.; Zeng, W.G.; Batzloff, M.R.; Jackson, D.C.; Good, M.F. New multi-determinant strategy for a group A streptococcal vaccine designed for the Australian Aboriginal population. Nat. Med. 2000, 6, 455–459. [Google Scholar] [CrossRef] [PubMed]
- McMillan, D.J.; Drèze, P.A.; Vu, T.; Bessen, D.E.; Guglielmini, J.; Steer, A.C.; Carapetis, J.R.; Melderen, L.; Sriprakash, K.S.; Smeesters, P.R. Updated model of group A Streptococcus M proteins based on a comprehensive worldwide study. Clin. Microbiol. Infect. 2013, 19, E222–E229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pruksakorn, S.; Currie, B.; Brandt, E.; Phornphutkul, C.; Hunsakunachai, S.; Manmontri, A.; Robinson, J.H.; Kehoe, M.A.; Galbraith, A.; Good, M.F. Identification of T cell autoepitopes that cross-react with the C-terminal segment of the M protein of group A streptococci. Int. Immunol. 1994, 6, 1235–1244. [Google Scholar] [CrossRef] [PubMed]
- Pruksakorn, S.; Brandt, E.; Good, M.; Currie, B.; Martin, D.; Galbraith, A.; Phornphutkul, C.; Hunsakunachai, S.; Manmontri, A. Towards a vaccine for rheumatic fever: Identification of a conserved target epitope on M protein of group A streptococci. Lancet 1994, 344, 639–642. [Google Scholar] [CrossRef]
- Sekuloski, S.; Batzloff, M.R.; Griffin, P.; Parsonage, W.; Elliott, S.; Hartas, J.; O‘Rourke, P.; Marquart, L.; Pandey, M.; Rubin, F.A.; et al. Evaluation of safety and immunogenicity of a group A streptococcus vaccine candidate (MJ8VAX) in a randomized clinical trial. PLoS ONE 2018, 13, e0198658. [Google Scholar] [CrossRef]
- Pandey, M.; Powell, J.; Calcutt, A.; Zaman, M.; Phillips, Z.N.; Ho, M.F.; Batzloff, M.R.; Good, M.F. Physicochemical characterisation, immunogenicity and protective efficacy of a lead streptococcal vaccine: Progress towards Phase I trial. Sci. Rep. 2017, 7, 13786. [Google Scholar] [CrossRef] [Green Version]
- Olive, C.; Sun, H.K.; Ho, M.F.; Dyer, J.; Horvath, A.; Toth, I.; Good, M.F. Intranasal administration is an effective mucosal vaccine delivery route for self-adjuvanting lipid core peptides targeting the group A streptococcal M protein. J. Infect. Dis. 2006, 194, 316–324. [Google Scholar] [CrossRef]
- Dunn, L.A.; McMillan, D.J.; Batzloff, M.; Zeng, W.; Jackson, D.C.; Upcroft, J.A.; Upcroft, P.; Olive, C. Parenteral and mucosal delivery of a novel multi-epitope M protein-based group A streptococcal vaccine construct: Investigation of immunogenicity in mice. Vaccine 2002, 20, 2635–2640. [Google Scholar] [CrossRef]
- Marasini, N.; Giddam, A.K.; Ghaffar, K.A.; Batzloff, M.R.; Good, M.F.; Skwarczynski, M.; Toth, I.J.N. Multilayer engineered nanoliposomes as a novel tool for oral delivery of lipopeptide-based vaccines against group A Streptococcus. Nanomedicine 2016, 11, 1223–1236. [Google Scholar] [CrossRef]
- Khongkow, M.; Liu, T.Y.; Bartlett, S.; Hussein, W.M.; Nevagi, R.; Jia, Z.F.; Monteiro, M.J.; Wells, J.W.; Ruktanonchai, U.R.; Skwarczynski, M.; et al. Liposomal formulation of polyacrylate-peptide conjugate as a new vaccine candidate against cervical cancer. Prec. Nanomed. 2018, 1, 183–193. [Google Scholar] [CrossRef]
- Chandrudu, S.; Bartlett, S.; Khalil, Z.G.; Jia, Z.; Hussein, W.M.; Capon, R.J.; Batzloff, M.R.; Good, M.F.; Monteiro, M.J.; Skwarczynski, M.; et al. Linear and branched polyacrylates as a delivery platform for peptide-based vaccines. Ther. Deliv. 2016, 7, 601–609. [Google Scholar] [CrossRef] [PubMed]
- Hussein, W.M.; Liu, T.Y.; Jia, Z.; McMillan, N.A.; Monteiro, M.J.; Toth, I.; Skwarczynski, M. Multiantigenic peptide-polymer conjugates as therapeutic vaccines against cervical cancer. Bioorg. Med. Chem. 2016, 24, 4372–4380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, T.Y.; Giddam, A.K.; Hussein, W.M.; Jia, Z.; McMillan, N.A.; Monteiro, M.J.; Toth, I.; Skwarczynski, M. Self-Adjuvanting Therapeutic Peptide-Based Vaccine Induce CD8+ Cytotoxic T Lymphocyte Responses in a Murine Human Papillomavirus Tumor Model. Curr. Drug Deliv. 2015, 12, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, T.Y.; Hussein, W.M.; Giddam, A.K.; Jia, Z.F.; Reiman, J.M.; Zaman, M.; McMillan, N.A.J.; Good, M.F.; Monteiro, M.J.; Toth, I.; et al. Polyacrylate-Based Delivery System for Self-adjuvanting Anticancer Peptide Vaccine. J. Med. Chem. 2015, 58, 888–896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuaad, A.A.H.A.; Jia, Z.F.; Zaman, M.; Hartas, J.; Ziora, Z.M.; Lin, I.C.; Moyle, P.M.; Batzloff, M.R.; Good, M.F.; Monteiro, M.J.; et al. Polymer-peptide hybrids as a highly immunogenic single-dose nanovaccine. Nanomedicine 2014, 9, 35–43. [Google Scholar] [CrossRef]
- Zaman, M.; Skwarczynski, M.; Malcolm, J.M.; Urbani, C.N.; Jia, Z.F.; Batzloff, M.R.; Good, M.F.; Monteiro, M.J.; Toth, I. Self-adjuvanting polyacrylic nanoparticulate delivery system for group A streptococcus (GAS) vaccine. Nanomed. Nanotechnol. Biol. Med. 2011, 7, 168–173. [Google Scholar] [CrossRef]
- Skwarczynski, M.; Zaman, M.; Urbani, C.N.; Lin, I.C.; Jia, Z.F.; Batzloff, M.R.; Good, M.F.; Monteiro, M.F.; Toth, I. Polyacrylate Dendrimer Nanoparticles: A Self-Adjuvanting Vaccine Delivery System. Angew. Chem. Int. Ed. 2010, 49, 5742–5745. [Google Scholar] [CrossRef]
- Marasini, N.; Giddam, A.K.; Khalil, Z.G.; Hussein, W.M.; Capon, R.J.; Batzloff, M.R.; Good, M.F.; Toth, I.; Skwarczynski, M.J.N. Double adjuvanting strategy for peptide-based vaccines: Trimethyl chitosan nanoparticles for lipopeptide delivery. Nanomedicine 2016, 11, 3223–3235. [Google Scholar] [CrossRef]
- Ahmad Fuaad, A.A.H.; Skwarczynski, M.; Toth, I. The Use of Microwave-Assisted Solid-Phase Peptide Synthesis and Click Chemistry for the Synthesis of Vaccine Candidates Against Hookworm Infection. Methods Mol. Biol. 2016, 1403, 639–653. [Google Scholar] [CrossRef]
- Nevagi, R.J.; Skwarczynski, M.; Toth, I. Polymers for subunit vaccine delivery. Eur. Polym. J. 2019, 114, 397–410. [Google Scholar] [CrossRef]
- Adams, J.R.; Haughney, S.L.; Mallapragada, S.K. Effective polymer adjuvants for sustained delivery of protein subunit vaccines. Acta Biomater. 2015, 14, 104–114. [Google Scholar] [CrossRef] [PubMed]
- Sahdev, P.; Ochyl, L.J.; Moon, J.J. Biomaterials for Nanoparticle Vaccine Delivery Systems. Pharm. Res. 2014, 2563–2582. [Google Scholar] [CrossRef]
- Skwarczynski, M.; Toth, I. Recent advances in peptide-based subunit nanovaccines. Nanomedicine 2014, 9, 2657–2669. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Skwarczynski, M.; Toth, I. Polyelectrolyte-Based Platforms for the Delivery of Peptides and Proteins. ACS Biomater. Sci. Eng. 2019, 5, 4937–4950. [Google Scholar] [CrossRef]
- Ghaffar, K.A.; Giddam, A.K.; Zaman, M.; Skwarczynski, M.; Toth, I. Liposomes as nanovaccine delivery systems. Curr. Top. Med. Chem. 2014, 14, 1194–1208. [Google Scholar] [CrossRef]
- Eyles, J.; Carpenter, Z.; Alpar, H.; Williamson, E. Immunological aspects of polymer microsphere vaccine delivery systems. J. Drug Target. 2003, 11, 509–514. [Google Scholar] [CrossRef]
- Perrie, Y.; Kirby, D.; Bramwell, V.W.; Mohammed, A.R. Recent developments in particulate-based vaccines. Recent Pat. Drug Deliv. Formul. 2007, 1, 117–129. [Google Scholar] [CrossRef]
- Galliverti, G.; Tichet, M.; Domingos-Pereira, S.; Hauert, S.; Nardelli-Haefliger, D.; Swartz, M.A.; Hanahan, D.; Wullschleger, S. Nanoparticle Conjugation of Human Papillomavirus 16 E7-long Peptides Enhances Therapeutic Vaccine Efficacy against Solid Tumors in Mice. Cancer Immunol. Res. 2018, 6, 1301–1313. [Google Scholar] [CrossRef]
- Nevagi, R.J.; Khalil, Z.G.; Hussein, W.M.; Powell, J.; Batzloff, M.R.; Capon, R.J.; Good, M.F.; Skwarczynski, M.; Toth, I. Polyglutamic acid-trimethyl chitosan-based intranasal peptide nano-vaccine induces potent immune responses against group A streptococcus. Acta Biomater. 2018, 80, 278–287. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.-Y.; Fuaad, A.A.H.A.; Toth, I.; Skwarczynski, M. Self-assembled peptide-polymer conjugates as vaccines. Chim. Oggi. 2014, 32, 18–22. [Google Scholar]
- Wilson, D.S.; Hirosue, S.; Raczy, M.M.; Bonilla-Ramirez, L.; Jeanbart, L.; Wang, R.; Kwissa, M.; Franetich, J.F.; Broggi, M.A.S.; Diaceri, G.; et al. Antigens reversibly conjugated to a polymeric glyco-adjuvant induce protective humoral and cellular immunity. Nat. Mater. 2019, 18, 175–185. [Google Scholar] [CrossRef] [PubMed]
- Alexander, J.; del Guercio, M.-F.; Maewal, A.; Qiao, L.; Fikes, J.; Chesnut, R.W.; Paulson, J.; Bundle, D.R.; DeFrees, S.; Sette, A. Linear PADRE T helper epitope and carbohydrate B cell epitope conjugates induce specific high titer IgG antibody responses. J. Immunol. 2000, 164, 1625–1633. [Google Scholar] [CrossRef] [PubMed]
- Moyle, P.M.; Toth, I. Modern Subunit Vaccines: Development, Components, and Research Opportunities. ChemMedChem 2013, 8, 360–376. [Google Scholar] [CrossRef]
- Liu, T.Y.; Hussein, W.M.; Jia, Z.F.; Ziora, Z.M.; McMillan, N.A.J.; Monteiro, M.J.; Toth, I.; Skwarczynski, M. Self-Adjuvanting Polymer-Peptide Conjugates As Therapeutic Vaccine Candidates against Cervical Cancer. Biomacromolecules 2013, 14, 2798–2806. [Google Scholar] [CrossRef] [Green Version]
- Chandy, A.G.; Hultkrantz, S.; Raghavan, S.; Czerkinsky, C.; Lebens, M.; Telemo, E.; Holmgren, J. Oral tolerance induction by mucosal administration of cholera toxin B-coupled antigen involves T-cell proliferation in vivo and is not affected by depletion of CD25(+) T cells. Immunology 2006, 118, 311–320. [Google Scholar] [CrossRef]
- Sun, J.-B.; Flach, C.-F.; Czerkinsky, C.; Holmgren, J. B Lymphocytes Promote Expansion of Regulatory T Cells in Oral Tolerance: Powerful Induction by Antigen Coupled to Cholera Toxin B Subunit. J. Immunol. 2008, 181, 8278. [Google Scholar] [CrossRef] [Green Version]
- Stratmann, T. Cholera Toxin Subunit B as Adjuvant—An Accelerator in Protective Immunity and a Break in Autoimmunity. Vaccines 2015, 3, 579. [Google Scholar] [CrossRef] [Green Version]
- Bartlett, S.; Eichenberger, R.M.; Nevagi, R.J.; Ghaffar, K.A.; Marasini, N.; Dai, Y.; Loukas, A.; Toth, I.; Skwarczynski, M. Lipopeptide-based oral vaccine against hookworm infection. J. Infect. Dis. 2019. [Google Scholar] [CrossRef]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Faruck, M.O.; Zhao, L.; Hussein, W.M.; Khalil, Z.G.; Capon, R.J.; Skwarczynski, M.; Toth, I. Polyacrylate–Peptide Antigen Conjugate as a Single-Dose Oral Vaccine against Group A Streptococcus. Vaccines 2020, 8, 23. https://doi.org/10.3390/vaccines8010023
Faruck MO, Zhao L, Hussein WM, Khalil ZG, Capon RJ, Skwarczynski M, Toth I. Polyacrylate–Peptide Antigen Conjugate as a Single-Dose Oral Vaccine against Group A Streptococcus. Vaccines. 2020; 8(1):23. https://doi.org/10.3390/vaccines8010023
Chicago/Turabian StyleFaruck, Mohammad Omer, Lili Zhao, Waleed M. Hussein, Zeinab G. Khalil, Robert J. Capon, Mariusz Skwarczynski, and Istvan Toth. 2020. "Polyacrylate–Peptide Antigen Conjugate as a Single-Dose Oral Vaccine against Group A Streptococcus" Vaccines 8, no. 1: 23. https://doi.org/10.3390/vaccines8010023