
Prophylactic Multi-Subunit Vaccine against Chlamydia trachomatis: In Vivo Evaluation in Mice

 , , ,
, , ,  and
and
Abstract
:1. Introduction
1.1. Chlamydia trachomatis and Diseases Caused by This Intracellular Bacterium
1.2. Former Attempts to Develop a Vaccine against Chlamydia trachomatis
1.3. Selection of Chlamydial Antigens, the Adjuvant and the Test System for Our New Vaccine
2. Materials and Methods
2.1. Chlamydial Strains and Their Growth in Cell Culture
2.2. Preparation and Purification of the Recombinant Antigens PmpA, PmpD, PmpG, PmpH, and Ctad1 Derived from C.tr. Serovar E/DK20

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pmp’s | C.tr. E E/DK20 | C.tr. D UW3/CX | C.tr. A A/HAR-13 | C.tr. LGV L2/434/Bu | C. muridarum Nigg 2 MCR |
|---|---|---|---|---|---|
| A | WP_009872639.1 (975aa) | WP_010725190.1 (975aa) | WP_009871764.1 (975aa) | WP_009872639.1 (975aa) | WP_010231239.1 (976aa) |
| D | WP_009872948.1 (1530aa) | WP_010725352.1 (1531aa) | WP_011324878.1 (1531aa) | WP_009873422.1 (1530aa) | WP_010229791.1 (1520aa) |
| G | WP_014541208.1 (1013aa) | WP_010725377.1 (1013aa) | WP_011324914.1 (1013aa) | WP_009873478.1 (1012aa) | WP_010229977.1 (986aa) |
| H | WP_014541209.1 (1014aa) | WP_010725378.1 (1016aa) | WP_011324915.1 (1018aa) | WP_009873479.1 (1006aa) | WP_010229979.1 (980aa) |
| Ctad1 | WP_010724985.1 (433aa) | WP_010724985.1 (433aa) | WP_011324521.1 (433aa) | AGJ64368.2 (477aa) | AID37821.1 (446aa) |
2.3. Immunoblotting and Coomassie-Staining
2.4. Preparation of the c-di-AMP-Adjuvanted Vaccine Directly before Application to Mice
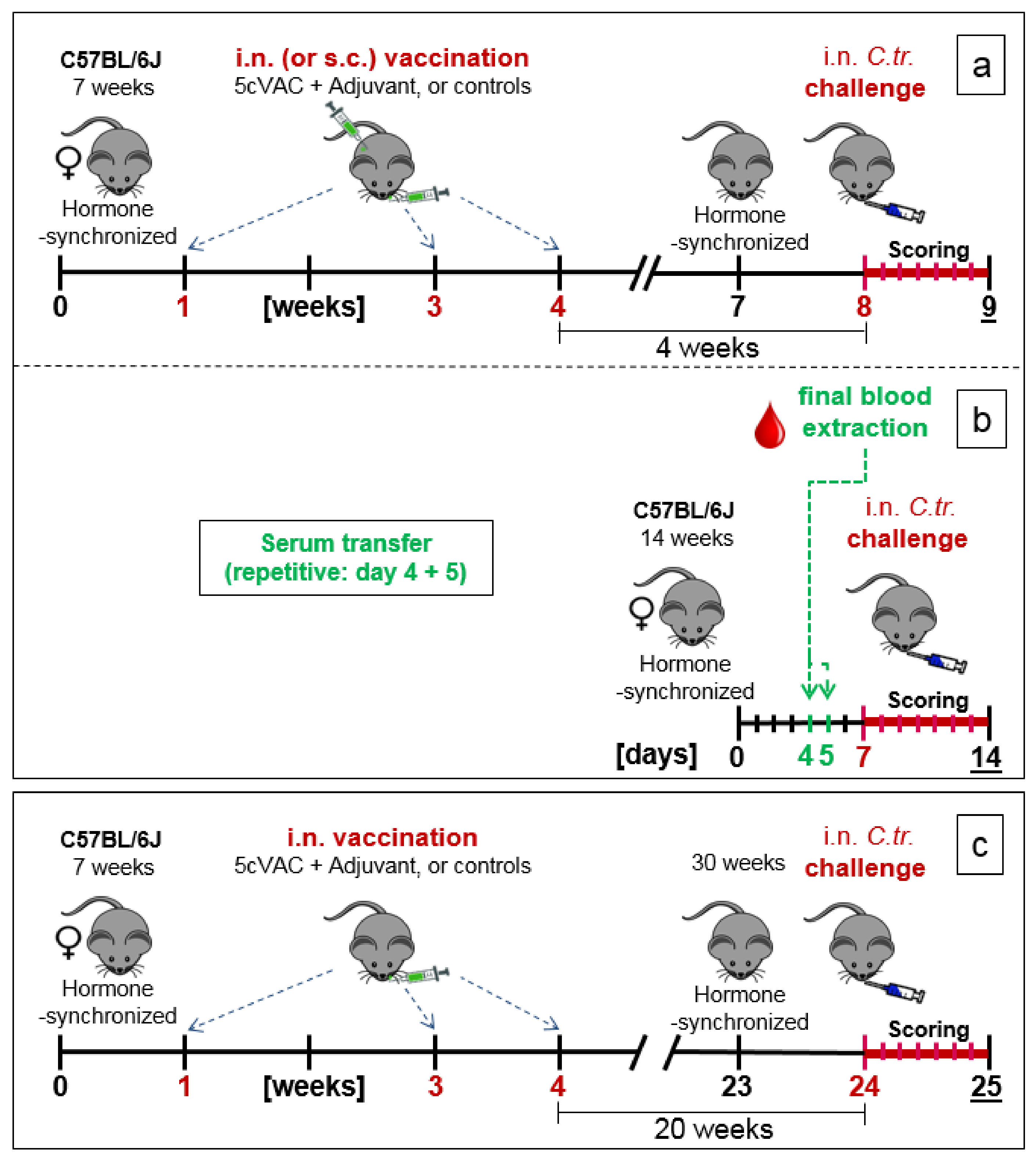
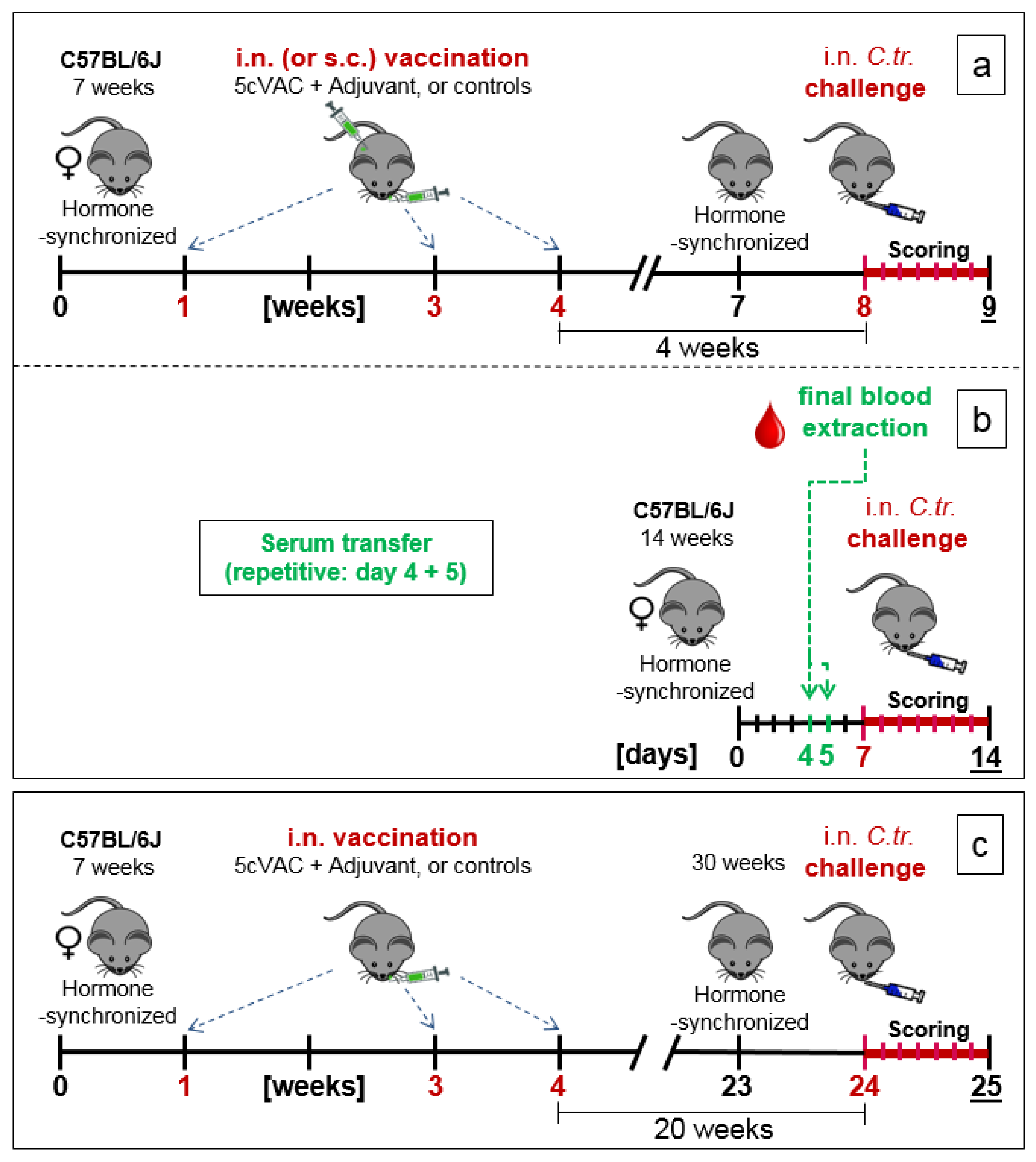
2.5. Three Experimental Setups of the Mouse Vaccination-Chlamydia Trachomatis Lung Challenge Infection Model: Short-Term, Serum-Transfer, and Long-Term Protection
- (a)
- Short-term protection model (Figure 1a): in week 7 of the experiment, the cycle of the female mice was hormone-synchronized again. In week 8, i.e., 4 weeks after the last booster vaccination, the 15-week-old mice were challenged by i.n. application of Chlamydia differently pretreated animals per group for the C.tr. E, D, and L2, and mice per group for C.tr. A, respectively). One group of animals received mock material instead of Chlamydia (usually n = 8, only in one experiment (as indicated) n = 4).
- (b)
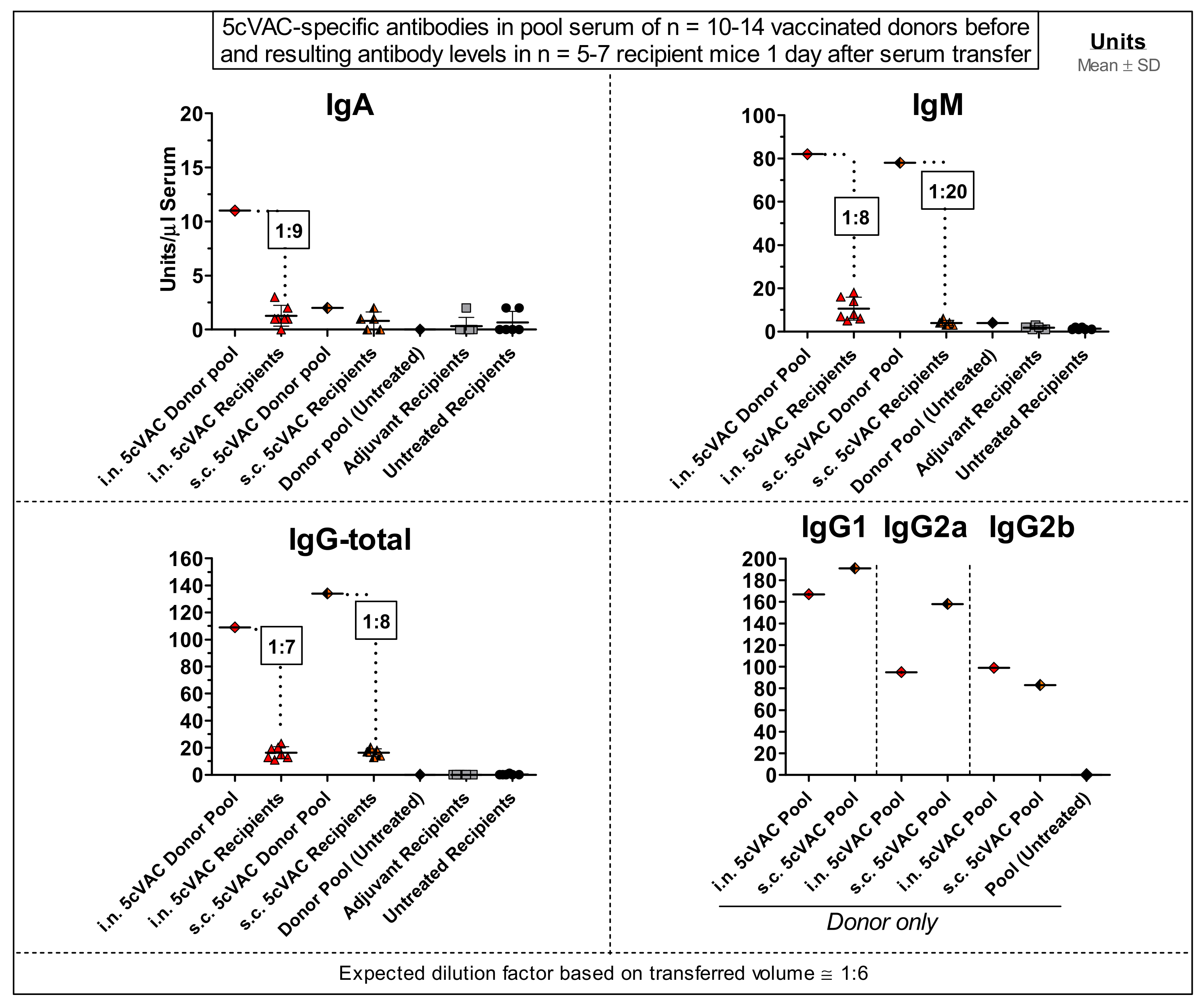
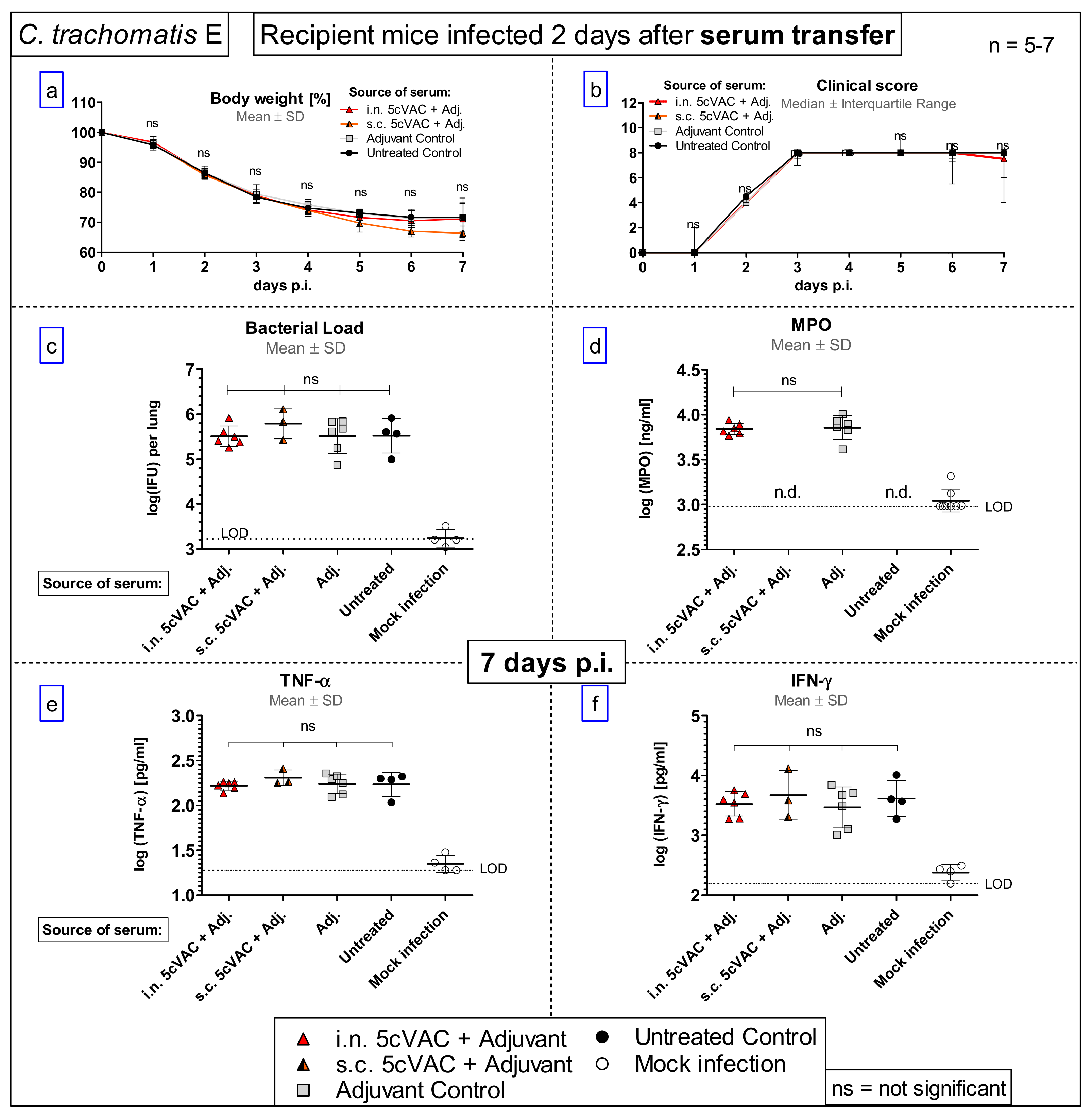
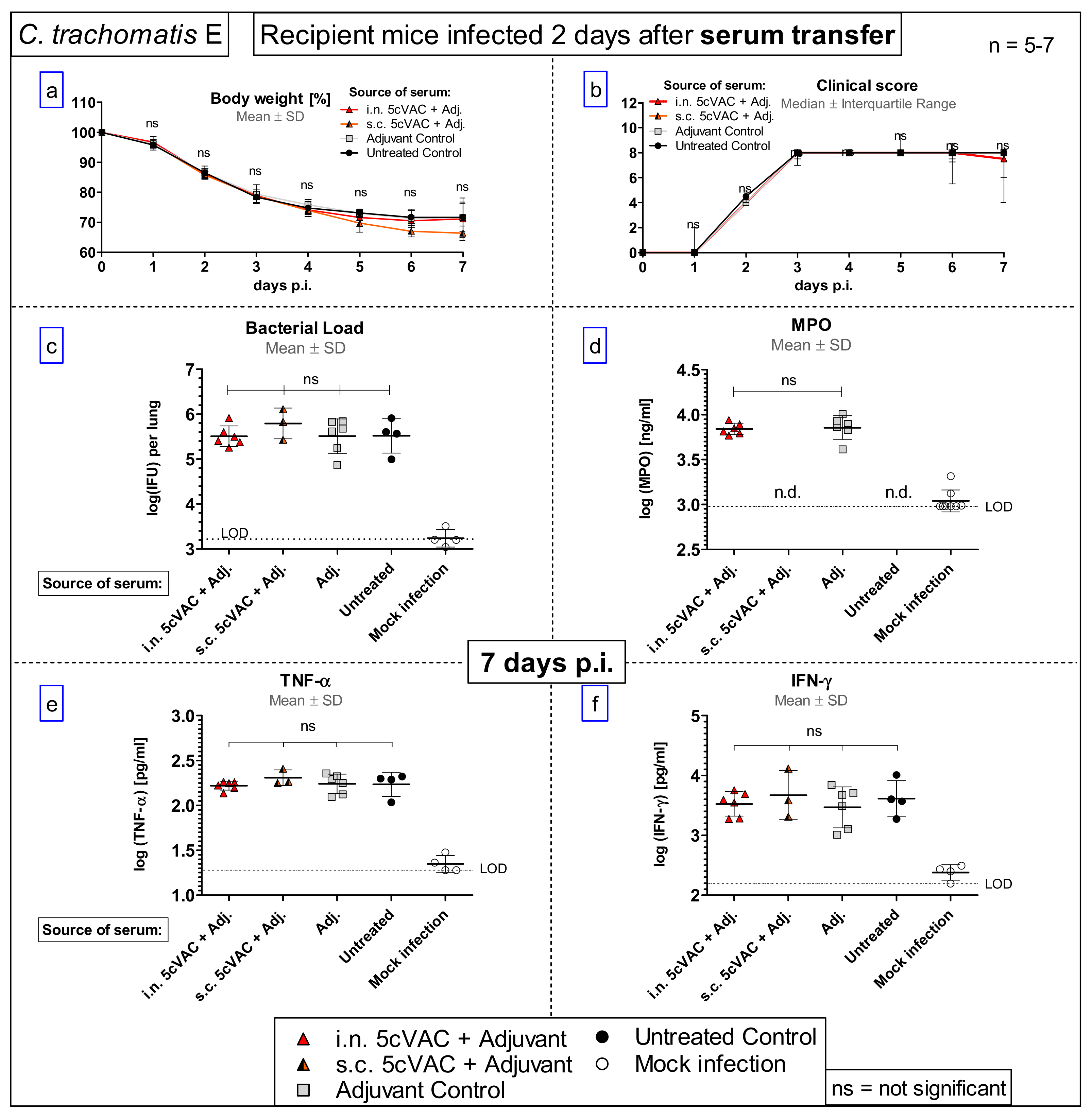
- Serum transfer-short-term protection model (Figure 1b): We wanted to clarify to which extent antibodies that are circulating in murine blood after vaccination with c-di-AMP-adjuvanted 5cVAC are actually conferring protection against C.tr. E. Four weeks after the last i.n. or s.c. application of the vaccine, or buffer, or c-di-AMP alone, respectively, i.e., exactly at the time point when the challenge infection was normally performed, the mice were sacrificed and their blood was collected by heart puncture. The sera of the four differently treated groups of animals (n = 10–14 per group) were pooled and 170 µL were transferred twice intravenously (i.v.) to non-vaccinated 14- to 15-week-old healthy animals (n = 5–7 per group) on days 4 and 5 after their hormone-synchronization. C.tr.-specific antibodies were determined in the four donor pool sera as well as in the sera of the individual recipient animals. For that purpose, small blood samples were collected on the day between serum-transfer and i.n. C.tr. E-challenge infection. One week after hormone-treatment and two days after the last serum-transfer, i.n. challenge infection was conducted with 1.3 × 106 IFU of C.tr. E, i.e., exactly like in the short-term protection model.
- (c)
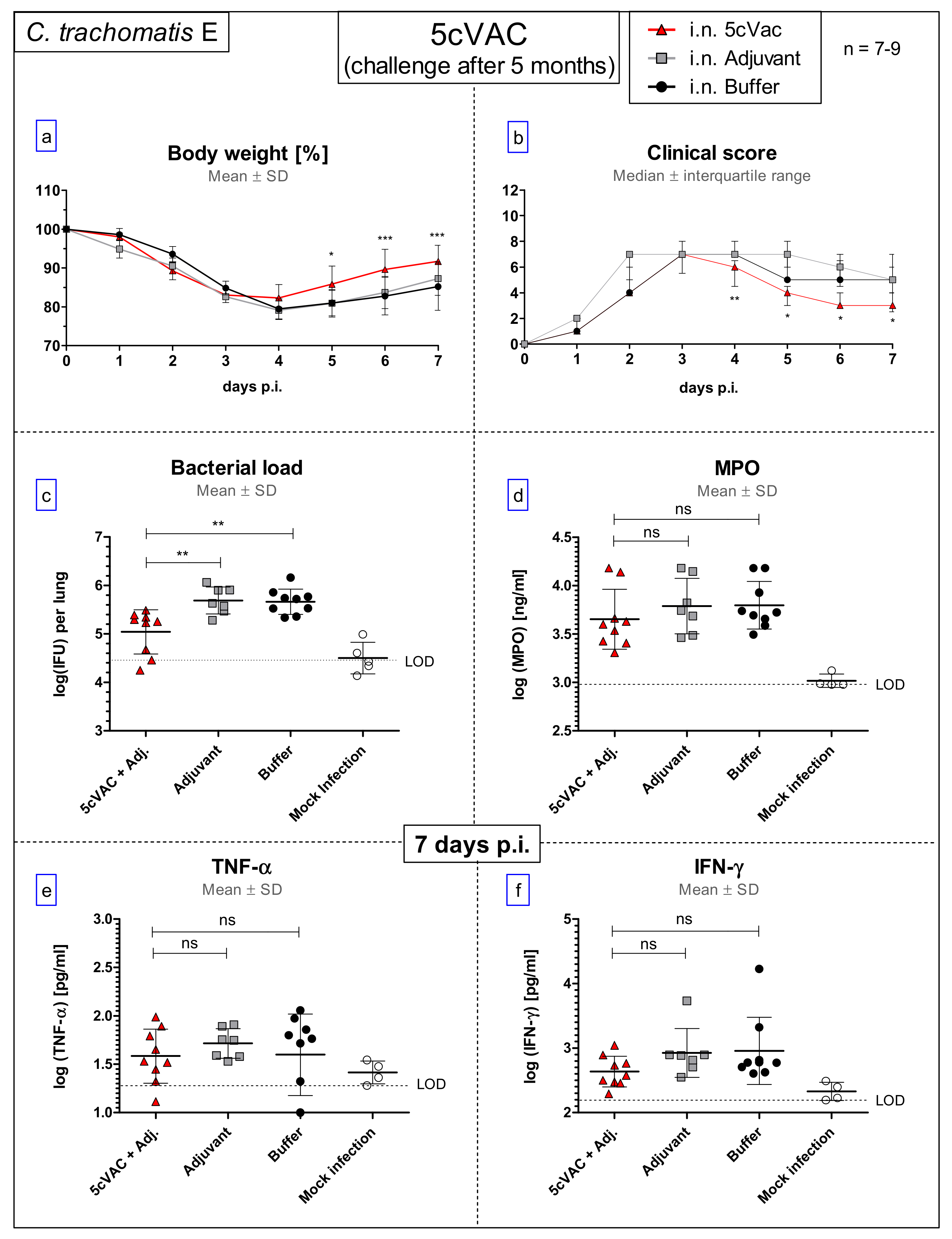
- Long-term protection model (Figure 1c): To determine the long-term protection achieved, mice that had been vaccinated i.n. with c-di-AMP-adjuvanted 5cVAC in week 1, 3, and 4, as described above, were hormone-synchronized again in week 23. Twenty weeks after the last booster vaccination, the mice were challenged i.n. with C.tr. E. According to preliminary titration experiments (Supplement Figure S4), 31-week-old mice react more sensitive to chlamydial infection, independent of body weight or vaccination. Thus, a more than three-times lower IFU (4 × 105) of C.tr. E had to be administered for challenge in this setting. Otherwise, a relevant portion of the older non-vaccinated, infected mice would not have achieved the desired seven-day-observation period after challenge infection, thereby reducing the group size of this essential control group.
2.6. Clinical Scoring
2.7. Determination of Bacterial Load in Mouse Lung Homogenate by Flow Cytometry
2.8. Determination of TNF-α and IFN-γ and Myeloperoxidase Levels in Lung Homogenate
2.9. ELISAs for Circulating 5cVAC-Specific IgA/M/G and IgG Subtypes in Blood
2.10. Functional Analysis of Mouse Splenocytes: Proliferation, ELISpot, Flow Cytometry
2.10.1. Measurement of Cellular Proliferation
2.10.2. ELISpot Assay
2.10.3. Flow Cytometric Determination of Multifunctional T Cells
2.11. Group Sizes and Statistical Analysis
3. Results
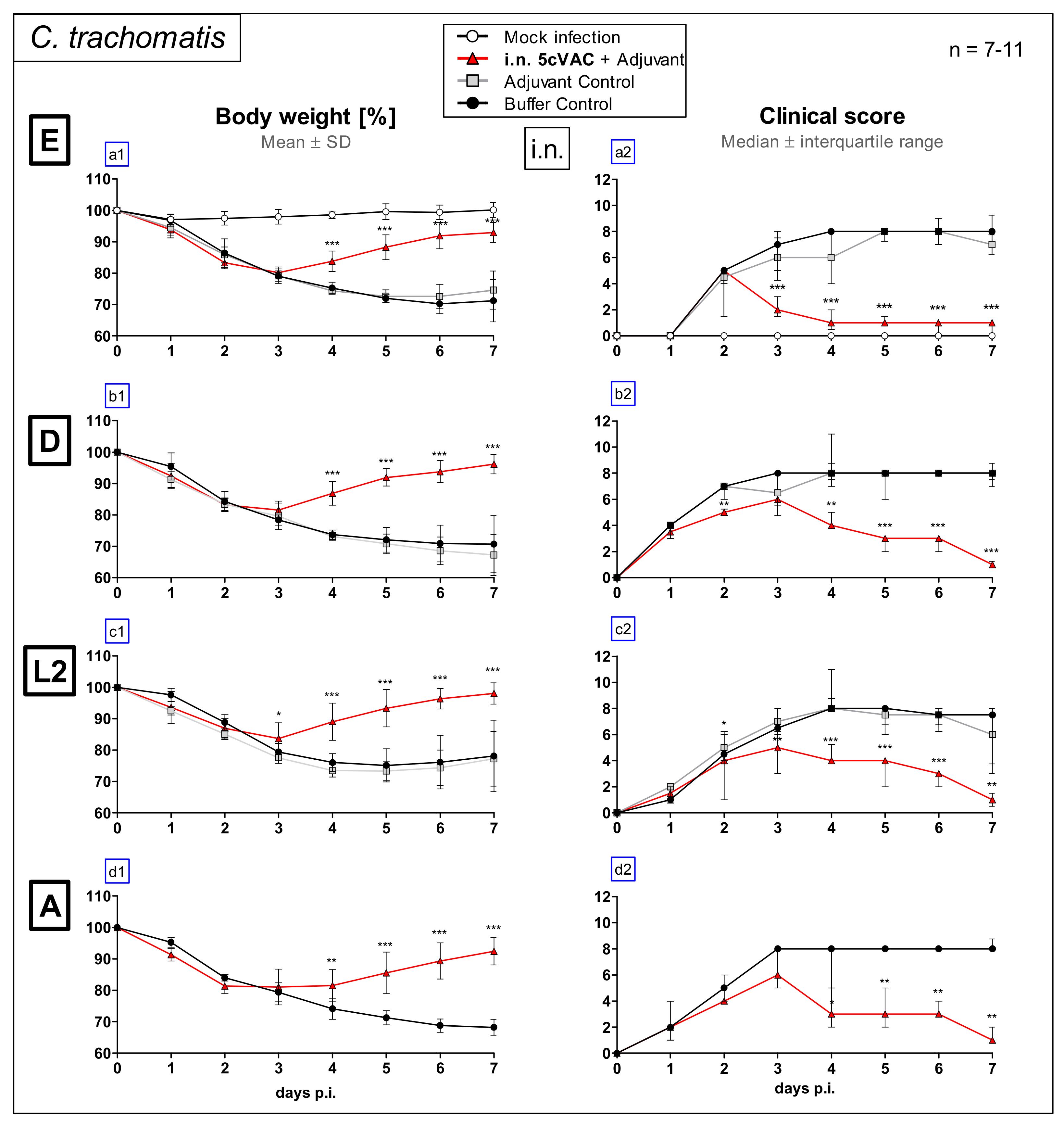
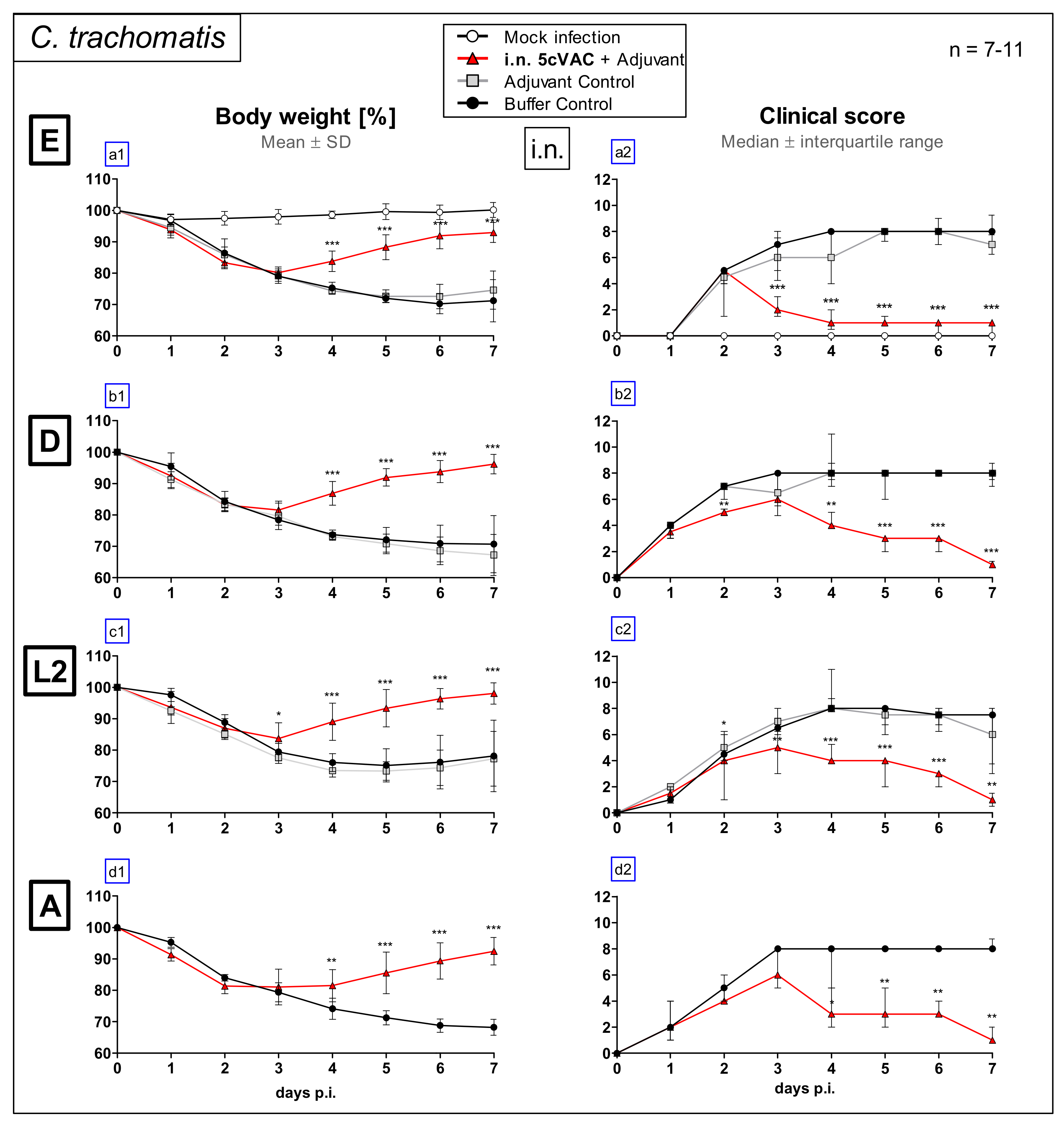
3.1. Intranasal Vaccination with the c-di-AMP-Adjuvanted 5cVAC-Formulation Improves Loss of Body Weight and Clinical Score in Lung Challenge Infection with Serovar E, D, L2, or A of C. Trachomatis
3.2. Intranasal Vaccination with c-di-AMP-Adjuvanted 5cVAC Improves Bacterial Clearance and Leads to Decreased Levels of the Granulocyte Marker MPO in the Lung after Challenge Infection with four Different Serovars of C. trachomatis
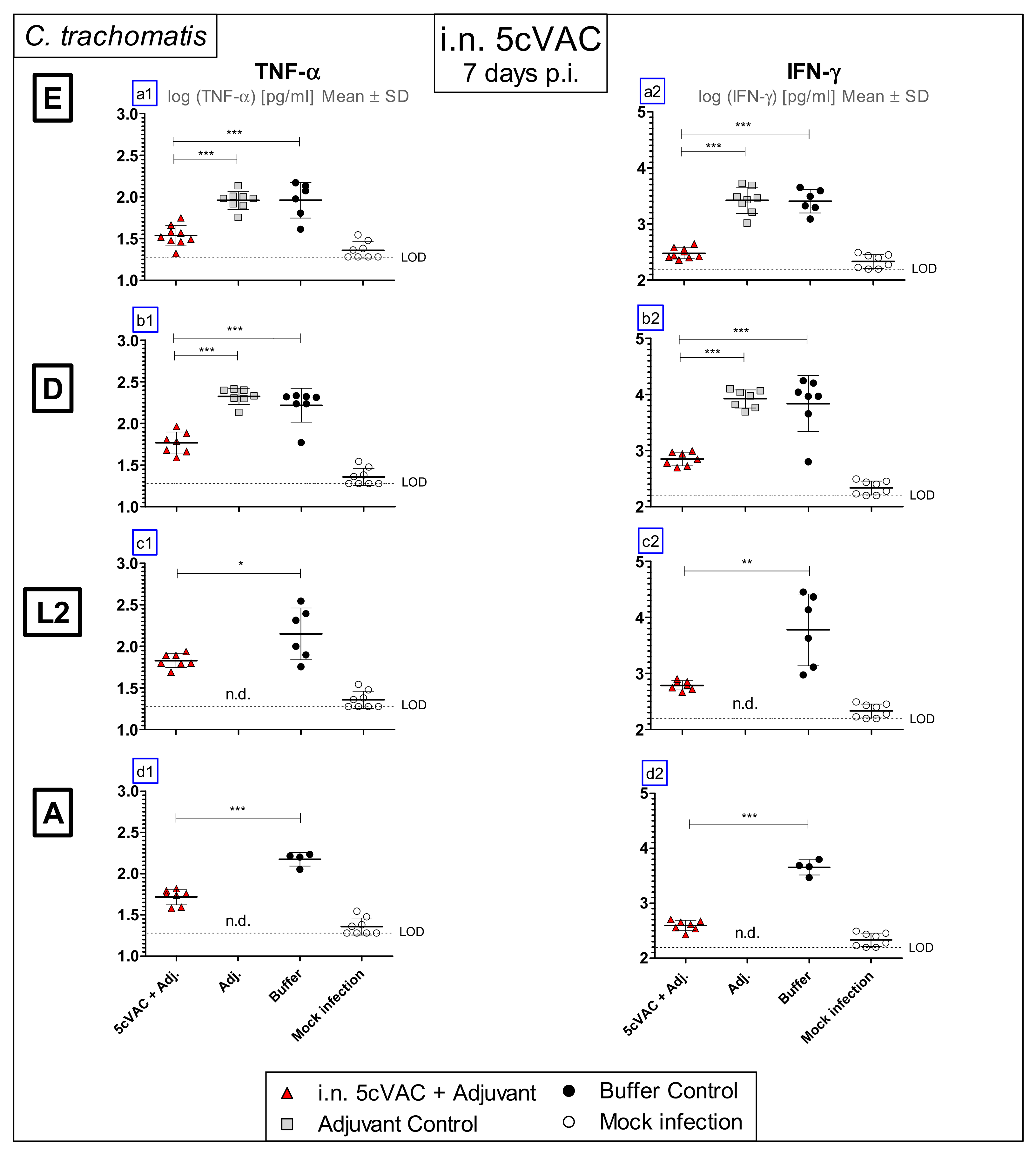
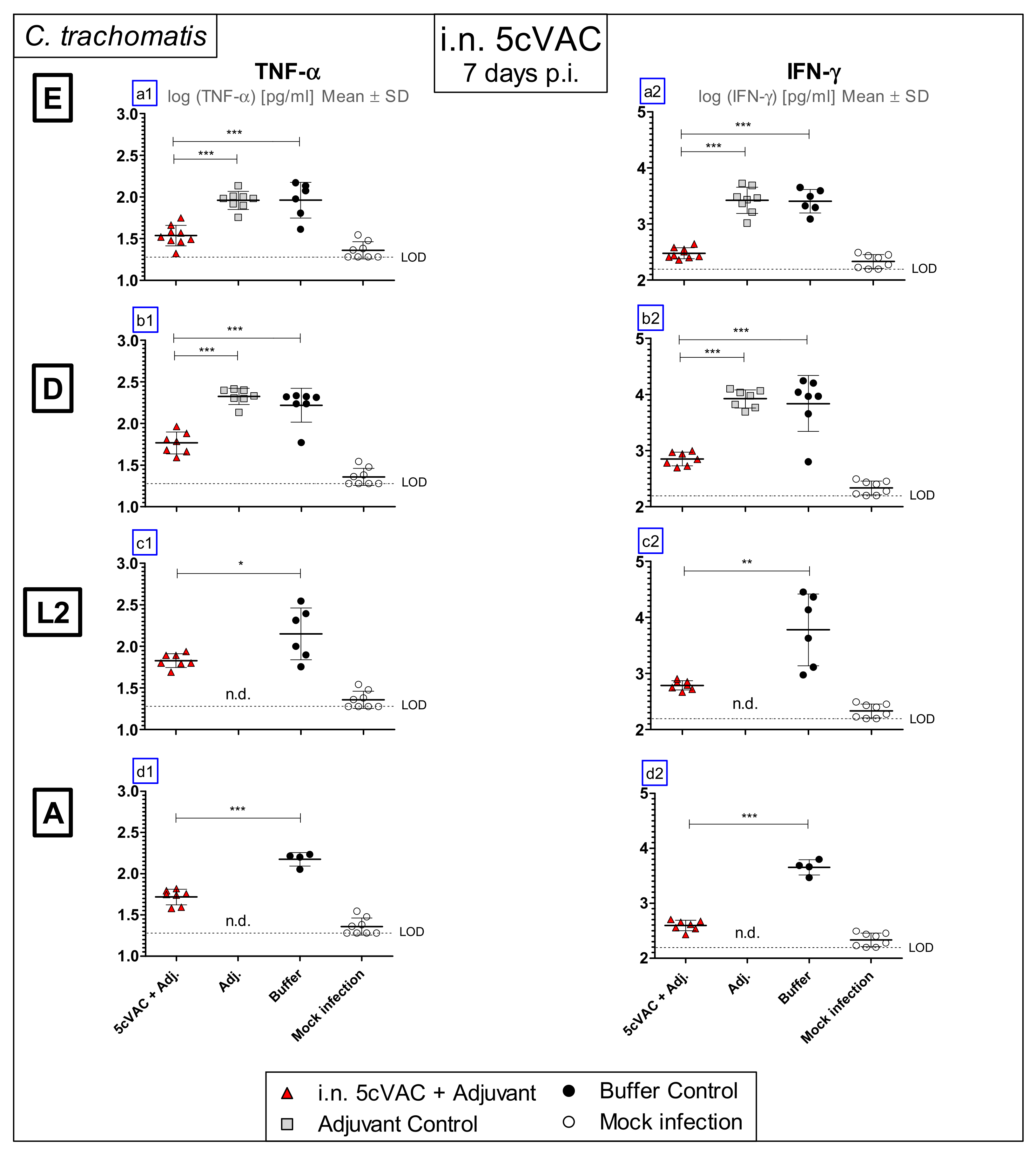
3.3. Intranasal Vaccination with Adjuvanted 5cVAC Leads to Decreased Levels of the Key Cytokines TNF-α and IFN-γ in Lung Homogenate after Challenge Infection with Different Serovars of C. trachomatis
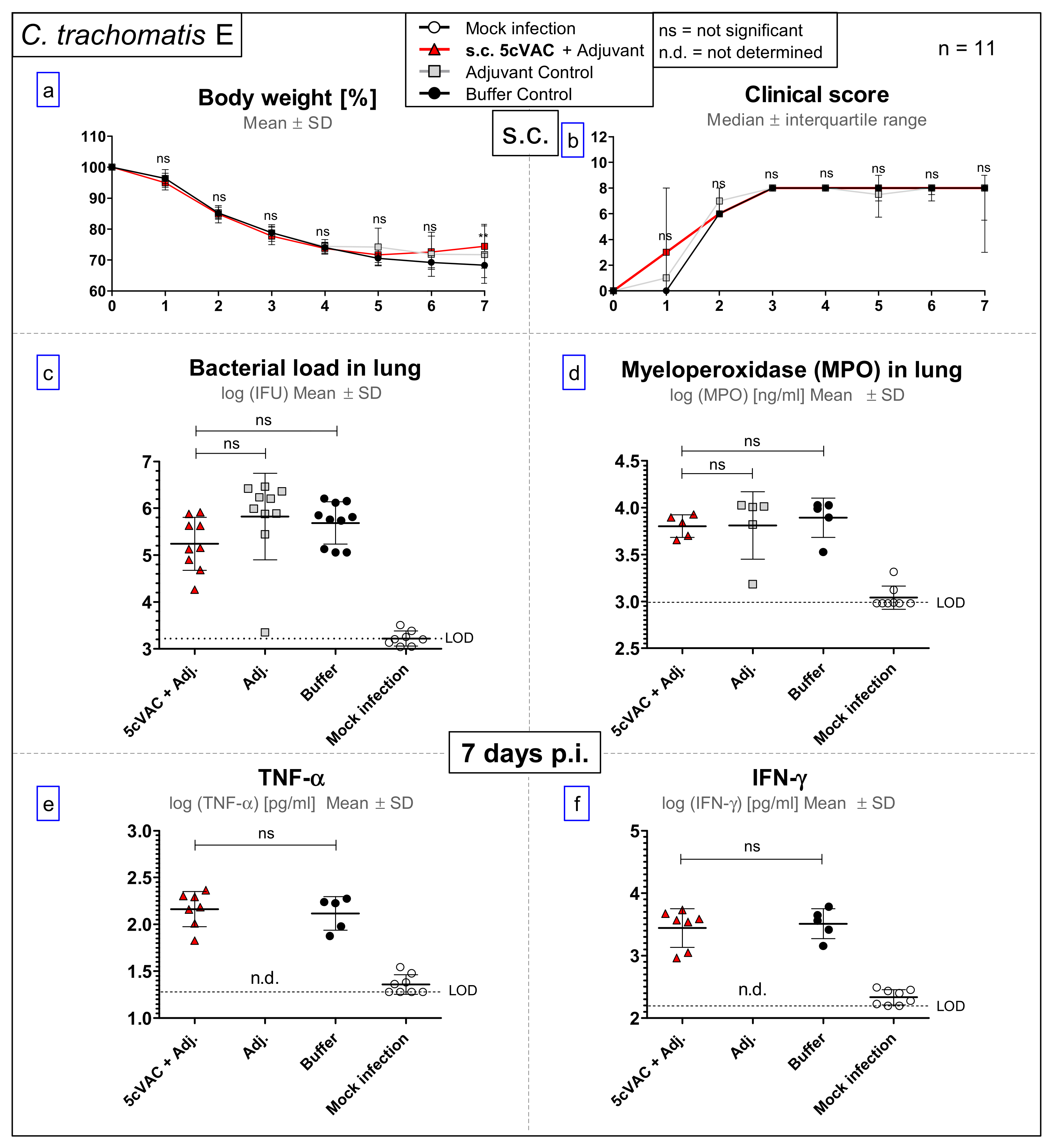
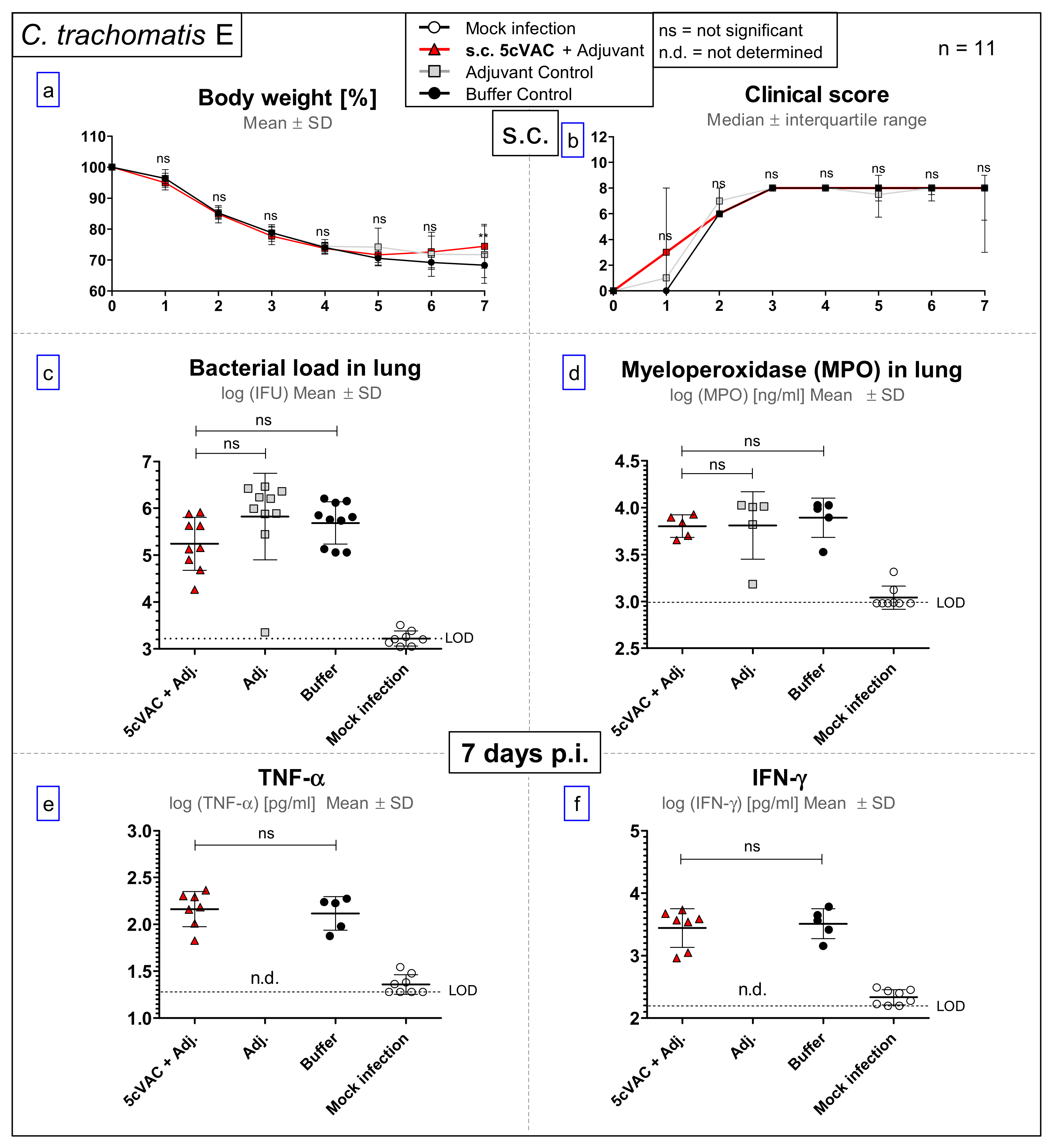
3.4. After Challenge Infection with Serovar E of C. trachomatis, s.c. Vaccination with c-di-AMP-Adjuvanted 5cVAC Does Not Lead to Relevant Protection in Regard to Body Weight, Clinical Score, Bacterial Load, the Levels of the Granulocyte Marker MPO, TNF-α or IFN-γ in Lung Homogenate
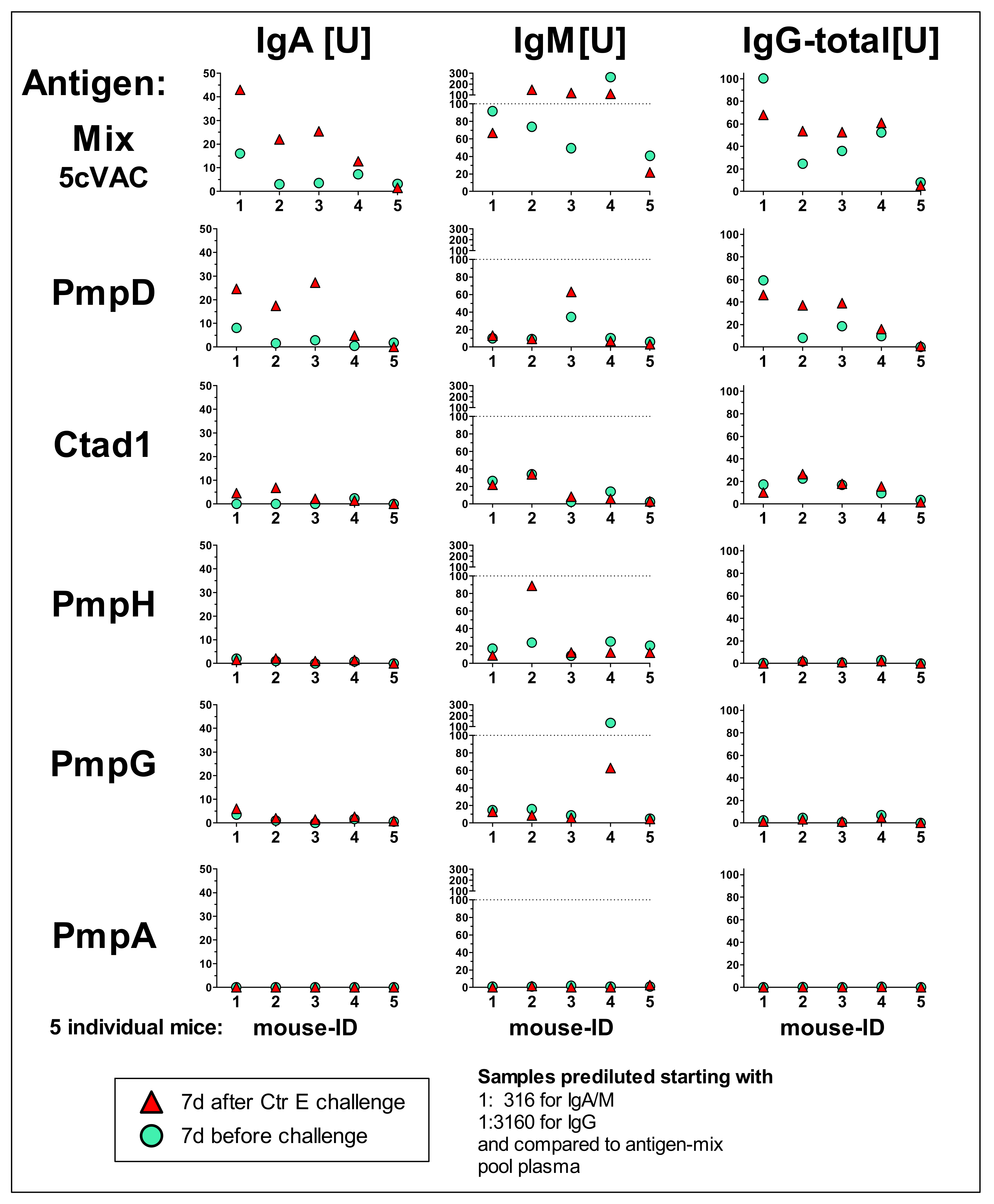
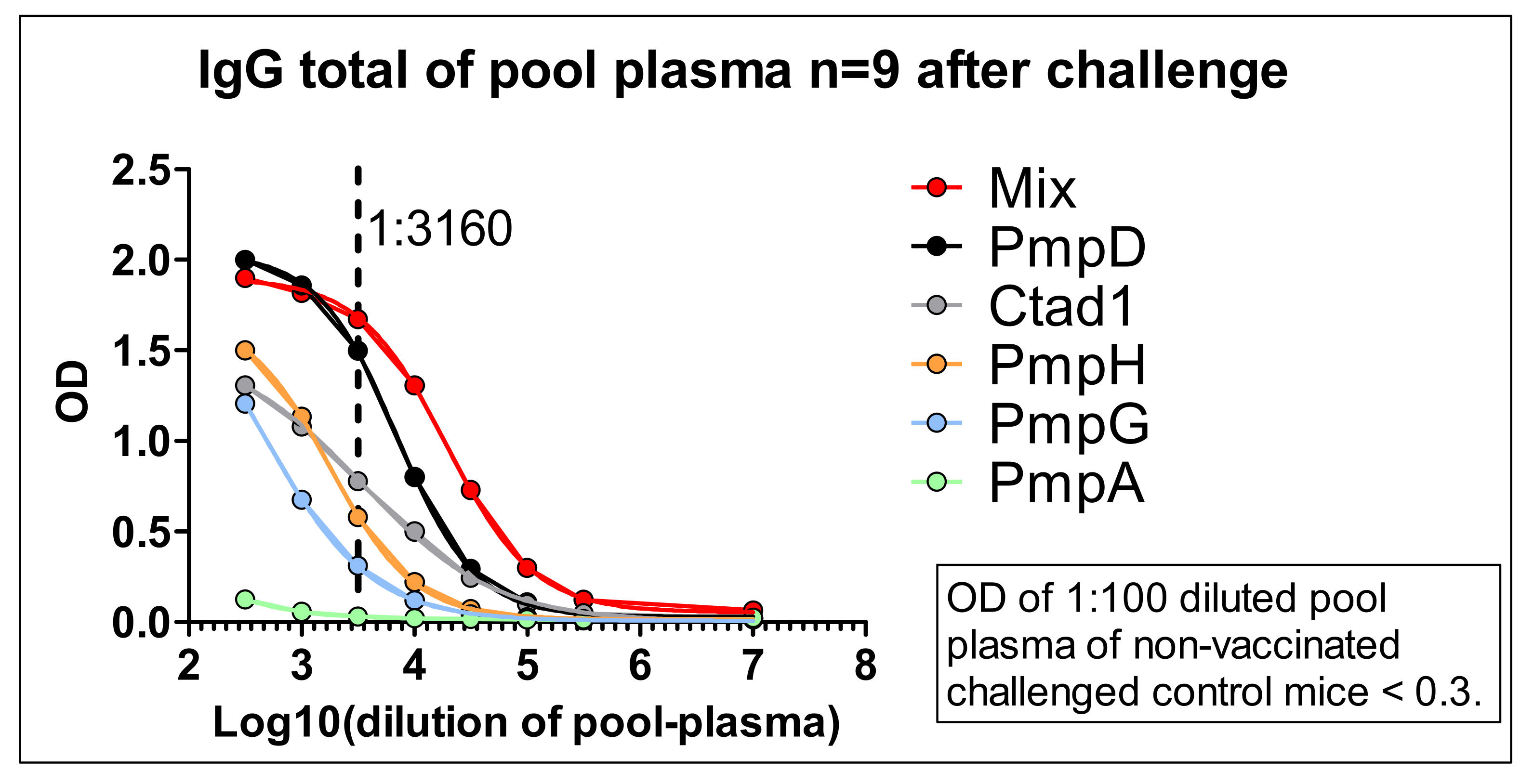
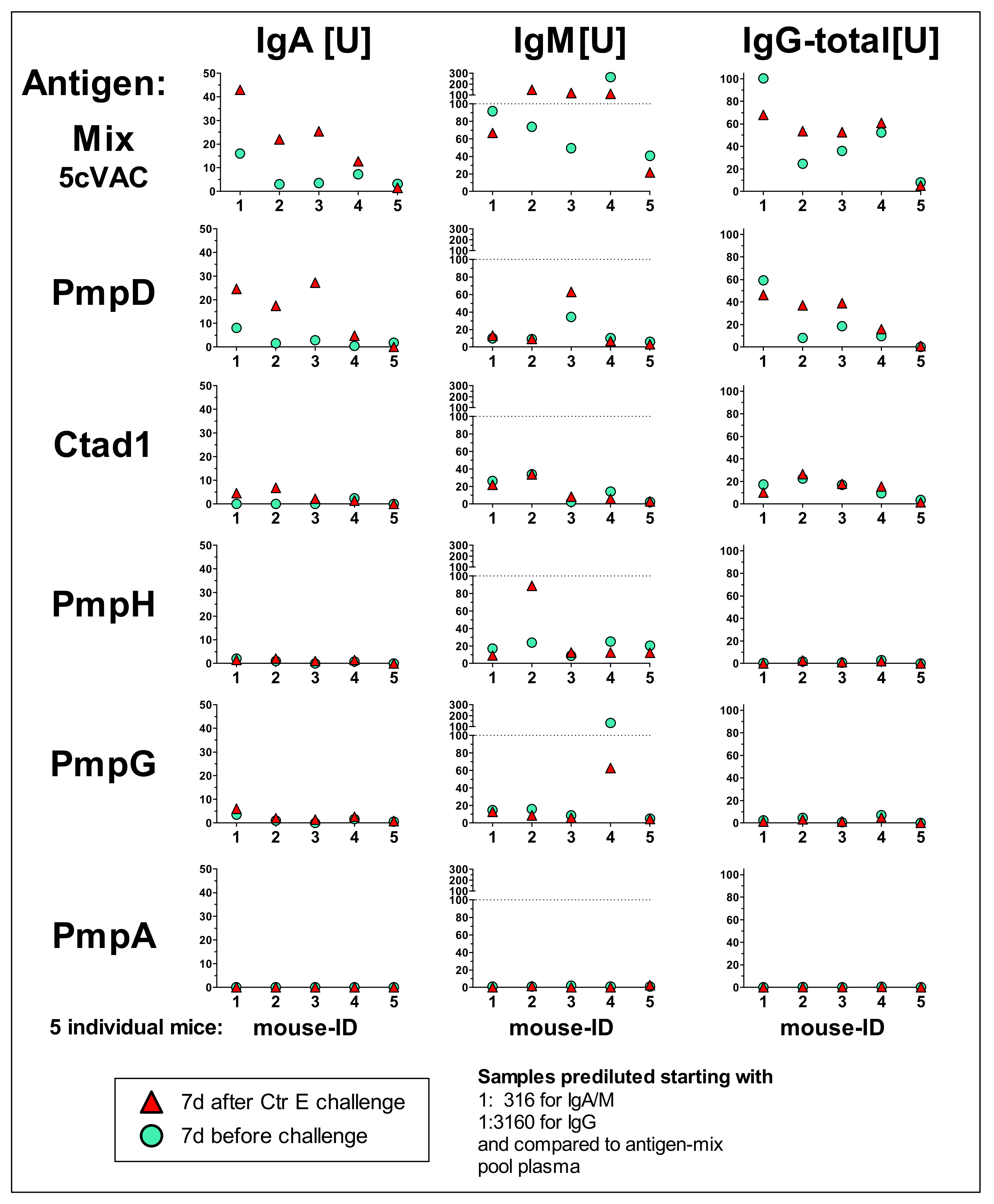
3.5. IgA, IgM, and Total IgG antibody Response Profiles towards the Adjuvanted 5cVAC Antigen-Mix or the Single Vaccine Components in Blood of Five Mice after Vaccination with 5cVAC, Seven Days before or after C.tr. E Challenge, Respectively
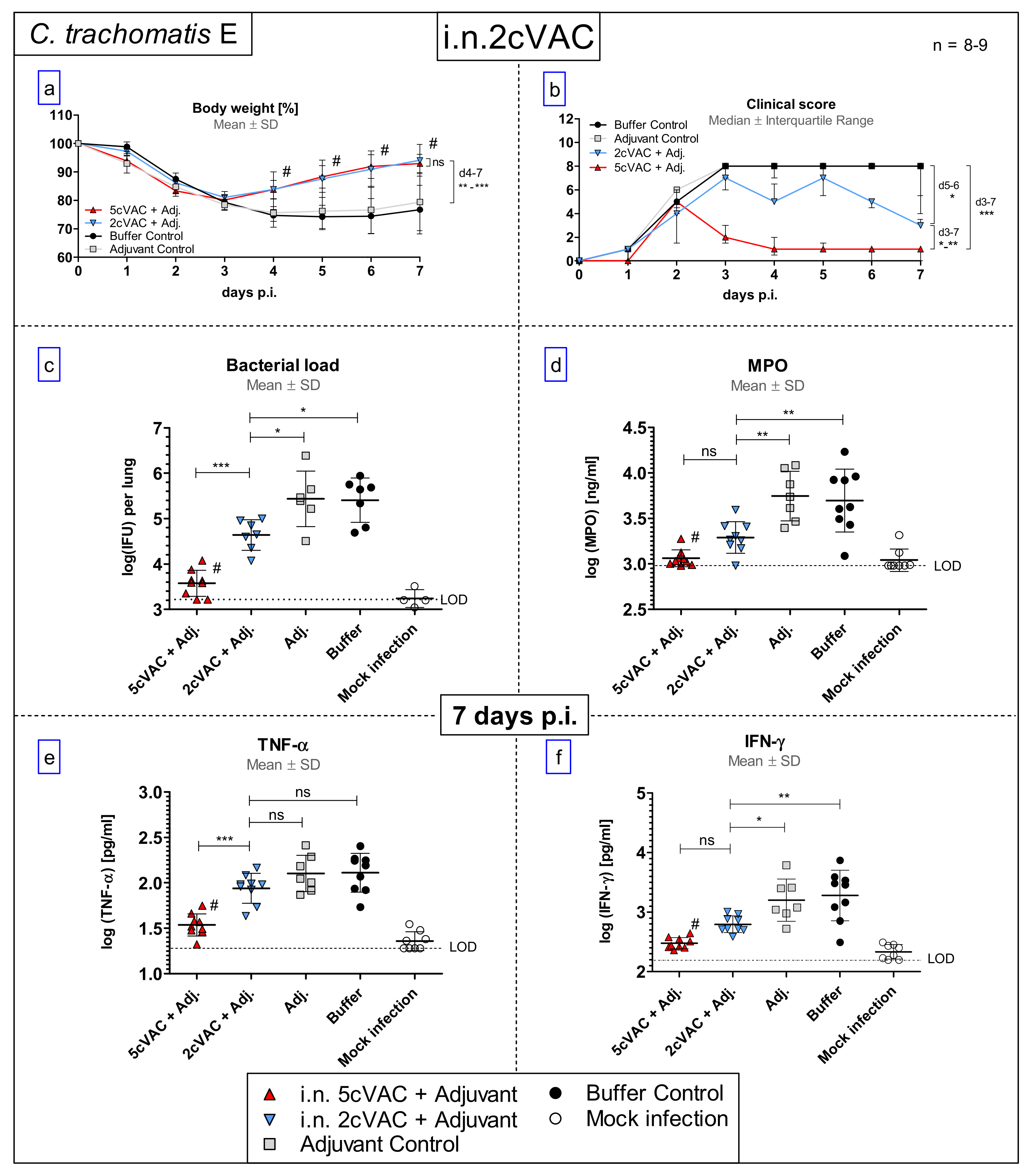
3.6. Intranasal Vaccination with c-di-AMP-Adjuvanted Two-Component 2cVAC Is Still Effective against C.tr. E, but with A Smaller Degree of Protection as Compared to 5cVAC
3.7. Serum-Transfer Demonstrates That Circulating Antibodies Raised by c-di-AMP-Adjuvanted 5cVAC Alone Are Not Protective against C. trachomatis E Lung Infection
3.8. Results of the Long-Term Protection Study
3.8.1. Diminished Weight Loss, Clinical Score, and Higher Bacterial Clearance from the Lung
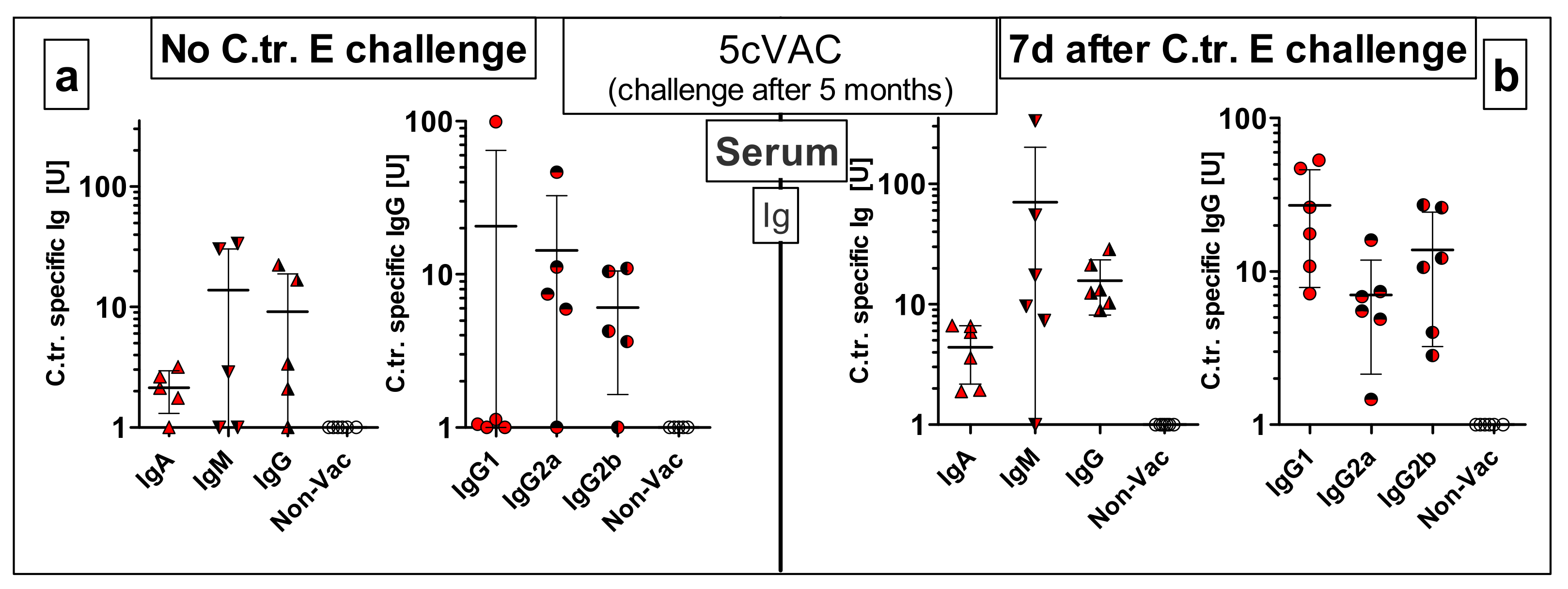
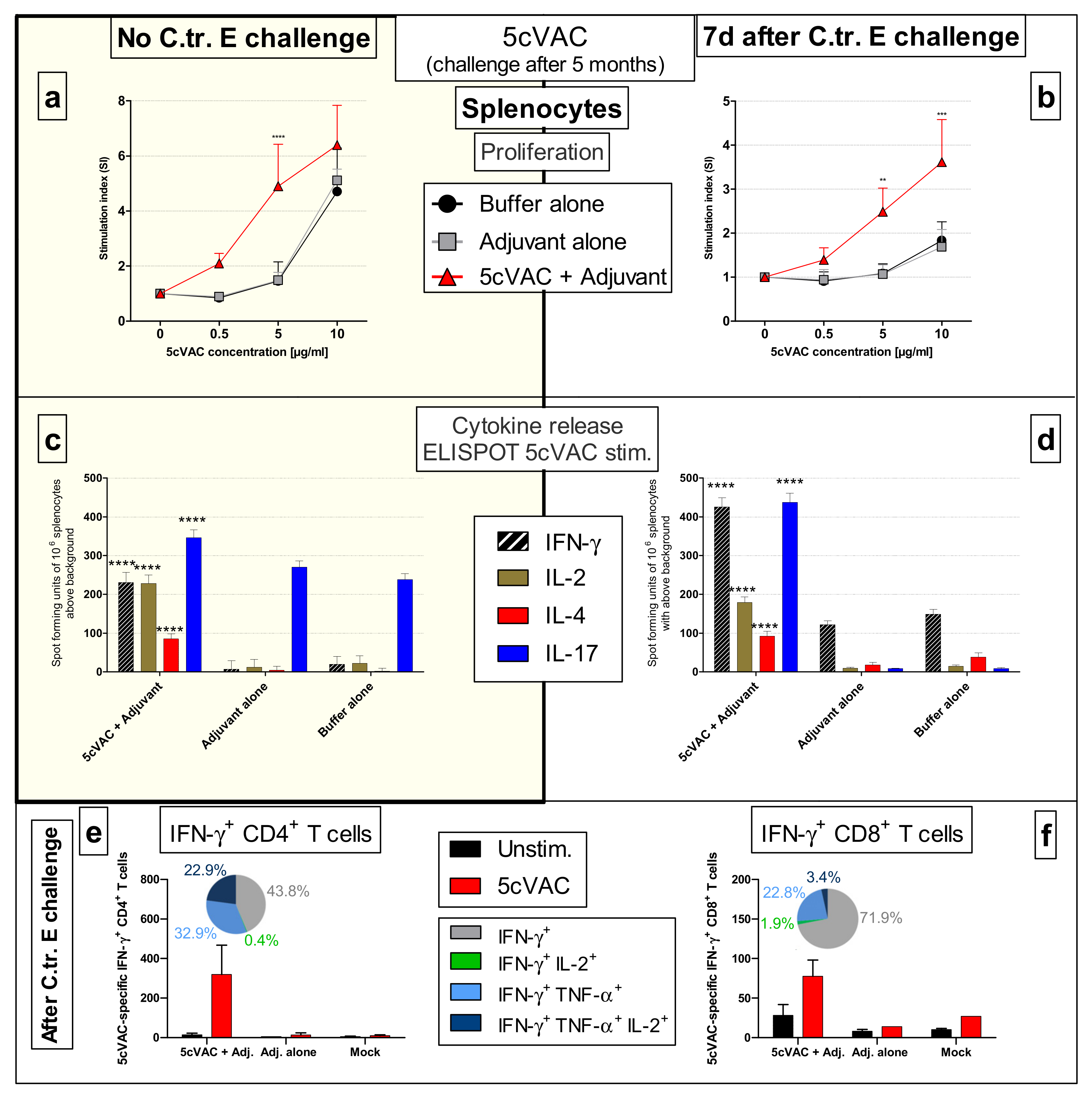
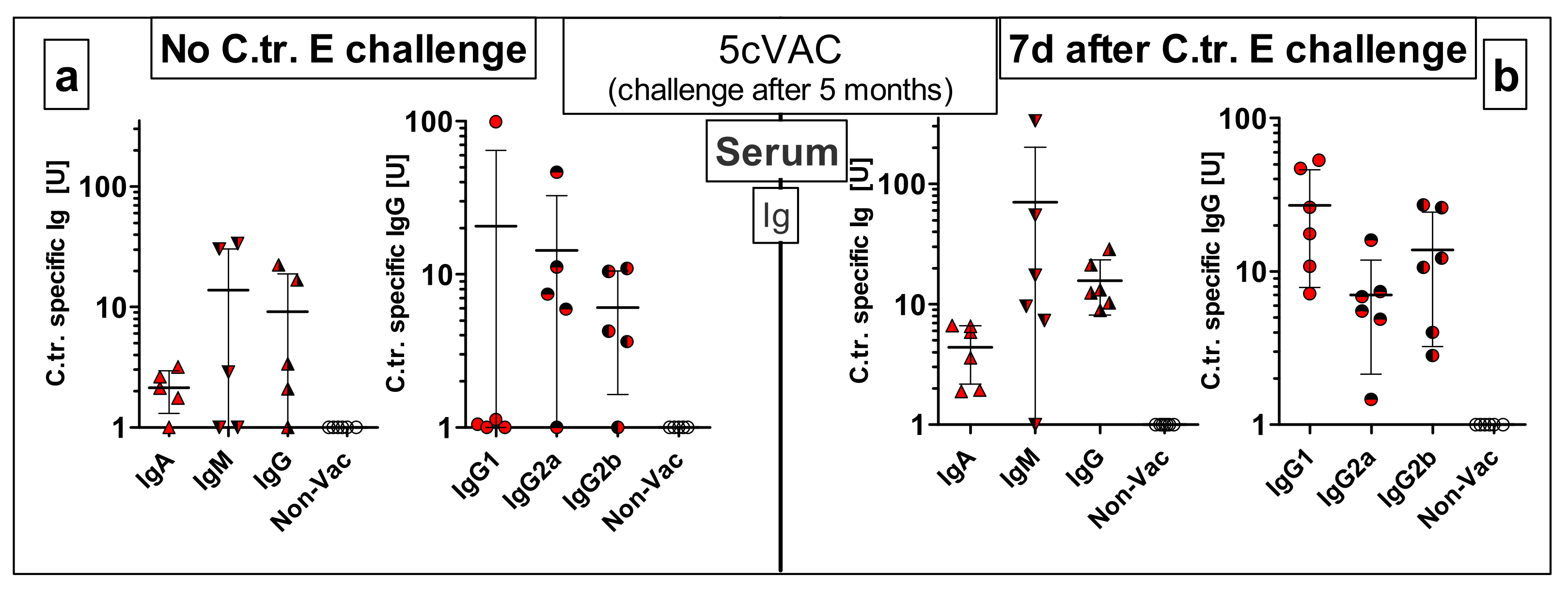
3.8.2. Vaccine-Specific Humoral Immune Responses in Blood and Cellular Responses in the Spleen Five Months after the Last Intranasal Booster Vaccination with c-di-AMP-Adjuvanted 5cVAC
4. Discussion
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bastidas, R.J.; Elwell, C.A.; Engel, J.N.; Valdivia, R.H. Chlamydial intracellular survival strategies. Cold Spring Harb. Perspect. Med. 2013, 3, a010256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smelov, V.; Vrbanac, A.; van Ess, E.F.; Noz, M.P.; Wan, R.; Eklund, C.; Morgan, T.; Shrier, L.A.; Sanders, B.; Dillner, J.; et al. Chlamydia trachomatis Strain Types Have Diversified Regionally and Globally with Evidence for Recombination across Geographic Divides. Front. Microbiol. 2017, 8, 2195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rowley, J.; Hoorn, S. Vander; Korenromp, E.; Low, N.; Unemo, M.; Abu-Raddad, L.J.; Chico, R.M.; Smolak, A.; Newman, L.; Gottlieb, S.; et al. Chlamydia, gonorrhoea, trichomoniasis and syphilis: Global prevalence and incidence estimates, 2016. Bull. World Health Organ. 2019, 97, 548. [Google Scholar] [CrossRef] [PubMed]
- Seedat, J.; Marcus, U.; Fehrmann, S.; Paape, C.; Petschelt, J. Robert Koch-Institut, Epidemiologisches Bulletin 46/2013. 2013. Available online: www.rki.de/epidbull (accessed on 15 May 2021).
- Phillips, S.; Quigley, B.L.; Timms, P. Seventy years of Chlamydia vaccine research - Limitations of the past and directions for the future. Front. Microbiol. 2019, 10, 70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunham, R.C.; Rey-Ladino, J. Immunology of Chlamydia infection: Implications for a Chlamydia trachomatis vaccine. Nat. Rev. Immunol. 2005, 5, 149–161. [Google Scholar] [CrossRef] [PubMed]
- Hoenderboom, B.M.; Van Benthem, B.H.B.; Van Bergen, J.E.A.M.; Dukers-Muijrers, N.H.T.M.; Götz, H.M.; Hoebe, C.J.P.A.; Hogewoning, A.A.; Land, J.A.; Van Der Sande, M.A.B.; Morré, S.A.; et al. Relation between Chlamydia trachomatis infection and pelvic inflammatory disease, ectopic pregnancy and tubal factor infertility in a Dutch cohort of women previously tested for chlamydia in a chlamydia screening trial. Sex. Transm. Infect. 2019, 95, 300–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darville, T. Chlamydia trachomatis infections in neonates and young children. Semin. Pediatr. Infect. Dis. 2005, 16, 235–244. [Google Scholar] [CrossRef]
- Gaston, J.S.H. Immunological basis of chlamydia induced reactive arthritis. Sex. Transm. Infect. 2000, 76, 156–161. [Google Scholar] [CrossRef] [Green Version]
- Schlott, T.; Eiffert, H.; Bohne, W.; Landgrebe, J.; Brunner, E.; Spielbauer, B.; Knight, B. Chlamydia trachomatis modulates expression of tumor suppressor gene caveolin-1 and oncogene C-myc in the transformation zone of non-neoplastic cervical tissue. Gynecol. Oncol. 2005, 98, 409–419. [Google Scholar] [CrossRef]
- Sethi, S.; Roy, A.; Garg, S.; Sree Venkatesan, L.; Bagga, R. Detection of Chlamydia trachomatis infections by polymerase chain reaction in asymptomatic pregnant women with special reference to the utility of the pooling of urine specimens. Indian J. Med. Res. Suppl. 2017, 146, 59–63. [Google Scholar] [CrossRef]
- Faris, R.; Andersen, S.E.; McCullough, A.; Gourronc, F.; Klingelhutz, A.J.; Weber, M.M. Chlamydia trachomatis Serovars Drive Differential Production of Proinflammatory Cytokines and Chemokines Depending on the Type of Cell Infected. Front. Cell. Infect. Microbiol. 2019, 9, 399. [Google Scholar] [CrossRef]
- Lesiak-Markowicz, I.; Schötta, A.M.; Stockinger, H.; Stanek, G.; Markowicz, M. Chlamydia trachomatis serovars in urogenital and ocular samples collected 2014–2017 from Austrian patients. Sci. Rep. 2019, 9, 18327. [Google Scholar] [CrossRef]
- World Health Organization Status of elimination of trachoma as a public health problem. Available online: https://apps.who.int/neglected_diseases/ntddata/trachoma/trachoma.html (accessed on 15 January 2021).
- Brunham, R.C.; Rappuoli, R. Chlamydia trachomatis control requires a vaccine. Vaccine 2013, 31, 1892–1897. [Google Scholar] [CrossRef] [Green Version]
- De La Maza, L.M.; Zhong, G.; Brunham, R.C. Update on Chlamydia trachomatis vaccinology. Clin. Vaccine Immunol. 2017, 24, e00543-16. [Google Scholar] [CrossRef] [Green Version]
- Gottlieb, S.L.; Johnston, C. Future prospects for new vaccines against sexually transmitted infections. Curr. Opin. Infect. Dis. 2017, 30, 77–86. [Google Scholar] [CrossRef]
- Poston, T.B.; Gottlieb, S.L.; Darville, T. Status of vaccine research and development of vaccines for Chlamydia trachomatis infection. Vaccine 2019, 37, 7289–7294. [Google Scholar] [CrossRef]
- De La Maza, L.M.; Pal, S.; Olsen, A.E.; Follmann, F. Chlamydia Vaccines. In Chlamydia Biology—From Genome to Disease; Tan M, J.H., Hegemann, S.C., Eds.; Caister Academic Press: Norfolk, UK, 2020. [Google Scholar]
- Qu, Y.; Frazer, L.C.; O’Connell, C.M.; Tarantal, A.F.; Andrews, C.W.; O’Connor, S.L.; Russell, A.N.; Sullivan, J.E.; Poston, T.B.; Vallejo, A.N.; et al. Comparable Genital Tract Infection, Pathology, and Immunity in Rhesus Macaques Inoculated with Wild-Type or Plasmid-Deficient Chlamydia trachomatis Serovar D. Infect. Immun. 2015, 83, 4056–4067. [Google Scholar] [CrossRef] [Green Version]
- Stary, G.; Olive, A.; Radovic-Moreno, A.F.; Gondek, D.; Alvarez, D.; Basto, P.A.; Perro, M.; Vrbanac, V.D.; Tager, A.M.; Shi, J.; et al. A mucosal vaccine against Chlamydia trachomatis generates two waves of protective memory T cells. Science 2015, 348, aaa8205. [Google Scholar] [CrossRef] [Green Version]
- Grimwood, J.; Stephens, R.S. Computational analysis of the polymorphic membrane protein superfamily of Chlamydia trachomatis and Chlamydia pneumoniae. Microb. Comp. Genom. 1999, 4, 187–201. [Google Scholar] [CrossRef]
- Luczak, S.E.T.; Smits, S.H.J.; Decker, C.; Nagel-Steger, L.; Schmitt, L.; Hegemann, J.H. The Chlamydia Pneumoniae Adhesin Pmp21 forms oligomers with adhesive properties. J. Biol. Chem. 2016, 291, 22806–22818. [Google Scholar] [CrossRef] [Green Version]
- Becker, E.; Hegemann, J.H. All subtypes of the Pmp adhesin family are implicated in chlamydial virulence and show species-specific function. Microbiologyopen 2014, 3, 544–556. [Google Scholar] [CrossRef] [PubMed]
- Kari, L.; Southern, T.R.; Downey, C.J.; Watkins, H.S.; Randall, L.B.; Taylor, L.D.; Sturdevant, G.L.; Whitmire, W.M.; Caldwell, H.D. Chlamydia trachomatis polymorphic membrane protein D is a virulence factor involved in early host-cell interactions. Infect. Immun. 2014, 82, 2756–2762. [Google Scholar] [CrossRef] [Green Version]
- Mölleken, K.; Becker, E.; Hegemann, J.H. The Chlamydia pneumoniae Invasin Protein Pmp21 Recruits the EGF Receptor for Host Cell Entry. PLoS Pathog. 2013, 9, e1003325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crane, D.D.; Carlson, J.H.; Fischer, E.R.; Bavoil, P.; Hsia, R.C.; Tan, C.; Kuo, C.C.; Caldwell, H.D. Chlamydia tracomatis polymorphic membrane protein D is a species-common pan-neutralizing antigen. Proc. Natl. Acad. Sci. USA 2006, 103, 1894–1899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.; Karunakaran, K.P.; Jiang, X.; Shen, C.; Andersen, P.; Brunham, R.C. Chlamydia muridarum T cell antigens and adjuvants that induce protective immunity in mice. Infect. Immun. 2012, 80, 1510–1518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karunakaran, K.P.; Yu, H.; Jiang, X.; Chan, Q.; Moon, K.M.; Foster, L.J.; Brunham, R.C. Outer membrane proteins preferentially load MHC class II peptides: Implications for a Chlamydia trachomatis T cell vaccine. Vaccine 2015, 33, 2159–2166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pal, S.; Favaroni, A.; Tifrea, D.F.; Hanisch, P.T.; Luczak, S.E.T.; Hegemann, J.H.; de la Maza, L.M. Comparison of the nine polymorphic membrane proteins of Chlamydia trachomatis for their ability to induce protective immune responses in mice against a C. muridarum challenge. Vaccine 2017, 35, 2543–2549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inic-Kanada, A.; Stojanovic, M.; Schlacher, S.; Stein, E.; Belij-Rammerstorfer, S.; Marinkovic, E.; Lukic, I.; Montanaro, J.; Schuerer, N.; Bintner, N.; et al. Delivery of a chlamydial adhesin N-PmpC subunit vaccine to the ocular mucosa using particulate carriers. PLoS ONE 2015, 10, e144380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, T.; Becker, E.; Stallmann, S.; Waldhuber, A.; Römmler-Dreher, F.; Albrecht, S.; Mohr, F.; Hegemann, J.H.; Miethke, T. Vaccination with the polymorphic membrane protein A reduces Chlamydia muridarum induced genital tract pathology. Vaccine 2017, 35, 2801–2810. [Google Scholar] [CrossRef] [PubMed]
- Stallmann, S.; Hegemann, J.H. The Chlamydia trachomatis Ctad1 invasin exploits the human integrin β1 receptor for host cell entry. Cell. Microbiol. 2016, 18, 761–775. [Google Scholar] [CrossRef] [Green Version]
- Uddowla, S.; Freytag, L.C.; Clements, J.D. Effect of adjuvants and route of immunizations on the immune response to recombinant plague antigens. Vaccine 2007, 25, 7984–7993. [Google Scholar] [CrossRef] [Green Version]
- Durando, P.; Iudici, R.; Alicino, C.; Alberti, M.; de Florentis, D.; Ansaldi, F.; Icardi, G. Adjuvants and alternative routes of administration towards the development of the ideal influenza vaccine. Hum. Vaccin. 2011, 7, 29–40. [Google Scholar] [CrossRef] [Green Version]
- Ebensen, T.; Debarry, J.; Pedersen, G.K.; Blazejewska, P.; Weissmann, S.; Schulze, K.; McCullough, K.C.; Cox, R.J.; Guzmán, C.A. Mucosal administration of cycle-di-nucleotide-adjuvanted virosomes efficiently induces protection against influenza H5N1 in mice. Front. Immunol. 2017, 8, 1223. [Google Scholar] [CrossRef]
- Landi, A.; Law, J.; Hockman, D.; Logan, M.; Crawford, K.; Chen, C.; Kundu, J.; Ebensen, T.; Guzman, C.A.; Deschatelets, L.; et al. Superior immunogenicity of HCV envelope glycoproteins when adjuvanted with cyclic-di-AMP, a STING activator or archaeosomes. Vaccine 2017, 35, 6949–6956. [Google Scholar] [CrossRef]
- Lirussi, D.; Ebensen, T.; Schulze, K.; Trittel, S.; Duran, V.; Liebich, I.; Kalinke, U.; Guzmán, C.A. Type I IFN and not TNF, is Essential for Cyclic Di-nucleotide-elicited CTL by a Cytosolic Cross-presentation Pathway. EBioMedicine 2017, 22, 100–111. [Google Scholar] [CrossRef] [Green Version]
- Sanchez Alberti, A.; Bivona, A.E.; Cerny, N.; Schulze, K.; Weißmann, S.; Ebensen, T.; Morales, C.; Padilla, A.M.; Cazorla, S.I.; Tarleton, R.L.; et al. Engineered trivalent immunogen adjuvanted with a sting agonist confers protection against trypanosoma cruzi infection. npj Vaccines 2017, 2, 9. [Google Scholar] [CrossRef]
- Dutow, P.; Wask, L.; Bothe, M.; Fehlhaber, B.; Laudeley, R.; Rheinheimer, C.; Yang, Z.; Zhong, G.; Glage, S.; Klos, A. An optimized, fast-to-perform mouse lung infection model with the human pathogen Chlamydia trachomatis for in vivo screening of antibiotics, vaccine candidates and modified host-pathogen interactions. Pathog. Dis. 2016, 74. [Google Scholar] [CrossRef] [Green Version]
- Rother, M.; Gonzalez, E.; Teixeira da Costa, A.R.; Wask, L.; Gravenstein, I.; Pardo, M.; Pietzke, M.; Gurumurthy, R.K.; Angermann, J.; Laudeley, R.; et al. Combined Human Genome-wide RNAi and Metabolite Analyses Identify IMPDH as a Host-Directed Target against Chlamydia Infection. Cell Host Microbe 2018, 23, 661–671. [Google Scholar] [CrossRef] [Green Version]
- Sommer, K.; Njau, F.; Wittkop, U.; Thalmann, J.; Bartling, G.; Wagner, A.; Klos, A. Identification of high- and low-virulent strains of Chlamydia pneumoniae by their characterization in a mouse pneumonia model. FEMS Immunol. Med. Microbiol. 2009, 55, 206–214. [Google Scholar] [CrossRef] [Green Version]
- Mölleken, K.; Schmidt, E.; Hegemann, J.H. Members of the Pmp protein family of Chlamydia pneumoniae mediate adhesion to human cells via short repetitive peptide motifs. Mol. Microbiol. 2010, 78, 1004–1017. [Google Scholar] [CrossRef] [Green Version]
- Bethesda (MD): National Library of Medicine (US), National Center for Biotechnology Information. Available online: https://www.ncbi.nlm.nih.gov/ (accessed on 11 March 2021).
- Sambrook, J.; Maniatis, T. Molecular Cloning: A Laboratory Manual; Cold Spring Harbor Laboratory Press: New York, NY, USA, 1989. [Google Scholar]
- Gherardi, M.M.; Pérez-Jiménez, E.; Nájera, J.L.; Esteban, M. Induction of HIV Immunity in the Genital Tract After Intranasal Delivery of a MVA Vector: Enhanced Immunogenicity After DNA Prime-Modified Vaccinia Virus Ankara Boost Immunization Schedule. J. Immunol. 2004, 172, 6209–6220. [Google Scholar] [CrossRef] [PubMed]
- Plante, M.; Jerse, A.; Hamel, J.; Couture, F.; Rioux, C.R.; Brodeur, B.R.; Martin, D. Intranasal Immunization with Gonococcal Outer Membrane Preparations Reduces the Duration of Vaginal Colonization of Mice by Neisseria gonorrhoeae. J. Infect. Dis. 2000, 182, 848–855. [Google Scholar] [CrossRef] [PubMed]
- Bode, J.; Dutow, P.; Sommer, K.; Janik, K.; Glage, S.; Tümmler, B.; Munder, A.; Laudeley, R.; Sachse, K.W.; Klos, A. A New Role of the Complement System: C3 Provides Protection in a Mouse Model of Lung Infection with Intracellular Chlamydia psittaci. PLoS ONE 2012, 7, e50327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohn, M.; Lanfermann, C.; Laudeley, R.; Glage, S.; Rheinheimer, C.; Klos, A. Complement and Chlamydia psittaci: Non-myeloid derived C3 predominantly induces protective adaptive immune responses in mouse lung infection. Front. Immunol. 2021, 12, 485. [Google Scholar] [CrossRef]
- Schulze, K.; Ebensen, T.; Chandrudu, S.; Skwarczynski, M.; Toth, I.; Olive, C.; Guzman, C.A. Bivalent mucosal peptide vaccines administered using the LCP carrier system stimulate protective immune responses against Streptococcus pyogenes infection. Nanomed. Nanotechnol. Biol. Med. 2017, 13, 2463–2474. [Google Scholar] [CrossRef]
- Tan, C.; Hsia, R. ching; Shou, H.; Carrasco, J.A.; Rank, R.G.; Bavoil, P.M. Variable expression of surface-exposed polymorphic membrane proteins in in vitro-grown Chlamydia trachomatis. Cell. Microbiol. 2010, 12, 174–187. [Google Scholar] [CrossRef] [Green Version]
- Pal, S.; Theodor, I.; Peterson, E.M.; De la Maza, L.M. Immunization with the Chlamydia trachomatis mouse pneumonitis major outer membrane protein can elicit a protective immune response against a genital challenge. Infect. Immun. 2001, 69, 6240–6247. [Google Scholar] [CrossRef] [Green Version]
- Pal, S.; Peterson, E.M.; De La Maza, L.M. Vaccination with the chlamydia trachomatis major outer membrane protein can elicit an immune response as protective as that resulting from inoculation with live bacteria. Infect. Immun. 2005, 73, 8153–8160. [Google Scholar] [CrossRef] [Green Version]
- Pal, S.; Tatarenkova, O.V.; de la Maza, L.M. A vaccine formulated with the major outer membrane protein can protect C3H/HeN, a highly susceptible strain of mice, from a Chlamydia muridarum genital challenge. Immunology 2015, 146, 432–443. [Google Scholar] [CrossRef] [Green Version]
- Wern, J.E.; Sorensen, M.R.; Olsen, A.W.; Andersen, P.; Follmann, F. Simultaneous subcutaneous and intranasal administration of a CAF01-adjuvanted Chlamydia vaccine elicits elevated IgA and protective Th1/Th17 responses in the genital tract. Front. Immunol. 2017, 8, 569. [Google Scholar] [CrossRef] [Green Version]
- Abraham, S.; Juel, H.B.; Bang, P.; Cheeseman, H.M.; Dohn, R.B.; Cole, T.; Kristiansen, M.P.; Korsholm, K.S.; Lewis, D.; Olsen, A.W.; et al. Safety and immunogenicity of the chlamydia vaccine candidate CTH522 adjuvanted with CAF01 liposomes or aluminium hydroxide: A first-in-human, randomised, double-blind, placebo-controlled, phase 1 trial. Lancet Infect. Dis. 2019, 19, 1091–1100. [Google Scholar] [CrossRef]
- Joseph, S.J.; Didelot, X.; Rothschild, J.; De Vries, H.J.C.; Morré, S.A.; Read, T.D.; Dean, D. Population genomics of chlamydia trachomatis: Insights on drift, selection, recombination, and population structure. Mol. Biol. Evol. 2012, 29, 3933–3946. [Google Scholar] [CrossRef] [Green Version]
- Borges, V.; Cordeiro, D.; Salas, A.I.; Lodhia, Z.; Correia, C.; Isidro, J.; Fernandes, C.; Rodrigues, A.M.; Azevedo, J.; Alves, J.; et al. Chlamydia trachomatis: When the virulence-associated genome backbone imports a prevalence-associated major antigen signature. Microb. Genom. 2019, 5, e000313. [Google Scholar] [CrossRef] [Green Version]
- Ebensen, T.; Libanova, R.; Schulze, K.; Yevsa, T.; Morr, M.; Guzmán, C.A. Bis-(3’,5’)-cyclic dimeric adenosine monophosphate: Strong Th1/Th2/Th17 promoting mucosal adjuvant. Vaccine 2011, 29, 5210–5220. [Google Scholar] [CrossRef]
- Gill, N.; Wlodarska, M.; Finlay, B.B. The future of mucosal immunology: Studying an integrated system-wide organ. Nat. Immunol. 2010, 11, 558–560. [Google Scholar] [CrossRef]
- Käser, T.; Pasternak, J.A.; Delgado-Ortega, M.; Hamonic, G.; Lai, K.; Erickson, J.; Walker, S.; Dillon, J.R.; Gerdts, V.; Meurens, F. Chlamydia suis and Chlamydia trachomatis induce multifunctional CD4 T cells in pigs. Vaccine 2017, 35, 91–100. [Google Scholar] [CrossRef]
- Ebensen, T.; Schulze, K.; Riese, P.; Link, C.; Morr, M.; Guzmán, C.A. The bacterial second messenger cyclic diGMP exhibits potent adjuvant properties. Vaccine 2007, 25, 1464–1469. [Google Scholar] [CrossRef]
- Crooke, S.N.; Ovsyannikova, I.G.; Poland, G.A.; Kennedy, R.B. Immunosenescence and human vaccine immune responses. Immun. Ageing 2019, 16, 25. [Google Scholar] [CrossRef] [Green Version]






| C.tr. E/DK20 Pf Antigen | C.tr. E Cp vs. C.tr. D Cp | C.tr. E Pf vs. C.tr. D Pf | C.tr. E Cp vs. C.tr. A Cp | C.tr. E Pf vs. C.tr. A Pf | C.tr. E Cp vs. C.tr. L2 Cp | C.tr. E Pf vs. C.tr. L2 Pf | C.tr. E Cp vs. C.mu. Cp | C.tr. E Pf vs. C.mu. Pf | |
|---|---|---|---|---|---|---|---|---|---|
| PmpA | 53aa-665aa | 99.8% | 99.7% | 99.8% | 99.8% | 100.0% | 100.0% | 76.8% | 81.2% |
| PmpD | 34aa-619aa | 99.1% | 99.5% | 99.0% | 99.5% | 99.4% | 99.7% | 71.7% | 68.6% |
| PmpG | 29aa-673aa | 99.4% | 99.5% | 99.9% | 99.7% | 97.5% | 97.7% | 72.8% | 67.2% |
| PmpH | 26aa-690aa | 99.7% | 99.6% | 94.7% | 92.5% | 94.7% | 92.9% | 75.4% | 69.1% |
| Ctad1 | 1aa-433aa | 100.0% | 100.0% | 98.6% | 98.6% | 98.4% | 98.4% | 82.0% | 82.0% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lanfermann, C.; Wintgens, S.; Ebensen, T.; Kohn, M.; Laudeley, R.; Schulze, K.; Rheinheimer, C.; Hegemann, J.H.; Guzmán, C.A.; Klos, A. Prophylactic Multi-Subunit Vaccine against Chlamydia trachomatis: In Vivo Evaluation in Mice. Vaccines 2021, 9, 609. https://doi.org/10.3390/vaccines9060609
Lanfermann C, Wintgens S, Ebensen T, Kohn M, Laudeley R, Schulze K, Rheinheimer C, Hegemann JH, Guzmán CA, Klos A. Prophylactic Multi-Subunit Vaccine against Chlamydia trachomatis: In Vivo Evaluation in Mice. Vaccines. 2021; 9(6):609. https://doi.org/10.3390/vaccines9060609
Chicago/Turabian StyleLanfermann, Christian, Sebastian Wintgens, Thomas Ebensen, Martin Kohn, Robert Laudeley, Kai Schulze, Claudia Rheinheimer, Johannes H. Hegemann, Carlos Alberto Guzmán, and Andreas Klos. 2021. "Prophylactic Multi-Subunit Vaccine against Chlamydia trachomatis: In Vivo Evaluation in Mice" Vaccines 9, no. 6: 609. https://doi.org/10.3390/vaccines9060609