
The Interplay between Cannabinoid Receptors and Microglia in the Pathophysiology of Alzheimer’s Disease

 , ,
, , 
Abstract
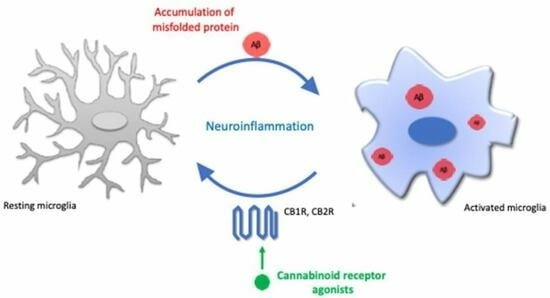
:
1. Introduction
1.1. A Glimpse into the Architecture of the Endocannabinoid System
1.2. Focus on Cannabinoid Receptors CB1R and CB2R
2. Microglia: Functions and Phenotypes
3. Alzheimer’s Disease and Microglia
4. Endocannabinoid Signaling and Microglia Activation
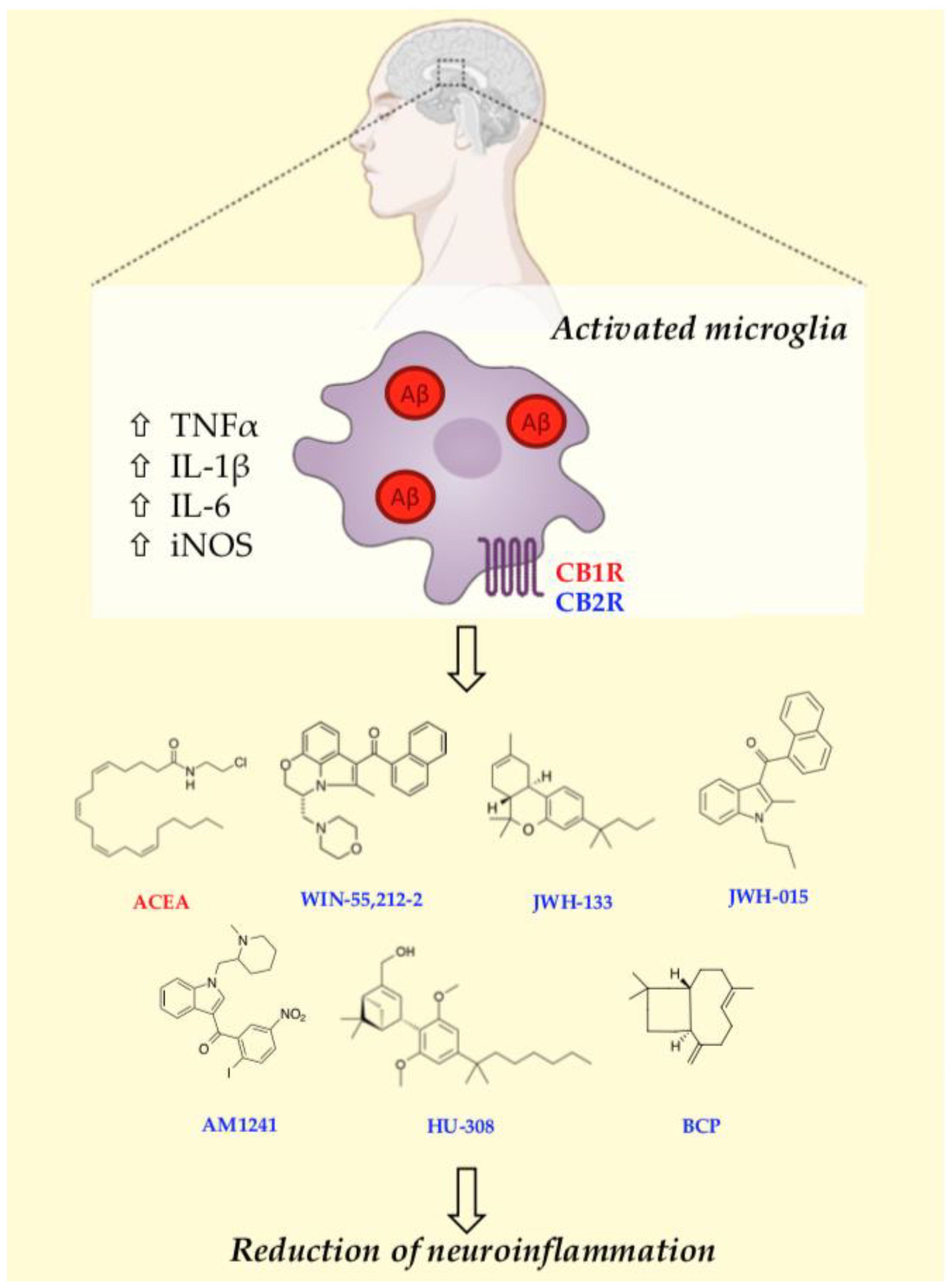
5. Cannabinoid Receptors-Microglia Communication: Therapeutic Implications for Alzheimer’s Disease
5.1. CB1R-Mediated Effects on Microglia Activation In Vitro and In Vivo Models

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | CB1R Modulator | Outcome | Reference |
|---|---|---|---|
| Primary microglia cells isolated from P0-P2 Wistar rats | AM251 (antagonist) | ↓ Arg1 | [77] |
| Murine BV2 microglial cells | SR141716A (antagonist) | ↑ TNF-α, IL-1β, IL-6 ↓ IL-10, MCP-1, CX3CL1 | [82] |
| Co-culture of murine BV2 microglial cells and CD4+ T cells | SR141716A (antagonist) | ↑ IL-17, IFNγ ↓ IL-10, IL-4 | [82] |
| APP/PS1 mice Primary cultures of cortical neurons isolated from OF1 mice | Arachidonyl-2-chloroethylamide (ACEA) (agonist) | ↓ Cognitive impairment ↓ Cytotoxic effect of Aβ42 olygomers | [53] |
5.2. CB2R-Mediated Effects on Microglia Activation In Vitro and In Vivo Models
5.2.1. WIN55,212-2
5.2.2. JWH133 and JWH015
5.2.3. AM1241
5.2.4. HU-308
5.2.5. β-Caryophyllene
5.2.6. RO6866945
| Model | CB2R Modulator | Outcome | Reference |
|---|---|---|---|
| Rat (Aβ25-35inj) | WIN55,212-2 | ↓ Microglia activation | [88] |
| Tg2576 mice | WIN 55,212-2 and JWH-133 | ↓ Microglial cell density was decreased by continuous JWH-133 oral treatment. ↓ COX-2 and TNF-α ↓ Aβ cortical levels | [85] |
| APP/PS1 mice | JWH133 | ↓ Microglial activity ↓ IL-1β, IL-6, TNF-α, and IFNγ secretion | [89] |
| JWH015 | ↓ Expression of M1 microglia biomarkers (IL-6, iNOS and TNF-α) ↑ Expression of M2 microglia biomarkers Ym1/2 | [91] | |
| Murine N9 microglial cells APP/PS1 mice | AM1241 | ↑ Arg1/IL-10/BDNF/GDNF↓ iNOS/IL-1β/IL-6/TNFα ↓ Amyloid plaque deposition ↑ Aβ phagocytosis | [75] [92] |
| R6/2 mice | HU-308 | ↓ Proliferation of microglia and cytokine expression ↑ Neuroprotection | [93] |
| Primary microglia cells isolated from C57BL/6 mice | β-Caryophyllene | ↓ Expression of M1 microglia biomarkers (IL-1β, iNOS, TNF-α, NO, ROS) ↑ Expression of M2 microglia biomarkers (IL-10, Arg-1, and urea, GSH) | [95,97] |
| Microglial cells isolated from 5xFAD/CB2EGFP/f/fmice | RO6866945 | ↓ Iba1+, phagocytosis activity ↑ cAMP, CREB and p38MAPK | [97] |
5.3. Potential Influence of Microglial CB1R-CB2R Heteromer-Mediated Regulation in Alzheimer’s Disease
5.4. Current Hot Topics in CBR-Oriented Drug Discovery: Allosteric and Bitopic Modulation
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zou, S.; Kumar, U. Cannabinoid Receptors and the Endocannabinoid System: Signaling and Function in the Central Nervous System. Int. J. Mol. Sci. 2018, 19, 833. [Google Scholar] [CrossRef]
- Devane, W.A.; Hanuš, L.; Breuer, A.; Pertwee, R.G.; Stevenson, L.A.; Griffin, G.; Gibson, D.; Mandelbaum, A.; Etinger, A.; Mechoulam, R. Isolation and Structure of a Brain Constituent That Binds to the Cannabinoid Receptor. Science 1992, 258, 1946–1949. [Google Scholar] [CrossRef]
- Mechoulam, R.; Ben-Shabat, S.; Hanus, L.; Ligumsky, M.; Kaminski, N.E.; Schatz, A.R.; Gopher, A.; Almog, S.; Martin, B.R.; Compton, D.R.; et al. Identification of an Endogenous 2-Monoglyceride, Present in Canine Gut, That Binds to Cannabinoid Receptors. Biochem. Pharmacol. 1995, 50, 83–90. [Google Scholar] [CrossRef]
- Cravatt, B.F.; Giang, D.K.; Mayfield, S.P.; Boger, D.L.; Lerner, R.A.; Gilula, N.B. Molecular Characterization of an Enzyme That Degrades Neuromodulatory Fatty-Acid Amides. Nature 1996, 384, 83–87. [Google Scholar] [CrossRef]
- Dinh, T.P.; Carpenter, D.; Leslie, F.M.; Freund, T.F.; Katona, I.; Sensi, S.L.; Kathuria, S.; Piomelli, D. Brain Monoglyceride Lipase Participating in Endocannabinoid Inactivation. Proc. Natl. Acad. Sci. USA 2002, 99, 10819–10824. [Google Scholar] [CrossRef]
- Di Marzo, V.; Melck, D.; Bisogno, T.; De Petrocellis, L. Endocannabinoids: Endogenous Cannabinoid Receptor Ligands with Neuromodulatory Action. Trends Neurosci. 1998, 21, 521–528. [Google Scholar] [CrossRef]
- Bisogno, T.; Ligresti, A.; Dimarzo, V. The Endocannabinoid Signalling System: Biochemical Aspects. Pharmacol. Biochem. Behav. 2005, 81, 224–238. [Google Scholar] [CrossRef]
- Pacher, P.; Bátkai, S.; Kunos, G. The Endocannabinoid System as an Emerging Target of Pharmacotherapy. Pharmacol. Rev. 2006, 58, 389–462. [Google Scholar] [CrossRef]
- Bisogno, T.; Howell, F.; Williams, G.; Minassi, A.; Cascio, M.G.; Ligresti, A.; Matias, I.; Schiano-Moriello, A.; Paul, P.; Williams, E.-J.; et al. Cloning of the First Sn1-DAG Lipases Points to the Spatial and Temporal Regulation of Endocannabinoid Signaling in the Brain. J. Cell Biol. 2003, 163, 463–468. [Google Scholar] [CrossRef]
- Blankman, J.L.; Simon, G.M.; Cravatt, B.F. A Comprehensive Profile of Brain Enzymes That Hydrolyze the Endocannabinoid 2-Arachidonoylglycerol. Chem. Biol. 2007, 14, 1347–1356. [Google Scholar] [CrossRef]
- Matsuda, L.A.; Lolait, S.J.; Brownstein, M.J.; Young, A.C.; Bonner, T.I. Structure of a Cannabinoid Receptor and Functional Expression of the Cloned cDNA. Nature 1990, 346, 561–564. [Google Scholar] [CrossRef]
- Munro, S.; Thomas, K.L.; Abu-Shaar, M. Molecular Characterization of a Peripheral Receptor for Cannabinoids. Nature 1993, 365, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Howlett, A.C. International Union of Pharmacology. XXVII. Classification of Cannabinoid Receptors. Pharmacol. Rev. 2002, 54, 161–202. [Google Scholar] [CrossRef] [PubMed]
- Rozenfeld, R. Type I Cannabinoid Receptor Trafficking: All Roads Lead to Lysosome. Traffic 2011, 12, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Svizenska, I.; Dubovy, P.; Sulcova, A. Cannabinoid Receptors 1 and 2 (CB1 and CB2), Their Distribution, Ligands and Functional Involvement in Nervous System Structures—A Short Review. Pharmacol. Biochem. Behav. 2008, 90, 501–511. [Google Scholar] [CrossRef]
- Chanda, D.; Neumann, D.; Glatz, J.F.C. The Endocannabinoid System: Overview of an Emerging Multi-Faceted Therapeutic Target. Prostaglandins Leukot. Essent. Fat. Acids 2019, 140, 51–56. [Google Scholar] [CrossRef]
- Ye, L.; Cao, Z.; Wang, W.; Zhou, N. New Insights in Cannabinoid Receptor Structure and Signaling. CMP 2019, 12, 239–248. [Google Scholar] [CrossRef]
- Onaivi, E.S.; Ishiguro, H.; Gong, J.; Patel, S.; Meozzi, P.A.; Myers, L.; Perchuk, A.; Mora, Z.; Tagliaferro, P.A.; Gardner, E.; et al. Functional Expression of Brain Neuronal CB2 Cannabinoid Receptors Are Involved in the Effects of Drugs of Abuse and in Depression. Ann. N. Y. Acad. Sci. 2008, 1139, 434–449. [Google Scholar] [CrossRef]
- Gong, J.-P.; Onaivi, E.S.; Ishiguro, H.; Liu, Q.-R.; Tagliaferro, P.A.; Brusco, A.; Uhl, G.R. Cannabinoid CB2 Receptors: Immunohistochemical Localization in Rat Brain. Brain Res. 2006, 1071, 10–23. [Google Scholar] [CrossRef]
- Van Sickle, M.D.; Duncan, M.; Kingsley, P.J.; Mouihate, A.; Urbani, P.; Mackie, K.; Stella, N.; Makriyannis, A.; Piomelli, D.; Davison, J.S.; et al. Identification and Functional Characterization of Brainstem Cannabinoid CB2 Receptors. Science 2005, 310, 329–332. [Google Scholar] [CrossRef]
- Li, Y.; Kim, J. Neuronal Expression of CB2 Cannabinoid Receptor mRNAs in the Mouse Hippocampus. Neuroscience 2015, 311, 253–267. [Google Scholar] [CrossRef] [PubMed]
- Aracil-Fernández, A.; Trigo, J.M.; García-Gutiérrez, M.S.; Ortega-Álvaro, A.; Ternianov, A.; Navarro, D.; Robledo, P.; Berbel, P.; Maldonado, R.; Manzanares, J. Decreased Cocaine Motor Sensitization and Self-Administration in Mice Overexpressing Cannabinoid CB2 Receptors. Neuropsychopharmacology 2012, 37, 1749–1763. [Google Scholar] [CrossRef] [PubMed]
- Stella, N. Cannabinoid and Cannabinoid-like Receptors in Microglia, Astrocytes, and Astrocytomas. Glia 2010, 58, 1017–1030. [Google Scholar] [CrossRef]
- Cristino, L.; Bisogno, T.; Di Marzo, V. Cannabinoids and the Expanded Endocannabinoid System in Neurological Disorders. Nat. Rev. Neurol. 2020, 16, 9–29. [Google Scholar] [CrossRef] [PubMed]
- Yiangou, Y.; Facer, P.; Durrenberger, P.; Chessell, I.P.; Naylor, A.; Bountra, C.; Banati, R.R.; Anand, P. COX-2, CB2 and P2X7-Immunoreactivities Are Increased in Activated Microglial Cells/Macrophages of Multiple Sclerosis and Amyotrophic Lateral Sclerosis Spinal Cord. BMC Neurol. 2006, 6, 12. [Google Scholar] [CrossRef]
- López, A.; Aparicio, N.; Pazos, M.R.; Grande, M.T.; Barreda-Manso, M.A.; Benito-Cuesta, I.; Vázquez, C.; Amores, M.; Ruiz-Pérez, G.; García-García, E.; et al. Cannabinoid CB2 Receptors in the Mouse Brain: Relevance for Alzheimer’s Disease. J. Neuroinflamm. 2018, 15, 158. [Google Scholar] [CrossRef]
- Beltramo, M.; Bernardini, N.; Bertorelli, R.; Campanella, M.; Nicolussi, E.; Fredduzzi, S.; Reggiani, A. CB2 Receptor-mediated Antihyperalgesia: Possible Direct Involvement of Neural Mechanisms. Eur. J. Neurosci. 2006, 23, 1530–1538. [Google Scholar] [CrossRef]
- Jordan, C.J.; Xi, Z.-X. Progress in Brain Cannabinoid CB2 Receptor Research: From Genes to Behavior. Neurosci. Biobehav. Rev. 2019, 98, 208–220. [Google Scholar] [CrossRef]
- Ibsen, M.S.; Connor, M.; Glass, M. Cannabinoid CB1 and CB2 Receptor Signaling and Bias. Cannabis Cannabinoid Res. 2017, 2, 48–60. [Google Scholar] [CrossRef]
- Twitchell, W.; Brown, S.; Mackie, K. Cannabinoids Inhibit N- and P/Q-Type Calcium Channels in Cultured Rat Hippocampal Neurons. J. Neurophysiol. 1997, 78, 43–50. [Google Scholar] [CrossRef]
- Ho, B.Y.; Uezono, Y.; Takada, S.; Takase, I.; Izumi, F. Coupling of the Expressed Cannabinoid CB1 and CB2 Receptors to Phospholipase C and G Protein-Coupled Inwardly Rectifying K+ Channels. Recept. Channels 1999, 6, 363–374. [Google Scholar] [PubMed]
- Demuth, D.G.; Molleman, A. Cannabinoid Signalling. Life Sci. 2006, 78, 549–563. [Google Scholar] [CrossRef] [PubMed]
- Mallipeddi, S.; Janero, D.R.; Zvonok, N.; Makriyannis, A. Functional Selectivity at G-Protein Coupled Receptors: Advancing Cannabinoid Receptors as Drug Targets. Biochem. Pharmacol. 2017, 128, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Barres, B.A. Microglia and Macrophages in Brain Homeostasis and Disease. Nat. Rev. Immunol. 2018, 18, 225–242. [Google Scholar] [CrossRef]
- Song, G.J.; Suk, K. Pharmacological Modulation of Functional Phenotypes of Microglia in Neurodegenerative Diseases. Front. Aging Neurosci. 2017, 9, 139. [Google Scholar] [CrossRef]
- Etemad, S.; Zamin, R.M.; Ruitenberg, M.J.; Filgueira, L. A Novel in Vitro Human Microglia Model: Characterization of Human Monocyte-Derived Microglia. J. Neurosci. Methods 2012, 209, 79–89. [Google Scholar] [CrossRef]
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate Mapping Analysis Reveals That Adult Microglia Derive from Primitive Macrophages. Science 2010, 330, 841–845. [Google Scholar] [CrossRef]
- Ginhoux, F.; Lim, S.; Hoeffel, G.; Low, D.; Huber, T. Origin and Differentiation of Microglia. Front. Cell Neurosci. 2013, 7, 45. [Google Scholar] [CrossRef]
- Musiek, E.S.; Holtzman, D.M. Three Dimensions of the Amyloid Hypothesis: Time, Space and “Wingmen”. Nat. Neurosci. 2015, 18, 800–806. [Google Scholar] [CrossRef]
- Wong, W.T. Microglial Aging in the Healthy CNS: Phenotypes, Drivers, and Rejuvenation. Front. Cell Neurosci. 2013, 7, 22. [Google Scholar] [CrossRef]
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Resting Microglial Cells Are Highly Dynamic Surveillants of Brain Parenchyma In Vivo. Science 2005, 308, 1314–1318. [Google Scholar] [CrossRef] [PubMed]
- Leng, F.; Edison, P. Neuroinflammation and Microglial Activation in Alzheimer Disease: Where Do We Go from Here? Nat. Rev. Neurol. 2021, 17, 157–172. [Google Scholar] [CrossRef] [PubMed]
- Kigerl, K.A.; De Rivero Vaccari, J.P.; Dietrich, W.D.; Popovich, P.G.; Keane, R.W. Pattern Recognition Receptors and Central Nervous System Repair. Exp. Neurol. 2014, 258, 5–16. [Google Scholar] [CrossRef]
- Solé-Domènech, S.; Cruz, D.L.; Capetillo-Zarate, E.; Maxfield, F.R. The Endocytic Pathway in Microglia during Health, Aging and Alzheimer’s Disease. Ageing Res. Rev. 2016, 32, 89–103. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Wang, X.; Kalicki, C.; Menta, B.W.; Baumgardner, M.; Koppel, S.J.; Weidling, I.W.; Perez-Ortiz, J.; Wilkins, H.M.; Swerdlow, R.H. Effects of Microglial Cytokines on Alzheimer’s Disease-Related Phenomena. J. Alzheimer’s Dis. 2019, 67, 1021–1034. [Google Scholar] [CrossRef]
- Walker, D.G.; Lue, L.-F. Immune Phenotypes of Microglia in Human Neurodegenerative Disease: Challenges to Detecting Microglial Polarization in Human Brains. Alz Res. Ther. 2015, 7, 56. [Google Scholar] [CrossRef]
- Tang, Y.; Le, W. Differential Roles of M1 and M2 Microglia in Neurodegenerative Diseases. Mol. Neurobiol. 2016, 53, 1181–1194. [Google Scholar] [CrossRef]
- Diaz-Jimenez, D.; Kolb, J.P.; Cidlowski, J.A. Glucocorticoids as Regulators of Macrophage-Mediated Tissue Homeostasis. Front. Immunol. 2021, 12, 669891. [Google Scholar] [CrossRef]
- Zhao, F.; Li, P.; Chen, S.R.W.; Louis, C.F.; Fruen, B.R. Dantrolene Inhibition of Ryanodine Receptor Ca2+ Release Channels: Molecular Mechanism and Isoform Selectivity. J. Biol. Chem. 2001, 276, 13810–13816. [Google Scholar] [CrossRef]
- Khan, S.; Barve, K.H.; Kumar, M.S. Recent Advancements in Pathogenesis, Diagnostics and Treatment of Alzheimer’s Disease. Curr. Neuropharmacol. 2020, 18, 1106–1125. [Google Scholar] [CrossRef]
- Coles, M.; Steiner-Lim, G.Z.; Karl, T. Therapeutic Properties of Multi-Cannabinoid Treatment Strategies for Alzheimer’s Disease. Front. Neurosci. 2022, 16, 962922. [Google Scholar] [CrossRef]
- Citron, M. Alzheimer’s Disease: Strategies for Disease Modification. Nat. Rev. Drug Discov. 2010, 9, 387–398. [Google Scholar] [CrossRef] [PubMed]
- Aso, E.; Ferrer, I. Cannabinoids for Treatment of Alzheimer’s Disease: Moving toward the Clinic. Front. Pharmacol. 2014, 5, 37. [Google Scholar] [CrossRef] [PubMed]
- Puopolo, T.; Liu, C.; Ma, H.; Seeram, N.P. Inhibitory Effects of Cannabinoids on Acetylcholinesterase and Butyrylcholinesterase Enzyme Activities. Med. Cannabis Cannabinoids 2022, 5, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Eubanks, L.M.; Rogers, C.J.; Beuscher; Koob, G.F.; Olson, A.J.; Dickerson, T.J.; Janda, K.D. A Molecular Link between the Active Component of Marijuana and Alzheimer’s Disease Pathology. Mol. Pharm. 2006, 3, 773–777. [Google Scholar] [CrossRef]
- Scipioni, L.; Ciaramellano, F.; Carnicelli, V.; Leuti, A.; Lizzi, A.R.; De Dominicis, N.; Oddi, S.; Maccarrone, M. Microglial Endocannabinoid Signalling in AD. Cells 2022, 11, 1237. [Google Scholar] [CrossRef] [PubMed]
- Hanzel, C.E.; Pichet-Binette, A.; Pimentel, L.S.B.; Iulita, M.F.; Allard, S.; Ducatenzeiler, A.; Do Carmo, S.; Cuello, A.C. Neuronal Driven Pre-Plaque Inflammation in a Transgenic Rat Model of Alzheimer’s Disease. Neurobiol. Aging 2014, 35, 2249–2262. [Google Scholar] [CrossRef]
- Gratuze, M.; Leyns, C.E.G.; Holtzman, D.M. New Insights into the Role of TREM2 in Alzheimer’s Disease. Mol. Neurodegener. 2018, 13, 66. [Google Scholar] [CrossRef]
- European Alzheimer’s Disease Initiative (EADI); Genetic and Environmental Risk in Alzheimer’s Disease (GERAD); Alzheimer’s Disease Genetic Consortium (ADGC); Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE); Lambert, J.-C.; Ibrahim-Verbaas, C.A.; Harold, D.; Naj, A.C.; Sims, R.; Bellenguez, C.; et al. Meta-Analysis of 74,046 Individuals Identifies 11 New Susceptibility Loci for Alzheimer’s Disease. Nat. Genet. 2013, 45, 1452–1458. [Google Scholar] [CrossRef]
- Bisht, K.; Sharma, K.; Lacoste, B.; Tremblay, M.-È. Dark Microglia: Why Are They Dark? Commun. Integr. Biol. 2016, 9, e1230575. [Google Scholar] [CrossRef]
- Walter, L.; Franklin, A.; Witting, A.; Möller, T.; Stella, N. Astrocytes in Culture Produce Anandamide and Other Acylethanolamides. J. Biol. Chem. 2002, 277, 20869–20876. [Google Scholar] [CrossRef] [PubMed]
- Walter, L.; Franklin, A.; Witting, A.; Wade, C.; Xie, Y.; Kunos, G.; Mackie, K.; Stella, N. Nonpsychotropic Cannabinoid Receptors Regulate Microglial Cell Migration. J. Neurosci. 2003, 23, 1398–1405. [Google Scholar] [CrossRef]
- Carrier, E.J.; Kearn, C.S.; Barkmeier, A.J.; Breese, N.M.; Yang, W.; Nithipatikom, K.; Pfister, S.L.; Campbell, W.B.; Hillard, C.J. Cultured Rat Microglial Cells Synthesize the Endocannabinoid 2-Arachidonylglycerol, Which Increases Proliferation via a CB 2 Receptor-Dependent Mechanism. Mol. Pharmacol. 2004, 65, 999–1007. [Google Scholar] [CrossRef] [PubMed]
- Witting, A.; Chen, L.; Cudaback, E.; Straiker, A.; Walter, L.; Rickman, B.; Möller, T.; Brosnan, C.; Stella, N. Experimental Autoimmune Encephalomyelitis Disrupts Endocannabinoid-Mediated Neuroprotection. Proc. Natl. Acad. Sci. USA 2006, 103, 6362–6367. [Google Scholar] [CrossRef] [PubMed]
- Witting, A.; Walter, L.; Wacker, J.; Möller, T.; Stella, N. P2X7 Receptors Control 2-Arachidonoylglycerol Production by Microglial Cells. Proc. Natl. Acad. Sci. USA 2004, 101, 3214–3219. [Google Scholar] [CrossRef] [PubMed]
- Mecha, M.; Carrillo-Salinas, F.J.; Feliú, A.; Mestre, L.; Guaza, C. Microglia Activation States and Cannabinoid System: Therapeutic Implications. Pharmacol. Ther. 2016, 166, 40–55. [Google Scholar] [CrossRef]
- Núñez, E.; Benito, C.; Pazos, M.R.; Barbachano, A.; Fajardo, O.; González, S.; Tolón, R.M.; Romero, J. Cannabinoid CB2 Receptors Are Expressed by Perivascular Microglial Cells in the Human Brain: An Immunohistochemical Study: CB2 In Human Cerebellum. Synapse 2004, 53, 208–213. [Google Scholar] [CrossRef]
- Navarro, G.; Borroto-Escuela, D.; Angelats, E.; Etayo, Í.; Reyes-Resina, I.; Pulido-Salgado, M.; Rodríguez-Pérez, A.I.; Canela, E.I.; Saura, J.; Lanciego, J.L.; et al. Receptor-Heteromer Mediated Regulation of Endocannabinoid Signaling in Activated Microglia. Role of CB1 and CB2 Receptors and Relevance for Alzheimer’s Disease and Levodopa-Induced Dyskinesia. Brain Behav. Immun. 2018, 67, 139–151. [Google Scholar] [CrossRef]
- Cabral, G.A.; Marciano-Cabral, F. Cannabinoid Receptors in Microglia of the Central Nervous System: Immune Functional Relevance. J. Leukoc. Biol. 2005, 78, 1192–1197. [Google Scholar] [CrossRef]
- Stella, N. Endocannabinoid Signaling in Microglial Cells. Neuropharmacology 2009, 56, 244–253. [Google Scholar] [CrossRef]
- Maresz, K.; Carrier, E.J.; Ponomarev, E.D.; Hillard, C.J.; Dittel, B.N. Modulation of the Cannabinoid CB2 Receptor in Microglial Cells in Response to Inflammatory Stimuli. J. Neurochem. 2005, 95, 437–445. [Google Scholar] [CrossRef] [PubMed]
- Schmöle, A.-C.; Lundt, R.; Ternes, S.; Albayram, Ö.; Ulas, T.; Schultze, J.L.; Bano, D.; Nicotera, P.; Alferink, J.; Zimmer, A. Cannabinoid Receptor 2 Deficiency Results in Reduced Neuroinflammation in an Alzheimer’s Disease Mouse Model. Neurobiol. Aging 2015, 36, 710–719. [Google Scholar] [CrossRef] [PubMed]
- Komorowska-Müller, J.A.; Schmöle, A.-C. CB2 Receptor in Microglia: The Guardian of Self-Control. Int. J. Mol. Sci. 2020, 22, 19. [Google Scholar] [CrossRef] [PubMed]
- Ehrhart, J.; Obregon, D.; Mori, T.; Hou, H.; Sun, N.; Bai, Y.; Klein, T.; Fernandez, F.; Tan, J.; Shytle, R.D. Stimulation of Cannabinoid Receptor 2 (CB2) Suppresses Microglial Activation. J. Neuroinflamm. 2005, 2, 29. [Google Scholar] [CrossRef]
- Ma, L.; Jia, J.; Liu, X.; Bai, F.; Wang, Q.; Xiong, L. Activation of Murine Microglial N9 Cells Is Attenuated through Cannabinoid Receptor CB2 Signaling. Biochem. Biophys. Res. Commun. 2015, 458, 92–97. [Google Scholar] [CrossRef]
- Correa, F.; Hernangómez, M.; Mestre, L.; Loría, F.; Spagnolo, A.; Docagne, F.; Marzo, V.D.; Guaza, C. Anandamide Enhances IL-10 Production in Activated Microglia by Targeting CB2 Receptors: Roles of ERK1/2, JNK, and NF-κB. Glia 2010, 58, 135–147. [Google Scholar] [CrossRef]
- Mecha, M.; Feliú, A.; Carrillo-Salinas, F.J.; Rueda-Zubiaurre, A.; Ortega-Gutiérrez, S.; de Sola, R.G.; Guaza, C. Endocannabinoids Drive the Acquisition of an Alternative Phenotype in Microglia. Brain Behav. Immun. 2015, 49, 233–245. [Google Scholar] [CrossRef]
- Abate, G.; Uberti, D.; Tambaro, S. Potential and Limits of Cannabinoids in Alzheimer’s Disease Therapy. Biology 2021, 10, 542. [Google Scholar] [CrossRef]
- Todaro, B. Cannabinoids in the Treatment of Chemotherapy-Induced Nausea and Vomiting. J. Natl. Compr. Canc. Netw. 2012, 10, 487–492. [Google Scholar] [CrossRef]
- De Meij, J.; Alfanek, Z.; Morel, L.; Decoeur, F.; Leyrolle, Q.; Picard, K.; Carrier, M.; Aubert, A.; Séré, A.; Lucas, C.; et al. Microglial Cannabinoid Type 1 Receptor Regulates Brain Inflammation in a Sex-Specific Manner. Cannabis Cannabinoid Res. 2021, 6, 488–507. [Google Scholar] [CrossRef]
- Navarro, G.; Morales, P.; Rodríguez-Cueto, C.; Fernández-Ruiz, J.; Jagerovic, N.; Franco, R. Targeting Cannabinoid CB2 Receptors in the Central Nervous System. Medicinal Chemistry Approaches with Focus on Neurodegenerative Disorders. Front. Neurosci. 2016, 10, 406. [Google Scholar] [CrossRef] [PubMed]
- Lou, Z.-Y.; Cheng, J.; Wang, X.-R.; Zhao, Y.-F.; Gan, J.; Zhou, G.-Y.; Liu, Z.-G.; Xiao, B.-G. The Inhibition of CB1 Receptor Accelerates the Onset and Development of EAE Possibly by Regulating Microglia/Macrophages Polarization. J. Neuroimmunol. 2018, 317, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Facchinetti, F.; Del Giudice, E.; Furegato, S.; Passarotto, M.; Leon, A. Cannabinoids Ablate Release of TNFα in Rat Microglial Cells Stimulated with Lypopolysaccharide. Glia 2003, 41, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Marchalant, Y.; Brothers, H.M.; Wenk, G.L. Cannabinoid Agonist WIN-55,212-2 Partially Restores Neurogenesis in the Aged Rat Brain. Mol. Psychiatry 2009, 14, 1068–1069. [Google Scholar] [CrossRef] [PubMed]
- Martín-Moreno, A.M.; Brera, B.; Spuch, C.; Carro, E.; García-García, L.; Delgado, M.; Pozo, M.A.; Innamorato, N.G.; Cuadrado, A.; De Ceballos, M.L. Prolonged Oral Cannabinoid Administration Prevents Neuroinflammation, Lowers β-Amyloid Levels and Improves Cognitive Performance in Tg APP 2576 Mice. J. Neuroinflamm. 2012, 9, 8. [Google Scholar] [CrossRef] [PubMed]
- Martín-Moreno, A.M.; Reigada, D.; Ramirez, B.G.; Mechoulam, R.; Innamorato, N.G.; Cuadrado, A.; Ceballos, M.L. de Cannabidiol and Other Cannabinoids Reduce Microglial Activation In Vitro and In Vivo: Relevance to Alzheimer’s Disease. Mol. Pharmacol. 2011, 79, 964–973. [Google Scholar] [CrossRef]
- Abbassian, H.; Esmaeili, P.; Tahamtan, M.; Aghaei, I.; Vaziri, Z.; Sheibani, V.; Whalley, B.J.; Shabani, M. Cannabinoid Receptor Agonism Suppresses Tremor, Cognition Disturbances and Anxiety-like Behaviors in a Rat Model of Essential Tremor. Physiol. Behav. 2016, 164, 314–320. [Google Scholar] [CrossRef]
- Ramírez, B.G.; Blázquez, C.; Gómez del Pulgar, T.; Guzmán, M.; de Ceballos, M.L. Prevention of Alzheimer’s Disease Pathology by Cannabinoids: Neuroprotection Mediated by Blockade of Microglial Activation. J. Neurosci. 2005, 25, 1904–1913. [Google Scholar] [CrossRef]
- Aso, E.; Juvés, S.; Maldonado, R.; Ferrer, I. CB2 Cannabinoid Receptor Agonist Ameliorates Alzheimer-Like Phenotype in AβPP/PS1 Mice. J. Alzheimer’s Dis. 2013, 35, 847–858. [Google Scholar] [CrossRef]
- Chung, Y.C.; Shin, W.-H.; Baek, J.Y.; Cho, E.J.; Baik, H.H.; Kim, S.R.; Won, S.-Y.; Jin, B.K. CB2 Receptor Activation Prevents Glial-Derived Neurotoxic Mediator Production, BBB Leakage and Peripheral Immune Cell Infiltration and Rescues Dopamine Neurons in the MPTP Model of Parkinson’s Disease. Exp. Mol. Med. 2016, 48, e205. [Google Scholar] [CrossRef]
- Li, C.; Shi, J.; Wang, B.; Li, J.; Jia, H. CB2 Cannabinoid Receptor Agonist Ameliorates Novel Object Recognition but Not Spatial Memory in Transgenic APP/PS1 Mice. Neurosci. Lett. 2019, 707, 134286. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Huang, R.; Wang, D.; Yu, L.; Liu, Y.; Huang, R.; Yin, S.; He, X.; Chen, B.; Liu, Z.; et al. EVs-Mediated Delivery of CB2 Receptor Agonist for Alzheimer’s Disease Therapy. Asian J. Pharm. Sci. 2023, 18, 100835. [Google Scholar] [CrossRef] [PubMed]
- Palazuelos, J.; Aguado, T.; Pazos, M.R.; Julien, B.; Carrasco, C.; Resel, E.; Sagredo, O.; Benito, C.; Romero, J.; Azcoitia, I.; et al. Microglial CB2 Cannabinoid Receptors Are Neuroprotective in Huntington’s Disease Excitotoxicity. Brain 2009, 132, 3152–3164. [Google Scholar] [CrossRef] [PubMed]
- Ullah, H.; Di Minno, A.; Santarcangelo, C.; Khan, H.; Daglia, M. Improvement of Oxidative Stress and Mitochondrial Dysfunction by β-Caryophyllene: A Focus on the Nervous System. Antioxidants 2021, 10, 546. [Google Scholar] [CrossRef]
- Askari, V.R.; Baradaran Rahimi, V.; Shafiee-Nick, R. Low Doses of β-Caryophyllene Reduced Clinical and Paraclinical Parameters of an Autoimmune Animal Model of Multiple Sclerosis: Investigating the Role of CB2 Receptors in Inflammation by Lymphocytes and Microglial. Brain Sci. 2023, 13, 1092. [Google Scholar] [CrossRef]
- Askari, V.R.; Shafiee-Nick, R. The Protective Effects of β-Caryophyllene on LPS-Induced Primary Microglia M1/M2 Imbalance: A Mechanistic Evaluation. Life Sci. 2019, 219, 40–73. [Google Scholar] [CrossRef]
- Ruiz De Martín Esteban, S.; Benito-Cuesta, I.; Terradillos, I.; Martínez-Relimpio, A.M.; Arnanz, M.A.; Ruiz-Pérez, G.; Korn, C.; Raposo, C.; Sarott, R.C.; Westphal, M.V.; et al. Cannabinoid CB2 Receptors Modulate Microglia Function and Amyloid Dynamics in a Mouse Model of Alzheimer’s Disease. Front. Pharmacol. 2022, 13, 841766. [Google Scholar] [CrossRef]
- Reusch, N.; Ravichandran, K.A.; Olabiyi, B.F.; Komorowska-Müller, J.A.; Hansen, J.N.; Ulas, T.; Beyer, M.; Zimmer, A.; Schmöle, A. Cannabinoid Receptor 2 Is Necessary to Induce Toll-like Receptor-mediated Microglial Activation. Glia 2022, 70, 71–88. [Google Scholar] [CrossRef]
- Callén, L.; Moreno, E.; Barroso-Chinea, P.; Moreno-Delgado, D.; Cortés, A.; Mallol, J.; Casadó, V.; Lanciego, J.L.; Franco, R.; Lluis, C.; et al. Cannabinoid Receptors CB1 and CB2 Form Functional Heteromers in Brain. J. Biol. Chem. 2012, 287, 20851–20865. [Google Scholar] [CrossRef]
- Wisler, J.W.; Rockman, H.A.; Lefkowitz, R.J. Biased G Protein–Coupled Receptor Signaling: Changing the Paradigm of Drug Discovery. Circulation 2018, 137, 2315–2317. [Google Scholar] [CrossRef]
- Polini, B.; Cervetto, C.; Carpi, S.; Pelassa, S.; Gado, F.; Ferrisi, R.; Bertini, S.; Nieri, P.; Marcoli, M.; Manera, C. Positive Allosteric Modulation of CB1 and CB2 Cannabinoid Receptors Enhances the Neuroprotective Activity of a Dual CB1R/CB2R Orthosteric Agonist. Life 2020, 10, 333. [Google Scholar] [CrossRef]
- Haque, M.E.; Kim, I.-S.; Jakaria, M.; Akther, M.; Choi, D.-K. Importance of GPCR-Mediated Microglial Activation in Alzheimer’s Disease. Front. Cell. Neurosci. 2018, 12, 258. [Google Scholar] [CrossRef] [PubMed]
- Gado, F.; Di Cesare Mannelli, L.; Lucarini, E.; Bertini, S.; Cappelli, E.; Digiacomo, M.; Stevenson, L.A.; Macchia, M.; Tuccinardi, T.; Ghelardini, C.; et al. Identification of the First Synthetic Allosteric Modulator of the CB2 Receptors and Evidence of Its Efficacy for Neuropathic Pain Relief. J. Med. Chem. 2019, 62, 276–287. [Google Scholar] [CrossRef] [PubMed]
- Valant, C.; Sexton, P.M.; Christopoulos, A. Orthosteric/Allosteric Bitopic Ligands: Going Hybrid at GPCRs. Mol. Interv. 2009, 9, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Gado, F.; Ferrisi, R.; Polini, B.; Mohamed, K.A.; Ricardi, C.; Lucarini, E.; Carpi, S.; Domenichini, F.; Stevenson, L.A.; Rapposelli, S.; et al. Design, Synthesis, and Biological Activity of New CB2 Receptor Ligands: From Orthosteric and Allosteric Modulators to Dualsteric/Bitopic Ligands. J. Med. Chem. 2022, 65, 9918–9938. [Google Scholar] [CrossRef]
- Ferrisi, R.; Gado, F.; Polini, B.; Ricardi, C.; Mohamed, K.A.; Stevenson, L.A.; Ortore, G.; Rapposelli, S.; Saccomanni, G.; Pertwee, R.G.; et al. Design, Synthesis and Biological Evaluation of Novel Orthosteric-Allosteric Ligands of the Cannabinoid Receptor Type 2 (CB2R). Front. Chem. 2022, 10, 984069. [Google Scholar] [CrossRef]
- Ferrisi, R.; Polini, B.; Ricardi, C.; Gado, F.; Mohamed, K.A.; Baron, G.; Faiella, S.; Poli, G.; Rapposelli, S.; Saccomanni, G.; et al. New Insights into Bitopic Orthosteric/Allosteric Ligands of Cannabinoid Receptor Type 2. Int. J. Mol. Sci. 2023, 24, 2135. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferrisi, R.; Gado, F.; Ricardi, C.; Polini, B.; Manera, C.; Chiellini, G. The Interplay between Cannabinoid Receptors and Microglia in the Pathophysiology of Alzheimer’s Disease. J. Clin. Med. 2023, 12, 7201. https://doi.org/10.3390/jcm12237201
Ferrisi R, Gado F, Ricardi C, Polini B, Manera C, Chiellini G. The Interplay between Cannabinoid Receptors and Microglia in the Pathophysiology of Alzheimer’s Disease. Journal of Clinical Medicine. 2023; 12(23):7201. https://doi.org/10.3390/jcm12237201
Chicago/Turabian StyleFerrisi, Rebecca, Francesca Gado, Caterina Ricardi, Beatrice Polini, Clementina Manera, and Grazia Chiellini. 2023. "The Interplay between Cannabinoid Receptors and Microglia in the Pathophysiology of Alzheimer’s Disease" Journal of Clinical Medicine 12, no. 23: 7201. https://doi.org/10.3390/jcm12237201