
Trace Elements in Alzheimer’s Disease and Dementia: The Current State of Knowledge
 ,
,  , , , and
, , , and 
Abstract
:1. Introduction
2. Trace Elements in Alzheimer’s Disease
2.1. Iron (Fe)
2.2. Copper (Cu)
2.3. Zinc (Zn)
2.4. Selenium (Se)
2.5. Aluminium (Al)
2.6. Cadmium (Cd)
2.7. Calcium (Ca)
2.8. Lead (Pb)
2.9. Magnesium (Mg)
2.10. Mercury (Hg)
2.11. Arsenic (As)
2.12. Bromine (Br)
2.13. Chromium (Cr)
2.14. Manganese (Mn)
2.15. Nickel (Ni)
2.16. Rubidium (Rb)
2.17. Strontium (Sr)
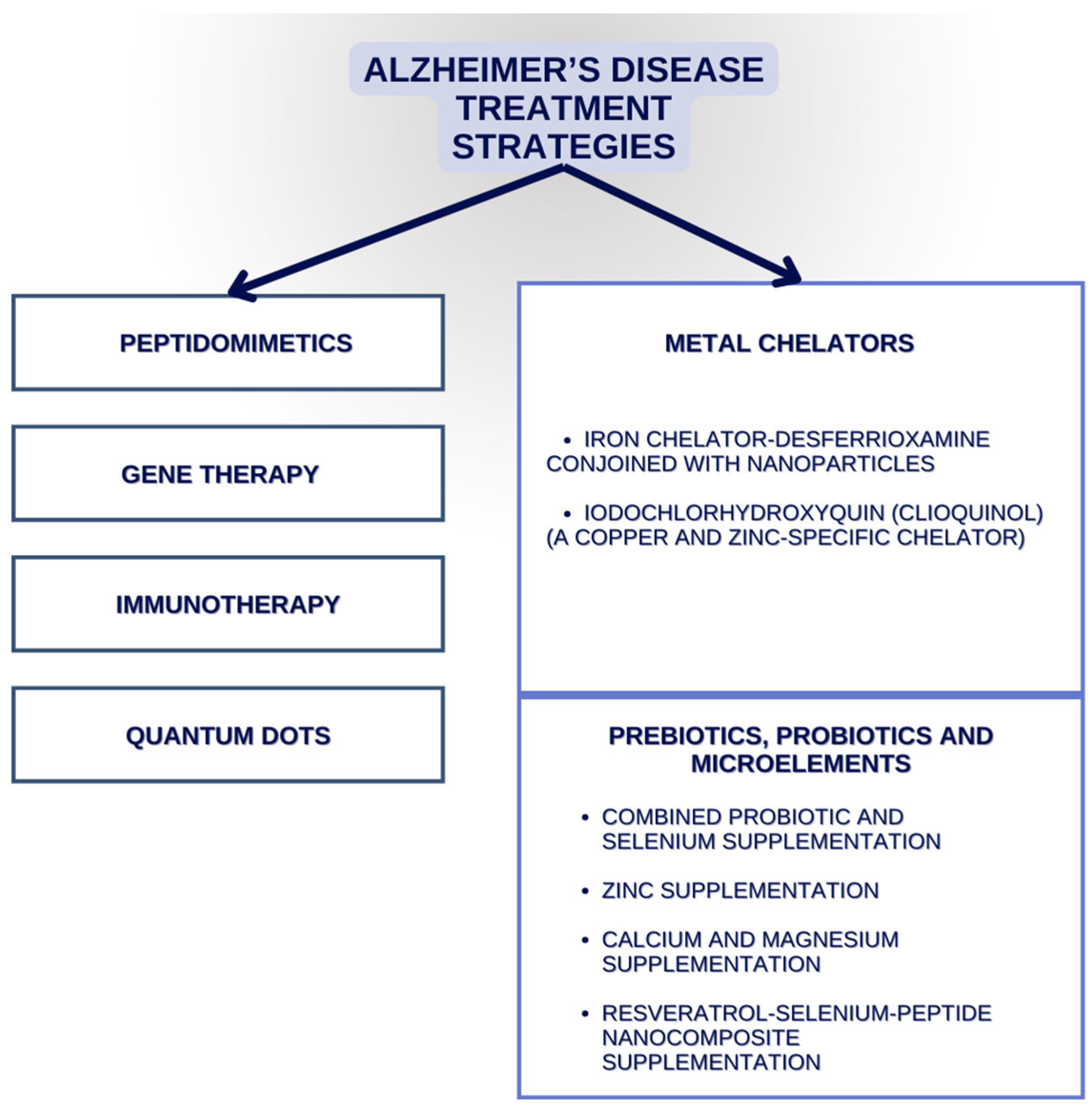
3. Alzheimer’s Disease Therapies That Might Target Elements
4. Trace Elements’ Abbreviations in Other Dementia Disorders
4.1. Lewy Bodies Dementias
4.2. Frontotemporal Dementia (FTD)
4.3. Amyotrophic Lateral Sclerosis (ALS)
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Gale, S.A.; Acar, D.; Daffner, K.R. Dementia. Am. J. Med. 2018, 131, 1161–1169. [Google Scholar] [CrossRef] [PubMed]
- Rostagno, A.A. Pathogenesis of Alzheimer’s Disease. Int. J. Mol. Sci. 2022, 24, 107. [Google Scholar] [CrossRef] [PubMed]
- Scheltens, P.; De Strooper, B.; Kivipelto, M.; Holstege, H.; Chetelat, G.; Teunissen, C.E.; Cummings, J.; Van der Flier, W.M. Alzheimer’s disease. Lancet 2021, 397, 1577–1590. [Google Scholar] [CrossRef] [PubMed]
- Eratne, D.; Loi, S.M.; Farrand, S.; Kelso, W.; Velakoulis, D.; Looi, J.C. Alzheimer’s disease: Clinical update on epidemiology, pathophysiology and diagnosis. Australas. Psychiatry 2018, 26, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Soria Lopez, J.A.; González, H.M.; Léger, G.C. Alzheimer’s disease. Handb. Clin. Neurol. 2019, 167, 231–255. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Men, P.; Perry, G.; Smith, M.A. Nanoparticle and iron chelators as a potential novel Alzheimer therapy. Methods Mol. Biol. 2010, 610, 123–144. [Google Scholar] [CrossRef] [PubMed]
- Baj, J.; Forma, A.; Sitarz, E.; Karakuła, K.; Flieger, W.; Sitarz, M.; Grochowski, C.; Maciejewski, R.; Karakula-Juchnowicz, H. Beyond the Mind—Serum Trace Element Levels in Schizophrenic Patients: A Systematic Review. Int. J. Mol. Sci. 2020, 21, 9566. [Google Scholar] [CrossRef] [PubMed]
- Baj, J.; Flieger, W.; Flieger, M.; Forma, A.; Sitarz, E.; Skórzyńska-Dziduszko, K.; Grochowski, C.; Maciejewki, R.; Karakuła-Juchnowicz, H. Autism spectrum disorder: Trace elements imbalances and the pathogenesis and severity of autistic symptoms. Neurosci. Biobehav. Rev. 2021, 129, 117–132. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Barve, K.H.; Kumar, M.S. Recent Advancements in Pathogenesis, Diagnostics and Treatment of Alzheimer’s Disease. Curr. Neuropharmacol. 2020, 18, 1106–1125. [Google Scholar] [CrossRef]
- Gunshin, H.; Mackenzie, B.; Berger, U.V.; Gunshin, Y.; Romero, M.F.; Boron, W.F.; Nussberger, S.; Gollan, J.L.; Hediger, M.A. Cloning and characterization of a mammalian proton-coupled metal-ion transporter. Nature 1997, 388, 482–488. [Google Scholar] [CrossRef]
- Zecca, L.; Youdim, M.B.; Riederer, P.; Connor, J.R.; Crichton, R.R. Iron, brain ageing and neurodegenerative disorders. Nat. Rev. Neurosci. 2004, 5, 863–873. [Google Scholar] [CrossRef]
- Popescu, B.F.; George, M.J.; Bergmann, U.; Garachtchenko, A.V.; Kelly, M.E.; McCrea, R.P.E.; Luning, K.; Devon, R.M.; George, G.N.; Handon, A.D.; et al. Mapping metals in Parkinson’s and normal brain using rapid-scanning x-ray fluorescence. Phys. Med. Biol. 2009, 54, 651–663. [Google Scholar] [CrossRef]
- Dexter, D.T.; Wells, F.R.; Lees, A.J.; Agid, F.; Agid, Y.; Jenner, P.; Marsden, C.D. Increased nigral iron content and alterations in other metal ions occurring in brain in Parkinson’s disease. J. Neurochem. 1989, 52, 1830–1836. [Google Scholar] [CrossRef] [PubMed]
- Grochowski, C.; Blicharska, E.; Krukow, P.; Jonak, K.; Maciejewski, M.; Szczepanek, D.; Jonak, K.; Flieger, J.; Maciejewski, R. Analysis of Trace Elements in Human Brain: Its Aim, Methods, and Concentration Levels. Front. Chem. 2019, 7, 115. [Google Scholar] [CrossRef] [PubMed]
- Lumsden, A.L.; Mulugeta, A.; Mäkinen, V.P.; Hyppönen, E. Metabolic profile-based subgroups can identify differences in brain volumes and brain iron deposition. Diabetes Obes. Metab. 2023, 25, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, S.; Berggren, K.L.; Marks, E.; Fox, J.H. Impact of high iron intake on cognition and neurodegeneration in humans and in animal models: A systematic review. Nutr. Rev. 2017, 75, 456–470. [Google Scholar] [CrossRef] [PubMed]
- Spence, H.; McNeil, C.J.; Waiter, G.D. The impact of brain iron accumulation on cognition: A systematic review. PLoS ONE 2020, 15, e0240697. [Google Scholar] [CrossRef] [PubMed]
- Tranchant, C.; Koob, M.; Anheim, M. Parkinsonian-Pyramidal syndromes: A systematic review. Park. Relat. Disord. 2017, 39, 4–16. [Google Scholar] [CrossRef] [PubMed]
- Hayflick, S.J.; Westaway, S.K.; Levinson, B.; Zhou, B.; Johnson, M.A.; Ching, K.H.L.; Gitschier, J. Genetic, clinical, and radiographic delineation of Hallervorden-Spatz syndrome. N. Engl. J. Med. 2003, 348, 33–40. [Google Scholar] [CrossRef]
- Hogarth, P. Neurodegeneration with brain iron accumulation: Diagnosis and management. J. Mov. Disord. 2015, 8, 1–13. [Google Scholar] [CrossRef]
- Lei, P.; Ayton, S.; Bush, A.I. The essential elements of Alzheimer’s disease. J. Biol. Chem. 2021, 296, 100105. [Google Scholar] [CrossRef] [PubMed]
- Pal, A.; Cerchiaro, G.; Rani, I.; Ventriglia, M.; Rongioletti, M.; Longobardi, A.; Squitti, R. Iron in Alzheimer’s Disease: From Physiology to Disease Disabilities. Biomolecules 2022, 12, 1248. [Google Scholar] [CrossRef] [PubMed]
- Gong, L.; Sun, J.; Cong, S. Levels of iron and iron-related proteins in Alzheimer’s disease: A systematic review and meta-analysis. J. Trace Elem. Med. Biol. 2023, 80, 127304. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Min, J.; Wang, F. Copper homeostasis and cuproptosis in health and disease. Signal Transduct. Target. Ther. 2022, 7, 378. [Google Scholar] [CrossRef] [PubMed]
- Aggett, P.J. An overview of the metabolism of copper. Eur. J. Med. Res. 1999, 4, 214–216. [Google Scholar] [PubMed]
- Pena, M.M.; Lee, J.; Thiele, D.J. A delicate balance: Homeostatic control of copper uptake and distribution. J. Nutr. 1999, 129, 1251–1260. [Google Scholar] [CrossRef] [PubMed]
- Ala, A.; Walker, A.P.; Ashkan, K.; Dooley, J.S.; Schilsky, M.L. Wilson’s disease. Lancet 2007, 369, 397–408. [Google Scholar] [CrossRef]
- Das, S.K.; Ray, K. Wilson’s disease: An update. Nat. Clin. Pract. Neurol. 2006, 2, 482–493. [Google Scholar] [CrossRef] [PubMed]
- Eskici, G.; Axelsen, P.H. Copper and oxidative stress in the pathogenesis of Alzheimer’s disease. Biochemistry 2012, 51, 6289–6311. [Google Scholar] [CrossRef]
- Squitti, R.; et al. Meta-analysis of serum non-ceruloplasmin copper in Alzheimer’s disease. J. Alzheimer’s Dis. 2014, 38, 809–822. [Google Scholar] [CrossRef]
- Atwood, C.S.; et al. Copper mediates dityrosine cross-linking of Alzheimer’s amyloid-beta. Biochemistry 2004, 43, 560–568. [Google Scholar] [CrossRef] [PubMed]
- Cherny, R.A.; et al. Treatment with a copper-zinc chelator markedly and rapidly inhibits beta-amyloid accumulation in Alzheimer’s disease transgenic mice. Neuron 2001, 30, 665–676. [Google Scholar] [CrossRef] [PubMed]
- Kitazawa, M.; Hsu, H.W.; Medeiros, R. Copper exposure perturbs brain inflammatory responses and impairs clearance of amyloid-beta. Toxicol. Sci. 2016, 152, 194–204. [Google Scholar] [CrossRef] [PubMed]
- Krall, R.F.; Tzounopoulos, T.; Aizenman, E. The Function and Regulation of Zinc in the Brain. Neuroscience 2021, 457, 235–258. [Google Scholar] [CrossRef]
- Wang, B.; Fang, T.; Chen, H. Zinc and Central Nervous System Disorders. Nutrients 2023, 15, 2140. [Google Scholar] [CrossRef] [PubMed]
- Cuesta, A.; Zambrano, A.; Royo, M.; Pascual, A. The tumour suppressor p53 regulates the expression of amyloid precursor protein (APP). Biochem. J. 2009, 418, 643–650. [Google Scholar] [CrossRef] [PubMed]
- Cristóvão, J.S.; Santos, R.; Gomes, C.M. Metals and Neuronal Metal Binding Proteins Implicated in Alzheimer’s Disease. Oxidative Med. Cell. Longev. 2016, 2016, 9812178. [Google Scholar] [CrossRef]
- Brewer, G.J.; Kanzer, S.H.; Zimmerman, E.A.; Molho, E.S.; Celmins, D.F.; Heckman, S.M.; Dick, R. Subclinical zinc deficiency in Alzheimer’s disease and Parkinson’s disease. Am. J. Alzheimer’s Dis. Other Dement. 2010, 25, 572–575. [Google Scholar] [CrossRef] [PubMed]
- Mocchegiani, E.; Bertoni-Freddari, C.; Marcellini, F.; Malavolta, M. Brain, aging and neurodegeneration: Role of zinc ion availability. Prog. Neurobiol. 2005, 75, 367–390. [Google Scholar] [CrossRef]
- Rivers-Auty, J.; Tapia, V.S.; White, C.S.; Daniels, M.J.D.; Drinkall, S.; Kennedy, P.T.; Spence, H.G.; Yu, S.; Green, J.P.; Hoyle, C.; et al. Zinc Status Alters Alzheimer’s Disease Progression through NLRP3-Dependent Inflammation. J. Neurosci. 2021, 41, 3025–3038. [Google Scholar] [CrossRef]
- Xie, Z.; Wu, H.; Zhao, J. Multifunctional roles of zinc in Alzheimer’s disease. Neurotoxicology 2020, 80, 112–123. [Google Scholar] [CrossRef] [PubMed]
- Pillai, R.; Uyehara-Lock, J.H.; Bellinger, F.P. Selenium and selenoprotein function in brain disorders. IUBMB Life 2014, 66, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Xue, Z.; Zheng, Y.; Li, S.; Zhou, L.; Cao, L.; Zou, Y. Selenium supplementation enhanced the expression of selenoproteins in hippocampus and played a neuroprotective role in LPS-induced neuroinflammation. Int. J. Biol. Macromol. 2023, 234, 123740. [Google Scholar] [CrossRef] [PubMed]
- Conrad, M.; Proneth, B. Selenium: Tracing Another Essential Element of Ferroptotic Cell Death. Cell Chem. Biol. 2020, 27, 409–419. [Google Scholar] [CrossRef] [PubMed]
- Santos, J.R.; Gois, A.M.; Mendonça, D.M.; Freire, M.A. Nutritional status, oxidative stress and dementia: The role of selenium in Alzheimer’s disease. Front. Aging Neurosci. 2014, 6, 206. [Google Scholar] [CrossRef] [PubMed]
- Xiong, S.; Markesbery, W.R.; Shao, C.; Lovell, M.A. Seleno-L-methionine protects against beta-amyloid and iron/hydrogen peroxide-mediated neuron death. Antioxid. Redox Signal. 2007, 9, 457–467. [Google Scholar] [CrossRef] [PubMed]
- Tung, Y.T.; Hsu, W.M.; Wang, B.J.; Wu, S.Y.; Yen, C.T.; Hu, M.K.; Liao, Y.F. Sodium selenite inhibits gamma-secretase activity through activation of ERK. Neurosci. Lett. 2008, 440, 38–43. [Google Scholar] [CrossRef] [PubMed]
- Corcoran, N.M.; Martin, D.; Hutter-Paier, B.; Windisch, M.; Nguyen, T.; Nheu, L.; Sundstrom, L.E.; Costello, A.J.; Hovens, C.M. Sodium selenate specifically activates PP2A phosphatase, dephosphorylates tau and reverses memory deficits in an Alzheimer’s disease model. J. Clin. Neurosci. 2010, 17, 1025–1033. [Google Scholar] [CrossRef] [PubMed]
- Strumylaite, L.; Kregzdyte, R.; Kucikiene, O.; Baranauskiene, D.; Simakauskiene, V.; Naginiene, R.; Damuleviciene, G.; Lesauskaite, V.; Zemaitiene, R. Alzheimer’s Disease Association with Metals and Metalloids Concentration in Blood and Urine. Int. J. Environ. Res. Public. Health 2022, 19, 7309. [Google Scholar] [CrossRef]
- Nampoothiri, M.; John, J.; Kumar, N.; Mudgal, J.; Nampurath, G.K.; Chamallamudi, M.R. Modulatory role of simvastatin against aluminium chloride-induced behavioural and biochemical changes in rats. Behav. Neurol. 2015, 2015, 210169. [Google Scholar] [CrossRef]
- Kawahara, M.; Kato-Negishi, M.; Tanaka, K.I. Dietary Trace Elements and the Pathogenesis of Neurodegenerative Diseases. Nutrients 2023, 15, 2067. [Google Scholar] [CrossRef] [PubMed]
- Doroszkiewicz, J.; Farhan, J.A.; Mroczko, J.; Winkel, I.; Perkowski, M.; Mroczko, B. Common and Trace Metals in Alzheimer’s and Parkinson’s Diseases. Int. J. Mol. Sci. 2023, 24, 15721. [Google Scholar] [CrossRef] [PubMed]
- Yokel, R.A.; O’Callaghan, J.P. An aluminum-induced increase in GFAP is attenuated by some chelators. Neurotoxicol. Teratol. 1998, 20, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Nehru, B.; Anand, P. Oxidative damage following chronic aluminium exposure in adult and pup rat brains. J. Trace Elem. Med. Biol. 2005, 19, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Dhapola, R.; Kumari, S.; Sharma, P.; HariKrishnaReddy, D. Insight into the emerging and common experimental in-vivo models of Alzheimer’s disease. Lab. Anim. Res. 2023, 39, 33. [Google Scholar] [CrossRef] [PubMed]
- Nabi, M.; Tabassum, N. Role of Environmental Toxicants on Neurodegenerative Disorders. Front. Toxicol. 2022, 4, 837579. [Google Scholar] [CrossRef]
- Dey, M.; Singh, R.K. Neurotoxic effects of aluminium exposure as a potential risk factor for Alzheimer’s disease. Pharmacol. Rep. 2022, 74, 439–450. [Google Scholar] [CrossRef] [PubMed]
- Sumathi, T.; Shobana, C.; Thangarajeswari, M.; Usha, R. Protective effect of L-Theanine against aluminium induced neurotoxicity in cerebral cortex, hippocampus and cerebellum of rat brain—Histopathological, and biochemical approach. Drug Chem. Toxicol. 2015, 38, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Maya, S.; Prakash, T.; Madhu, K.D.; Goli, D. Multifaceted effects of aluminium in neurodegenerative diseases: A review. Biomed. Pharmacother. 2016, 83, 746–754. [Google Scholar] [CrossRef]
- Woodruff, T.M.; Thundyil, J.; Tang, S.C.; Sobey, C.G.; Taylor, S.M.; Arumugam, T.V. Pathophysiology, treatment, and animal and cellular models of human ischemic stroke. Mol. Neurodegener. 2011, 6, 11. [Google Scholar] [CrossRef]
- Johnson, V.J.; Sharma, R.P. Aluminum disrupts the pro-inflammatory cytokine/neurotrophin balance in primary brain rotation-mediated aggregate cultures: Possible role in neurodegeneration. Neurotoxicology 2003, 24, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Kihira, T.; Yoshida, S.; Mitani, K.; Yasui, M.; Yase, Y. ALS in the Kii Peninsula of Japan, with special reference to neurofibrillary tangles and aluminum. Neuropathology 1993, 13, 125–136. [Google Scholar] [CrossRef]
- Kawahara, M.; Kato-Negishi, M. Link between Aluminum and the Pathogenesis of Alzheimer’s Disease: The Integration of the Aluminum and Amyloid Cascade Hypotheses. Int. J. Alzheimer’s Dis. 2011, 2011, 276393. [Google Scholar] [CrossRef] [PubMed]
- Haase, C.; Stieler, J.T.; Arendt, T.; Holzer, M. Pseudophosphorylation of tau protein alters its ability for self-aggregation. J. Neurochem. 2004, 88, 1509–1520. [Google Scholar] [CrossRef] [PubMed]
- Vacchi-Suzzi, C.; Kruse, D.; Harrington, J.; Levine, K.; Meliker, J.R. Is Urinary Cadmium a Biomarker of Long-term Exposure in Humans? A Review. Curr. Environ. Health Rep. 2016, 3, 450–458, Erratum in Curr. Environ. Health Rep. 2016, 3, 493–494. [Google Scholar] [CrossRef] [PubMed]
- Branca, J.J.V.; Morucci, G.; Pacini, A. Cadmium-induced neurotoxicity: Still much ado. Neural Regen. Res. 2018, 13, 1879–1882. [Google Scholar] [CrossRef] [PubMed]
- Ali, T.; Khan, A.; Alam, S.I.; Ahmad, S.; Ikram, M.; Park, J.S.; Lee, H.J.; Kim, M.O. Cadmium, an Environmental Contaminant, Exacerbates Alzheimer’s Pathology in the Aged Mice’s Brain. Front. Aging Neurosci. 2021, 13, 650930. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.; Matsushita, M.; Zhang, L.; Wang, H.; Shi, X.; Gu, H.; Xia, Z.; Cui, J.Y. Cadmium exposure modulates the gut-liver axis in an Alzheimer’s disease mouse model. Commun. Biol. 2021, 4, 1398. [Google Scholar] [CrossRef] [PubMed]
- Min, J.Y.; Min, K.B. Blood cadmium levels and Alzheimer’s disease mortality risk in older US adults. Environ. Health 2016, 15, 69. [Google Scholar] [CrossRef]
- Cascella, R.; Cecchi, C. Calcium Dyshomeostasis in Alzheimer’s Disease Pathogenesis. Int. J. Mol. Sci. 2021, 22, 4914. [Google Scholar] [CrossRef]
- Shah, D.; Gsell, W.; Wahis, J.; Luckett, E.S.; Jamoulle, T.; Vermaercke, B.; Preman, P.; Moechars, D.; Hendrickx, V.; Jaspers, T.; et al. Astrocyte calcium dysfunction causes early network hyperactivity in Alzheimer’s disease. Cell Rep. 2022, 40, 111280. [Google Scholar] [CrossRef]
- Chami, M. Calcium Signalling in Alzheimer’s Disease: From Pathophysiological Regulation to Therapeutic Approaches. Cells 2021, 10, 140. [Google Scholar] [CrossRef]
- Baracaldo-Santamaría, D.; Avendaño-Lopez, S.S.; Ariza-Salamanca, D.F.; Rodriguez-Giraldo, M.; Calderon-Ospina, C.A.; Gonzalez-Reyes, R.E.; Nava-Mesa, M.O. Role of Calcium Modulation in the Pathophysiology and Treatment of Alzheimer’s Disease. Int. J. Mol. Sci. 2023, 24, 9067. [Google Scholar] [CrossRef]
- Haraguchi, T.; Ishizu, H.; Kawai, K.; Tanabe, Y.; Uehira, K.; Takehisa, Y.; Terada, S.; Tsuchiya, K.; Ikeda, K.; Kuroda, S. Diffuse neurofibrillary tangles with calcification (a form of dementia): X-ray spectrometric evidence of lead accumulation in calcified regions. Neuroreport 2001, 12, 1257–1260. [Google Scholar] [CrossRef] [PubMed]
- Links, J.M.; Schwartz, B.S.; Simon, D.; Bandeen-Roche, K.; Stewart, W.F. Characterization of toxicokinetics and toxicodynamics with linear systems theory: Application to lead-associated cognitive decline. Environ. Health Perspect. 2001, 109, 361–368. [Google Scholar] [CrossRef]
- Kassy, C.W. Relationship and Accuracy of Urine Lead as an Alternative to Blood Lead Biomarker among Panel Beaters in Enugu Metropolis: Nigeria. Indian J. Occup. Environ. Med. 2023, 27, 351–354. [Google Scholar] [CrossRef]
- Schwartz, B.S.; Stewart, W.F.; Bolla, K.I.; Simon, P.D.; Bandeen-Roche, K.; Gordon, P.B.; Links, J.M.; Todd, A.C. Past adult lead exposure is associated with longitudinal decline in cognitive function. Neurology 2000, 55, 1144–1150, Erratum in Neurology 2001, 56, 283. [Google Scholar] [CrossRef] [PubMed]
- Gong, Z.; Little, A.R., Jr.; el-Fawal, H.; Evans, H.L. Trimethyl lead neurotoxicity in the rat: Changes in glial fibrillary acidic protein (GFAP). Arh. Hig. Rada Toksikol. 1995, 46, 381–390. [Google Scholar] [PubMed]
- Grandjean, P.; Nielsen, T. Organolead compounds: Environmental health aspects. Residue Rev. 1979, 72, 97–148. [Google Scholar] [CrossRef]
- Gutowski, M.; Altmann, L.; Sveinsson, K.; Wiegand, H. Synaptic plasticity in the CA1 and CA3 hippocampal region of pre- and postnatally lead-exposed rats. Toxicol. Lett. 1998, 95, 195–203. [Google Scholar] [CrossRef]
- Harry, G.J.; Schmitt, T.J.; Gong, Z.; Brown, H.; Zawia, N.; Evans, H.L. Lead-induced alterations of glial fibrillary acidic protein (GFAP) in the developing rat brain. Toxicol. Appl. Pharmacol. 1996, 139, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Niklowitz, W.J. Neurofibrillary changes after acute experimental lead poisoning. Neurology 1975, 25, 927–934. [Google Scholar] [CrossRef] [PubMed]
- Selvín-Testa, A.; Loidl, C.F.; López-Costa, J.J.; López, E.M.; Pecci-Saavedra, J. Chronic lead exposure induces astrogliosis in hippocampus and cerebellum. Neurotoxicology 1994, 15, 389–401. [Google Scholar] [PubMed]
- Brian SSchwartz Karen IBolla Walter Stewart, D. Patrick Ford, Jacqueline Agnew, Howard Frumkin, Decrements in Neurobehavioral Performance Associated with Mixed Exposure to Organic and Inorganic Lead. Am. J. Epidemiol. 1993, 137, 1006–1021. [Google Scholar] [CrossRef]
- Haraguchi, T.; Ishizu, H.; Takehisa, Y.; Kawai, K.; Yokota, O.; Terada, S.; Tsuchiya, K.; Ikeda, K.; Morita, K.; Horike, T.; et al. Lead content of brain tissue in diffuse neurofibrillary tangles with calcification (DNTC): The possibility of lead neurotoxicity. Neuroreport 2001, 12, 3887–3890, Erratum in Neuroreport 2002, 13, 3887–3890. [Google Scholar] [CrossRef] [PubMed]
- Bakulski, K.M.; Seo, Y.A.; Hickman, R.C.; Brandt, D.; Vadari, H.S.; Hu, H.; Park, S.K. Heavy Metals Exposure and Alzheimer’s Disease and Related Dementias. J. Alzheimer’s Dis. 2020, 76, 1215–1242. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.C.; Gao, Z.Y.; Wang, J.; Wu, M.Q.; Hu, S.; Chen, F.; Liu, J.X.; Pan, H.; Yan, C.H. Lead exposure induces alzheimers’s disease (ad)-like pathology and disturbes cholesterol metabolism in the young rat brain. Toxicol. Lett. 2018, 296, 173–183. [Google Scholar] [CrossRef]
- Ashok, A.; Rai, N.K.; Tripathi, S.; Bandyopadhyay, S. Exposure to as-, cd-, and pb-mixture induces abeta, amyloidogenic app processing and cognitive impairments via oxidative stress-dependent neuroinflammation in young rats. Toxicol. Sci. 2015, 143, 64–80. [Google Scholar] [CrossRef] [PubMed]
- Huat, T.J.; Camats-Perna, J.; Newcombe, E.A.; Valmas, N.; Kitazawa, M.; Medeiros, R. Metal Toxicity Links to Alzheimer’s Disease and Neuroinflammation. J. Mol. Biol. 2019, 431, 1843–1868. [Google Scholar] [CrossRef]
- Kirkland, A.E.; Sarlo, G.L.; Holton, K.F. The Role of Magnesium in Neurological Disorders. Nutrients 2018, 10, 730. [Google Scholar] [CrossRef]
- Maier, J.A.M.; Locatelli, L.; Fedele, G.; Cazzaniga, A.; Mazur, A. Magnesium and the Brain: A Focus on Neuroinflammation and Neurodegeneration. Int. J. Mol. Sci. 2022, 24, 223. [Google Scholar] [CrossRef] [PubMed]
- Chui, D.; Chen, Z.; Yu, J.; Zhang, H.; Wang, W.; Song, Y.; Yang, H.; Zhou, L.; Vink, R.; Nechifor, M. Magnesium in Alzheimer’s disease. In Magnesium in the Central Nervous System; Vink, R., Nechifor, M., Eds.; University of Adelaide Press: Adelaide, Australia, 2011. [Google Scholar]
- Lei, D.Y.; Sun, J. Magnesium may be an effective therapy for Alzheimer’s disease. World J. Psychiatry 2022, 12, 1261–1263. [Google Scholar] [CrossRef] [PubMed]
- Slutsky, I.; Abumaria, N.; Wu, L.J.; Huang, C.; Zhang, L.; Li, B.; Zhao, X.; Govindarajan, A.; Zhao, M.G.; Zhuo, M.; et al. Enhancement of learning and memory by elevating brain magnesium. Neuron 2010, 65, 165–177. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Ruan, Y.T.; Zhao, J.; Yang, Y.W.; Chen, L.P.; Mai, Y.R.; Yu, Q.; Cao, Z.Y.; Liu, F.F.; Liao, W.; et al. Magnesium-L-threonate exhibited a neuroprotective effect against oxidative stress damage in HT22 cells and Alzheimer’s disease mouse model. World J. Psychiatry 2022, 12, 410–424. [Google Scholar] [CrossRef] [PubMed]
- Azar, J.; Yousef, M.H.; El-Fawal, H.A.N.; Abdelnaser, A. Mercury and Alzheimer’s disease: A look at the links and evidence. Metab. Brain Dis. 2021, 36, 361–374. [Google Scholar] [CrossRef] [PubMed]
- Bjørklund, G.; Tinkov, A.A.; Dadar, M.; Rahman, M.M.; Chirumbolo, S.; Skalny, A.V.; Skalnaya, M.G.; Haley, B.E.; Ajsuvakova, O.P.; Aaseth, J. Insights into the Potential Role of Mercury in Alzheimer’s Disease. J. Mol. Neurosci. 2019, 67, 511–533. [Google Scholar] [CrossRef] [PubMed]
- Ehmann, W.; Markesbery, W.; Alauddin, M.; Hossain, T.; Brubaker, E. Brain trace elements in Alzheimer’s disease. Neurotoxicology 1986, 7, 195–206. [Google Scholar] [PubMed]
- Wenstrup, D.; Ehmann, W.D.; Markesbery, W.R. Trace element imbalances in isolated subcellular fractions of Alzheimer’s disease. Brain Res. 1990, 533, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Basun, H.; Forssell, L.; Wetterberg, L.; Winblad, B. Metals and trace elements in plasma and cerebrospinal fluid in normal aging and Alzheimer’s disease. J. Neural Transm. Park. Dis. Dement. Sect. 1991, 3, 231–258. [Google Scholar]
- Paglia, G.; Miedico, O.; Cristofano, A.; Vitale, M.; Angiolillo, A.; Chiaravalle, A.E.; Corso, G.; Costanzo, D. Distinctive Pattern of Serum Elements During the Progression of Alzheimer’s Disease. Sci. Rep. 2016, 6, 22769. [Google Scholar] [CrossRef]
- Paduraru, E.; Iacob, D.; Rarinca, V.; Rusu, A.; Jijie, R.; Ilie, O.D.; Ciobica, A.; Nicoara, M.; Doroftei, B. Comprehensive Review Regarding Mercury Poisoning and Its Complex Involvement in Alzheimer’s Disease. Int. J. Mol. Sci. 2022, 23, 1992. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.A.; Hannan, M.A.; Uddin, M.J.; Rahman, M.S.; Rashid, M.M.; Kim, B. Exposure to Environmental Arsenic and Emerging Risk of Alzheimer’s Disease: Perspective Mechanisms, Management Strategy, and Future Directions. Toxics 2021, 9, 188. [Google Scholar] [CrossRef] [PubMed]
- Salkov, V.N.; Voronkov, D.N.; Khudoerkov, R.M. Rol’ rtuti i mysh’yaka v etiologii i patogeneze boleznei Parkinsona i Al’tsgeimera [The role of mercury and arsenic in the etiology and pathogenesis of Parkinson’s and Alzheimer’s diseases]. Arkhiv. Patol. 2022, 84, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.; Abubakar, M.D.; Bisht, P.; Rachamalla, M.; Kumar, A.; Murti, K.; Ravichandiran, V.; Kumar, M. Arsenic Exposure and Amyloid Precursor Protein Processing: A Focus on Alzheimer’s Disease. Curr. Mol. Pharmacol. 2023. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, M.K.; Kartawy, M.; Ginzburg, S.; Amal, H. Arsenic alters nitric oxide signaling similar to autism spectrum disorder and Alzheimer’s disease-associated mutations. Transl. Psychiatry 2022, 12, 127. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, P.; Brockman, J.D.; Wang, Y.; Schneider, J.A.; Morris, M.C. Brain Bromine Levels Associated with Alzheimer’s Disease Neuropathology. J. Alzheimer’s Dis. 2020, 73, 327–332. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Zeng, L.; Li, Y.; Shi, C.; Peng, Y.; Pan, R.; Huang, M.; Wang, S.; Zhang, J.; Li, H. Decabromodiphenyl ethane induces locomotion neurotoxicity and potential Alzheimer’s disease risks through intensifying amyloid-beta deposition by inhibiting transthyretin/transthyretin-like proteins. Environ. Int. 2022, 168, 107482. [Google Scholar] [CrossRef]
- Xu, L.; Qiu, X.; Wang, S.; Wang, Q.; Zhao, X.L. NMDA Receptor Antagonist MK801 Protects Against 1-Bromopropane-Induced Cognitive Dysfunction. Neurosci. Bull. 2019, 35, 347–361. [Google Scholar] [CrossRef]
- Mohideen, S.S.; Ichihara, G.; Ichihara, S.; Nakamura, S. Exposure to 1-bromopropane causes degeneration of noradrenergic axons in the rat brain. Toxicology 2011, 285, 67–71. [Google Scholar] [CrossRef]
- Vincent, J.B.; Lukaski, H.C. Chromium. Adv. Nutr. 2018, 9, 505–506. [Google Scholar] [CrossRef]
- Akhtar, A.; Dhaliwal, J.; Saroj, P.; Uniyal, A.; Bishnoi, M.; Sah, S.P. Chromium picolinate attenuates cognitive deficit in ICV-STZ rat paradigm of sporadic Alzheimer’s-like dementia via targeting neuroinflammatory and IRS-1/PI3K/AKT/GSK-3β pathway. Inflammopharmacology 2020, 28, 385–400. [Google Scholar] [CrossRef] [PubMed]
- Chib, S.; Singh, S. Manganese and related neurotoxic pathways: A potential therapeutic target in neurodegenerative diseases. Neurotoxicol. Teratol. 2022, 94, 107124. [Google Scholar] [CrossRef]
- Mezzaroba, L.; Alfieri, D.F.; Colado Simão, A.N.; Vissoci Reiche, E.M. The role of zinc, copper, manganese and iron in neurodegenerative diseases. Neurotoxicology 2019, 74, 230–241. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Yang, X. The Essential Element Manganese, Oxidative Stress, and Metabolic Diseases: Links and Interactions. Oxidative Med. Cell. Longev. 2018, 2018, 7580707. [Google Scholar] [CrossRef] [PubMed]
- Reyes, M.B.; Martínez-Oyanedel, J.; Navarrete, C.; Mardones, E.; Martinez, I.; Salas, M.; Lopez, V.; Garcia-Robles, M.; Tarifeno-Saldivia, E.; Figueroa, M.; et al. Insights into the Mn2+ Binding Site in the Agmatinase-Like Protein (ALP): A Critical Enzyme for the Regulation of Agmatine Levels in Mammals. Int. J. Mol. Sci. 2020, 21, 4132. [Google Scholar] [CrossRef] [PubMed]
- Peres, T.V.; Schettinger, M.R.; Chen, P.; Carvalho, F.; Avila, D.S.; Bowman, A.B.; Aschner, M. Manganese-induced neurotoxicity: A review of its behavioral consequences and neuroprotective strategies. BMC Pharmacol. Toxicol. 2016, 17, 57. [Google Scholar] [CrossRef]
- Erikson, K.M.; John, C.E.; Jones, S.R.; Aschner, M. Manganese accumulation in striatum of mice exposed to toxic doses is dependent upon a functional dopamine transporter. Environ. Toxicol. Pharmacol. 2005, 20, 390–394. [Google Scholar] [CrossRef] [PubMed]
- Soto-Verdugo, J.; Ortega, A. Critical Involvement of Glial Cells in Manganese Neurotoxicity. Biomed. Res. Int. 2021, 2021, 1596185. [Google Scholar] [CrossRef] [PubMed]
- Martins, A.C., Jr.; Gubert, P.; Villas Boas, G.R.; Paes, M.M.; Santamaria, A.; Lee, E.; Tinkov, A.A.; Bowman, A.B.; Aschner, M. Manganese-induced neurodegenerative diseases and possible therapeutic approaches. Expert. Rev. Neurother. 2020, 20, 1109–1121. [Google Scholar] [CrossRef]
- Guilarte, T.R.; Yeh, C.L.; McGlothan, J.L.; Perez, J.; Finley, P.; Zhou, Y.; Wong, D.F.; Dydak, U.; Schneider, J.S. PET imaging of dopamine release in the frontal cortex of manganese-exposed non-human primates. J. Neurochem. 2019, 150, 188–201. [Google Scholar] [CrossRef]
- Anyachor, C.P.; Dooka, D.B.; Orish, C.N.; Amadi, C.N.; Bocca, B.; Ruggieri, F.; Senofonte, M.; Frazzoli, C.; Orisakwe, O.E. Mechanistic considerations and biomarkers level in nickel-induced neurodegenerative diseases: An updated systematic review. IBRO Neurosci. Rep. 2022, 13, 136–146. [Google Scholar] [CrossRef] [PubMed]
- Gorantla, N.V.; Das, R.; Balaraman, E.; Chinnathambi, S. Transition metal nickel prevents Tau aggregation in Alzheimer’s disease. Int. J. Biol. Macromol. 2020, 156, 1359–1365. [Google Scholar] [CrossRef] [PubMed]
- Benoit, S.L.; Maier, R.J. The nickel-chelator dimethylglyoxime inhibits human amyloid beta peptide in vitro aggregation. Sci. Rep. 2021, 11, 6622. [Google Scholar] [CrossRef] [PubMed]
- Kısa, D.; Korkmaz, N.; Taslimi, P.; Tuzun, B.; Tekin, S.; Karadag, A.; Sen, F. Bioactivity and molecular docking studies of some nickel complexes: New analogues for the treatment of Alzheimer, glaucoma and epileptic diseases. Bioorg. Chem. 2020, 101, 104066. [Google Scholar] [CrossRef] [PubMed]
- Amerikanou, C.; Kleftaki, S.A.; Karavoltsos, S.; Tagkouli, D.; Sakellari, A.; Valsamidou, E.; Gioxari, A.; Kalogeropoulos, N.; Kaliora, A.C. Vanadium, cobalt, zinc, and rubidium are associated with markers of inflammation and oxidative stress in a Greek population with obesity. Front. Endocrinol. 2023, 14, 1265310. [Google Scholar] [CrossRef] [PubMed]
- Roberts, B.R.; Doecke, J.D.; Rembach, A.; Yevenes, L.F.; Fowler, C.J.; McLean, C.A.; Lind, M.; Volitakis, I.; Masters, C.L.; Bush, A.I.; et al. Rubidium and potassium levels are altered in Alzheimer’s disease brain and blood but not in cerebrospinal fluid. Acta Neuropathol. Commun. 2016, 4, 119. [Google Scholar] [CrossRef] [PubMed]
- Cornett, C.R.; Ehmann, W.D.; Wekstein, D.R.; Markesbery, W.R. Trace elements in Alzheimer’s disease pituitary glands. Biol. Trace Elem. Res. 1998, 62, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Purdey, M. Elevated levels of ferrimagnetic metals in foodchains supporting the Guam cluster of neurodegeneration: Do metal nucleated crystal contaminants [corrected] evoke magnetic fields that initiate the progressive pathogenesis of neurodegeneration? Med. Hypotheses 2004, 63, 793–809, Erratum in Med. Hypotheses 2005, 65, 1207. [Google Scholar] [CrossRef] [PubMed]
- Sadeghi, L.; Marefat, A. Investigation of the Iron Oxide Nanoparticle Effects on Amyloid Precursor Protein Processing in Hippocampal Cells. Basic Clin. Neurosci. 2023, 14, 203–212. [Google Scholar] [CrossRef]
- Wang, J.; Fu, J.; Zhao, Y.; Liu, Q.; Yan, X.; Su, J. Iron and Targeted Iron Therapy in Alzheimer’s Disease. Int. J. Mol. Sci. 2023, 24, 16353. [Google Scholar] [CrossRef]
- Roy, R.G.; Mandal, P.K.; Maroon, J.C. Oxidative Stress Occurs Prior to Amyloid Aβ Plaque Formation and Tau Phosphorylation in Alzheimer’s Disease: Role of Glutathione and Metal Ions. ACS Chem. Neurosci. 2023, 14, 2944–2954. [Google Scholar] [CrossRef] [PubMed]
- Squitti, R.; Pal, A.; Picozza, M.; Avan, A.; Ventriglia, M.; Rongioletti, M.C.; Hoogenraad, T. Zinc Therapy in Early Alzheimer’s Disease: Safety and Potential Therapeutic Efficacy. Biomolecules 2020, 10, 1164. [Google Scholar] [CrossRef] [PubMed]
- Brewer, G.J.; Kaur, S. Zinc deficiency and zinc therapy efficacy with reduction of serum free copper in Alzheimer’s disease. Int. J. Alzheimer’s Dis. 2013, 2013, 586365. [Google Scholar] [CrossRef] [PubMed]
- Tamtaji, O.R.; Heidari-Soureshjani, R.; Mirhosseini, N.; Kouchaki, E.; Bahmani, F.; Aghadavod, E.; Tajabadi-Ebrahimi, M.; Asemi, Z. Probiotic and selenium co-supplementation, and the effects on clinical, metabolic and genetic status in Alzheimer’s disease: A randomized, double-blind, controlled trial. Clin. Nutr. 2019, 38, 2569–2575. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Wang, N.; Zheng, G.; Yang, L. Oral Administration of Resveratrol-Selenium-Peptide Nanocomposites Alleviates Alzheimer’s Disease-like Pathogenesis by Inhibiting Aβ Aggregation and Regulating Gut Microbiota. ACS Appl. Mater. Interfaces 2021, 13, 46406–46420. [Google Scholar] [CrossRef] [PubMed]
- Antoniadou, F.; Papamitsou, T.; Kavvadas, D.; Kapoukranidou, D.; Sioga, A.; Papaliagkas, V. Toxic Environmental Factors and their Association with the Development of Dementia: A Mini Review on Heavy Metals and Ambient Particulate Matter. Mater. Sociomed. 2020, 32, 299–306. [Google Scholar] [CrossRef] [PubMed]
- Mateo, D.; Marquès, M.; Torrente, M. Metals linked with the most prevalent primary neurodegenerative dementias in the elderly: A narrative review. Environ. Res. 2023, 236, 116722. [Google Scholar] [CrossRef]
- Koski, L.; Ronnevi, C.; Berntsson, E.; Wärmländer, S.K.T.S.; Roos, P.M. Metals in ALS TDP-43 Pathology. Int. J. Mol. Sci. 2021, 22, 12193. [Google Scholar] [CrossRef] [PubMed]
- Peters, T.L.; Beard, J.D.; Umbach, D.M.; Allen, K.; Keller, J.; Mariosa, D.; Sandler, D.P.; Schmidt, S.; Fang, F.; Ye, W.; et al. Blood levels of trace metals and amyotrophic lateral sclerosis. Neurotoxicology 2016, 54, 119–126. [Google Scholar] [CrossRef]
- Pesch, B.; Casjens, S.; Woitalla, D.; Dharmadhikari, S.; Edmondson, D.A.; Zella, M.A.S.; Lehnert, M.; Lotz, A.; Herrmann, L.; Muhlack, S.; et al. Impairment of Motor Function Correlates with Neurometabolite and Brain Iron Alterations in Parkinson’s Disease. Cells 2019, 8, 96. [Google Scholar] [CrossRef]
- Thomas, G.E.C.; Leyland, L.A.; Schrag, A.E.; Lees, A.J.; Acosta-Cabronero, J.; Weil, R.S. Brain iron deposition is linked with cognitive severity in Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 2020, 91, 418–425. [Google Scholar] [CrossRef] [PubMed]
- Guan, X.; Guo, T.; Zhou, C.; Wu, J.; Gao, T.; Bai, X.; Wei, H.; Zhang, Y.; Xuan, M.; Gu, Q.; et al. Asymmetrical nigral iron accumulation in Parkinson’s disease with motor asymmetry: An explorative, longitudinal and test-retest study. Aging 2020, 12, 18622–18634. [Google Scholar] [CrossRef] [PubMed]
- Thomas, G.E.C.; Zarkali, A.; Ryten, M.; Schmueli, K.; Gil-Martinez, A.L.; Leyland, L.A.; McColgan, P.; Acosta-Cabronero, J.; Lees, A.J.; Weil, R.S. Regional brain iron and gene expression provide insights into neurodegeneration in Parkinson’s disease. Brain 2021, 144, 1787–1798. [Google Scholar] [CrossRef] [PubMed]
- Ajsuvakova, O.P.; Tinkov, A.A.; Willkommen, D.; Skalnaya, A.A.; Danilov, A.B.; Pilipovich, A.A.; Aschner, M.; Skalny, A.V.; Michalke, B.; Skalnaya, M.G. Assessment of copper, iron, zinc and manganese status and speciation in patients with Parkinson’s disease: A pilot study. J. Trace Elem. Med. Biol. 2020, 59, 126423. [Google Scholar] [CrossRef] [PubMed]
- Willkommen, D.; Lucio, M.; Schmitt-Kopplin, P.; Gazzaz, M.; Schroeter, M.; Sigaroudi, A.; Michalke, B. Species fractionation in a case-control study concerning Parkinson’s disease: Cu-amino acids discriminate CSF of PD from controls. J. Trace Elem. Med. Biol. 2018, 49, 164–170. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.J.; Oh, S.B.; Kim, J.; Kim, K.; Ryu, H.S.; Kim, M.S.; Ayton, S.; Bush, A.I.; Lee, J.Y.; Chung, S.J. Association of metals with the risk and clinical characteristics of Parkinson’s disease. Park. Relat. Disord. 2018, 55, 117–121. [Google Scholar] [CrossRef] [PubMed]
- dos Santos, A.B.; Kohlmeier, K.A.; Rocha, M.E.; Barreto, G.E.; Barreto, J.A.; de Souza, A.C.A.; Bezerra, M.A. Hair in Parkinson’s disease patients exhibits differences in Calcium, Iron and Zinc concentrations measured by flame atomic absorption spectrometry—FAAS. J. Trace Elem. Med. Biol. 2018, 47, 134–139. [Google Scholar] [CrossRef]
- Dos Santos, A.B.; Bezerra, M.A.; Rocha, M.E.; Barreto, G.E.; Kohlmeier, K.A. Higher zinc concentrations in hair of Parkinson’s disease are associated with psychotic complications and depression. J. Neural Transm. 2019, 126, 1291–1301. [Google Scholar] [CrossRef] [PubMed]
- Dlamini, W.W.; Nelson, G.; Nielsen, S.S.; Racette, B.A. Manganese exposure, parkinsonian signs, and quality of life in South African mine workers. Am. J. Ind. Med. 2020, 63, 36–43. [Google Scholar] [CrossRef]
- Racette, B.A.; Nelson, G.; Dlamini, W.W.; Prathibha, P.; Turner, J.R.; Ushe, M.; Checkoway, H.; Sheppard, L.; Nielsen, S.S. Severity of parkinsonism associated with environmental manganese exposure. Environ. Health 2021, 20, 27. [Google Scholar] [CrossRef]
- Lee, C.P.; Zhu, C.H.; Su, C.C. Increased prevalence of Parkinson’s disease in soils with high arsenic levels. Park. Relat. Disord. 2021, 88, 19–23. [Google Scholar] [CrossRef] [PubMed]
- Pamphlett, R.; Bishop, D.P. Mercury is present in neurons and oligodendrocytes in regions of the brain affected by Parkinson’s disease and co-localises with Lewy bodies. PLoS ONE 2022, 17, e0262464. [Google Scholar] [CrossRef] [PubMed]
- Paul, K.C.; Horvath, S.; Del Rosario, I.; Bronstein, J.M.; Ritz, B. DNA methylation biomarker for cumulative lead exposure is associated with Parkinson’s disease. Clin. Epigenet. 2021, 13, 59. [Google Scholar] [CrossRef] [PubMed]
- Bang, J.; Spina, S.; Miller, B.L. Frontotemporal dementia. Lancet 2015, 386, 1672–1682. [Google Scholar] [CrossRef] [PubMed]
- Squitti, R.; Fostinelli, S.; Siotto, M.; Ferrari, C.; Binetti, G.; Benussi, L.; Rongioletti, M.; Ghidoni, R. Serum Copper is not Altered in Frontotemporal Lobar Degeneration. J. Alzheimer’s Dis. 2018, 63, 1427–1432. [Google Scholar] [CrossRef]
- Adani, G.; Filippini, T.; Garuti, C.; Malavolti, M.; Vinceti, G.; Zamboni, G.; Tondelli, M.; Galli, C.; Costa, M.; Vincenti, M.; et al. Environmental Risk Factors for Early-Onset Alzheimer’s Dementia and Frontotemporal Dementia: A Case-Control Study in Northern Italy. Int. J. Environ. Res. Public. Health 2020, 17, 7941. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Concentrations of Trace Elements in Alzheimer’s Disease | |||
|---|---|---|---|
| Element | Brain Tissue | Blood | Other (Cerebrospinal Fluid/Urine/Hair/Nails) |
| Iron | ↑ In vivo | ↓ | ↑ Ferritin in cerebrospinal fluid |
| Copper | Deficient in the brain tissue cells, increased in the extracellular plaques | ↑ | n/a |
| Zinc | ↓ | ↓ | n/a |
| Selenium | ↓ | ↓ | n/a |
| Aluminium | ↑ | ↑ | n/a |
| Cadmium | ↑ | ↑ | ↑ urine |
| Calcium | Ca2+ signalling disturbance | n/a | n/a |
| Lead | ↑ | ↑ | ↑ urine |
| Magnesium | ↓ | ↓ | ↓ Cerebrospinal fluid |
| Mercury | ↑ | ↑ ↓ | ↓ Hair and nails |
| Arsenic | ↑ | n/a | ↑ Urine, hair and nails |
| Bromine | ↑ | n/a | n/a |
| Manganese | ↑ | ↑ | n/a |
| Nickel | ↑ | ↑ | ↑ Cerebrospinal fluid |
| Rubidium | ↓ | ↓ | ↓ Cerebrospinal fluid |
| Strontium | ↑ | ↑ | n/a |
| Chromium | ↑ | ↑ | n/a |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tyczyńska, M.; Gędek, M.; Brachet, A.; Stręk, W.; Flieger, J.; Teresiński, G.; Baj, J. Trace Elements in Alzheimer’s Disease and Dementia: The Current State of Knowledge. J. Clin. Med. 2024, 13, 2381. https://doi.org/10.3390/jcm13082381
Tyczyńska M, Gędek M, Brachet A, Stręk W, Flieger J, Teresiński G, Baj J. Trace Elements in Alzheimer’s Disease and Dementia: The Current State of Knowledge. Journal of Clinical Medicine. 2024; 13(8):2381. https://doi.org/10.3390/jcm13082381
Chicago/Turabian StyleTyczyńska, Magdalena, Marta Gędek, Adam Brachet, Wojciech Stręk, Jolanta Flieger, Grzegorz Teresiński, and Jacek Baj. 2024. "Trace Elements in Alzheimer’s Disease and Dementia: The Current State of Knowledge" Journal of Clinical Medicine 13, no. 8: 2381. https://doi.org/10.3390/jcm13082381