
Carbon Monoxide Poisoning: From Occupational Health to Emergency Medicine

 , ,
, ,  , , ,
, , ,
Abstract
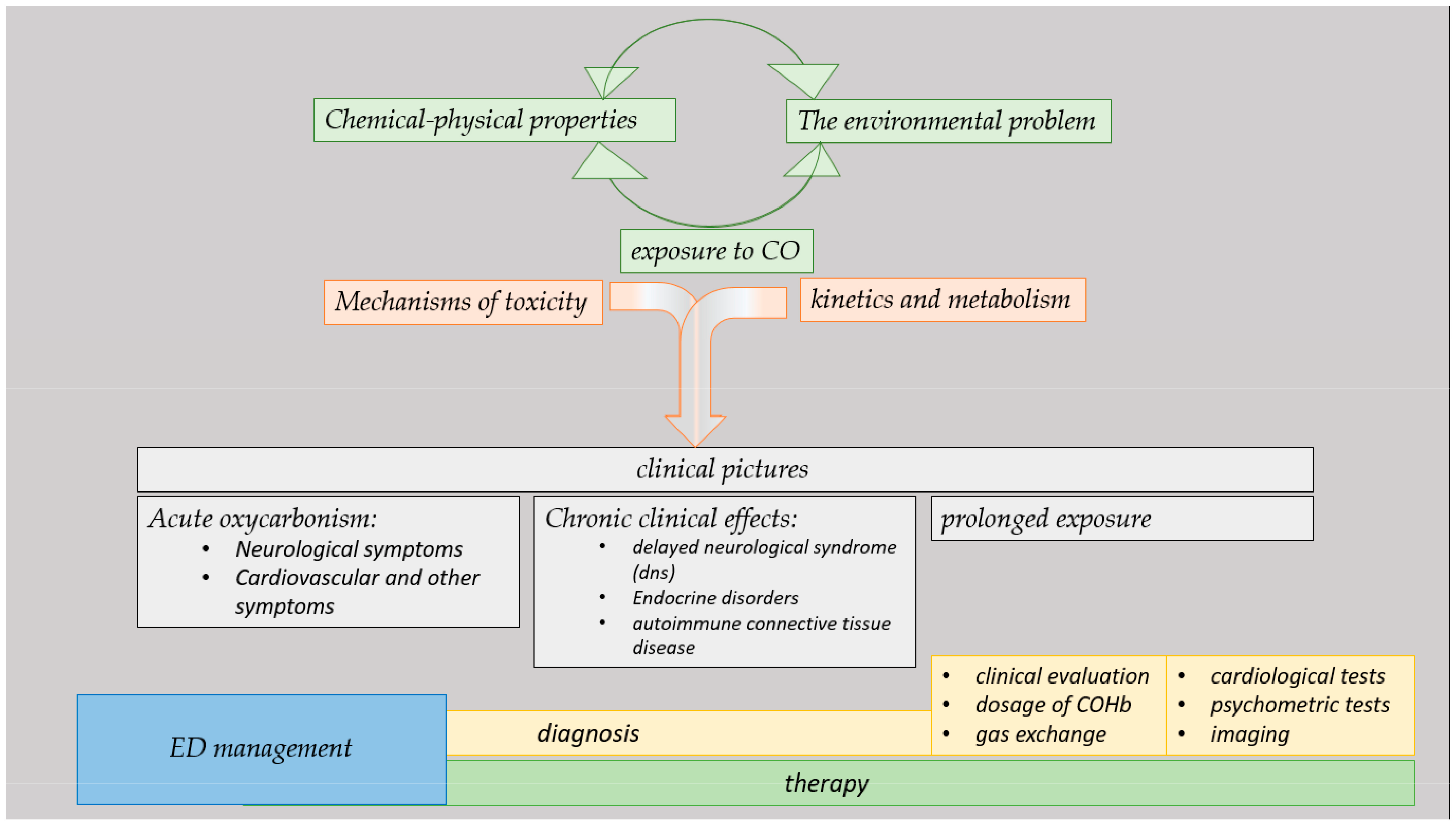
:1. Introduction
2. Materials and Methods
3. Results
3.1. Chemical–Physical Properties and Exposure
3.2. The Environmental Problem
3.3. Kinetics and Metabolism
3.4. Mechanisms of Toxicity
3.5. Clinical Pictures
3.6. Acute Oxycarbonism
3.6.1. Neurological Symptoms
3.6.2. Cardiovascular and Other Symptoms
3.7. Presentation and Management in EDs
3.8. Chronic Clinical Effects
3.9. Delayed Neurological Syndrome (DNS)
3.10. Endocrine Disorders
3.11. Autoimmune Connective Tissue Disease
3.12. Effects of Prolonged Exposure
3.13. Diagnosis
3.13.1. Clinical Evaluation
3.13.2. Dosage of COHb: The Fundamental Diagnostic Test
3.13.3. Arterial Hemogram Analysis: Evaluation of Gas Exchange
3.13.4. The Role of Blood Chemistry Tests
3.13.5. Recommended Cardiological Tests
3.13.6. Psychometric Tests: Post-Interval Syndrome
3.13.7. The Role of Brain Imaging
3.13.8. Differential Diagnosis and the Evaluation of Associated Intoxications
3.14. Therapy
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Megas, I.F.; Beier, J.P.; Grieb, G. The History of Carbon Monoxide Intoxication. Medicina 2021, 57, 400. [Google Scholar] [CrossRef] [PubMed]
- Ehrenfreund, P.; Spaans, M.; Holm, N.G. The evolution of organic matter in space. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 2011, 369, 538–554. [Google Scholar] [CrossRef] [PubMed]
- Abel, T.; Bryan, G.L.; Norman, M.L. The Formation of the first star in the universe. Science 2002, 295, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Cruickshank, W. Some observations on different hydrocarbonates and combinations of carbone with oxygen, ect. In reply to some of Dr. Priestley’s late objections to the new system of chemistry. J. Nat. Philos. Chem. Arts 1801, 5, 1–9. [Google Scholar]
- Petrolini, V.; Bigi, S.; Vecchio, S.; Lonati, D.; Giampreti, A.; Locatelli, C.; Butera, R.; Manzo, L. Il monossido di carbonio: “killer silenzioso” e “grande imitatore”. Emerg. Care J. 2008, 4, 6–13. [Google Scholar] [CrossRef]
- Kim, H.H.; Choi, S. Therapeutic Aspects of Carbon Monoxide in Cardiovascular Disease. Int. J. Mol. Sci. 2018, 19, 2381. [Google Scholar] [CrossRef] [PubMed]
- ATSDR, Agency for Toxic Substances and Disease Registry. Toxicological Profile for Carbon Monoxide; US Department of Health and Human Services, Public Health Service: Atlanta, GA, USA, 2012. [Google Scholar]
- Candura, F.; Candura, S.M. Elementi di Tecnologia Industriale a Uso Dei Cultori di Medicina Del Lavoro; CELT: Piacenza, Italy, 2002. [Google Scholar]
- Chenoweth, J.A.; Albertson, T.E.; Greer, M.R. Carbon Monoxide Poisoning. Crit. Care Clin. 2021, 37, 657–672. [Google Scholar] [CrossRef]
- Candura, S.M.; Verni, P.; Minelli, C.M.; Rosso, G.L.; Cappelli, M.I.; Strambi, S.; Martellosio, V. Rischi professionali nelle Forze dell’Ordine. G. Ital. Med. Lav. Erg. 2006, 28, 53–62. [Google Scholar]
- Sancini, A.; Tomei, F.T.G.; Caciari, T.; Di Giorgio, V.; Andrè, J.C.; Palermmo, P.; Andreozzi, G.; Nardone, N.; Schifano, M.P.; Fiaschetti, M.; et al. Urban pollution. G. Ital. Med. Lav. Erg. 2012, 34, 187–196. [Google Scholar]
- Bernard, C. Leçon Sur Les Effects des Substances Toxiques et Medicamenteuses. Baillière et Fils: Paris, France, 1857. Available online: https://gallica.bnf.fr/ark:/12148/bpt6k773289.texteImage (accessed on 15 January 2024).
- Haldane, J.S. The reation of the action of catabolic oxide to O2 tension. J. Physiol. 1895, 18, 201–217. [Google Scholar] [CrossRef]
- Chang, S.S.; Chen, Y.Y.; Yip, P.S.F.; Lee, W.J.; Hagihara, A.; Gunnell, D. Regional changes in charcoal-burning suicide rates in East/South East Asia from 1995 to 2011: A time trend analysis. PLoS Med. 2014, 11, e1001622. [Google Scholar] [CrossRef] [PubMed]
- Valent, F.; McGwin, G., Jr.; Bovenzi, M.; Barbone, F. Fatal work-related inhalation of harmful substances in the United States. Chest 2002, 121, 969–975. [Google Scholar] [CrossRef] [PubMed]
- Byard, R.W. Commercial fishing industry deaths—Forensic issues. J. Forensic Leg. Med. 2013, 20, 129–132. [Google Scholar] [CrossRef] [PubMed]
- McDermott, J.H.; Reynard, C.; Perry, J.; Dear, J.W.; Child, F.; Jenner, R. Acute carbon monoxide toxicity in a paediatric cohort: Analysis of 10 boys poisoned during a scuba diving lesson. Clin. Toxicol. 2018, 56, 856–859. [Google Scholar] [CrossRef] [PubMed]
- Allen, H. Carbon monoxide poisoning in a diver. Arch. Emerg. Med. J. 1992, 9, 65–66. [Google Scholar] [CrossRef] [PubMed]
- Tomaszewski, C. Carbon monoxide. In Goldfrank’s Toxicologic Emergencies, 8th ed.; Flomenbaum, N.E., Howland, M.A., Goldfrank, L.R., Lewin, N.A., Hoffman, R.S., Nelson, L.S., Eds.; McGraw-Hill: New York, NY, USA, 2006. [Google Scholar]
- Kales, S.N. Carbon monoxide intoxication. Am. Fam. Physician 1993, 48, 1100–1104. [Google Scholar]
- Pelham, T.W.; Holt, L.E.; Moss, M.A. Exposure to carbon monoxide and nitrogen dioxide in enclosed ice arenas. Occup. Environ. Med. 2002, 59, 224–233. [Google Scholar] [CrossRef] [PubMed]
- Locatelli, C.; Candura, S.M.; Maccarini, D.; Butera, R.; Manzo, L. Carbon monoxide poisoning in fire victims. Indoor Environ. 1994, 3, 16–21. [Google Scholar] [CrossRef]
- Anseeuw, K.; Delvau, N.; Burillo-Putze, G.; De Iaco, F.; Geldner, G.; Holmstrom, P.; Lambert, Y.; Sabbe, M. Cyanide poisoning by fire smoke inhalation: A European expert consensus. Eur. J. Emerg. Med. 2013, 20, 2–9. [Google Scholar] [CrossRef]
- Huzar, T.F.; George, T.; Cross, J.M. Carbon monoxide and cyanide toxicity: Etiology, pathophysiology and treatment in inhalation injury. Expert Rev. Respir. Med. 2013, 7, 159–170. [Google Scholar] [CrossRef]
- Guzman, J.A. Carbon monoxide poisoning. Crit. Care Clin. 2012, 28, 537–548. [Google Scholar] [CrossRef] [PubMed]
- Coburn, R.F.; Blakemore, W.S.; Forster, R.E. Endogenous carbon monoxide production in man. J. Clin. Investig. 1963, 42, 1172–1178. [Google Scholar] [CrossRef] [PubMed]
- Sjöstrand, T. Endogenous formation of carbon monoxide in man under normal and pathological conditions. Scand. J. Clin. Lab. Investig. 1949, 1, 201–214. [Google Scholar] [CrossRef]
- Sjöstrand, T. Endogenous formation of carbon monoxide. the co concentration in the inspired and expired air of hospital patients. Acta Physiol. Scand. 1951, 22, 137–141. [Google Scholar] [CrossRef] [PubMed]
- Baranano, D.E.; Ferris, C.D.; Snyder, S.H. Atypical neural messengers. Trends Neurosci. 2001, 24, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Eichhorn, L.; Thudium, M.; Jüttner, B. The Diagnosis and Treatment of Carbon Monoxide Poisoning. Dtsch. Arztebl. Int. 2018, 115, 863–870. [Google Scholar] [CrossRef] [PubMed]
- Widdop, B. Analysis of carbon monoxide. Ann. Clin. Biochem. 2002, 39, 378–391. [Google Scholar] [CrossRef] [PubMed]
- Nager, E.C.; O’Connor, R.E. Carbon monoxide poisoning from spray paint inhalation. Acad. Emerg. Med. 1998, 5, 84–86. [Google Scholar] [CrossRef]
- Zegdi, R.; Perrin, D.; Burdin, M.; Boiteau, R.; Tenaillon, A. Increased endogenous carbon monoxide production in severe sepsis. Intensiv. Care Med. 2002, 28, 793–796. [Google Scholar] [CrossRef]
- Marks, G.S.; Vreman, H.J.; McLaughlin, B.E.; Brien, J.F.; Nakatsu, K. Measurement of endogenous carbon monoxide formation in biological systems. Antioxid. Redox Signal. 2002, 4, 271–277. [Google Scholar] [CrossRef]
- Ahmed, H.; McLaughlin, B.E.; Soong, J.; Marks, G.S.; Brien, J.F.; Nakatsu, K. The source of endogenous carbon monoxide formation in human placental chorionic villi. Cell Mol. Biol. 2005, 51, 447–451. [Google Scholar] [PubMed]
- McLaughlin, B.E.; Lash, G.E.; Graham, C.H.; Smith, G.N.; Vreman, H.J.; Stevenson, D.K.; Marks, G.S.; Nakatsu, K.; Brien, J.F. Endogenous carbon monoxide formation by chorionic villi of term human placenta. Placenta 2001, 22, 886–888. [Google Scholar] [CrossRef] [PubMed]
- Fenn, W.O. The burning of CO in tissues. Ann. N. Y. Acad. Sci. 1970, 174, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Peterson, J.E.; Stewart, R.D. Absorption and elimination of carbon monoxide by inactive young man. Arch. Environ. Health 1970, 21, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, F.O.; Scott, M.G. The effect of CO on tissue respiration. Am. J. Physiol. 1934, 107, 85–93. [Google Scholar] [CrossRef]
- Nañagas, K.A.; Penfound, S.J.; Kao, L.W. Carbon Monoxide Toxicity. Emerg. Med. Clin. N. Am. 2022, 40, 283–312. [Google Scholar] [CrossRef] [PubMed]
- Kao, L.W.; Nañagas, K.A. Toxicity associated with carbon monoxide. Clin. Lab. Med. 2006, 26, 99–125. [Google Scholar] [CrossRef] [PubMed]
- Raub, J.A.; Mathieu-Nolf, M.; Hampson, N.B.; Thom, S.R. Carbon monoxide poisoning—A public health perspective. Toxicology 2000, 145, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Boehning, D.; Moon, C.; Sharma, S.; Hurt, K.; Hester, L.D.; Ronnett, G.V.; Shugar, D.; Snyder, S.H. Carbon monoxide neurotransmission activated by Ck2 phosphorylation of heme oxygenase-2. Neuron 2003, 40, 129–137. [Google Scholar] [CrossRef]
- Mannaioni, P.F.; Vannacci, A.; Masini, E. Carbon monoxide: The bad and the good side of the coin, from neuronal death to anti-inflammatory activity. Inflamm. Res. 2006, 55, 261–273. [Google Scholar] [CrossRef]
- Zhang, X.; Shan, P.; Otterbein, L.E.; Alam, J.; Flavell, R.A.; Davis, R.J.; Choi, A.M.K.; Lee, P.J. Carbon monoxide inhibition of apoptosis during ischemia-reperfusion lung injury is dependent on the p38 mitogen-activated protein kinase pathway and involves caspase 3. J. Biol. Chem. 2003, 278, 1248–1258. [Google Scholar] [CrossRef] [PubMed]
- Taillé, C.; Almolki, A.; Benhamed, M.; Zedda, C.; Mégret, J.; Berger, P.; Lesèche, G.; Fadel, E.; Yamaguchi, T.; Marthan, R.; et al. Heme oxygenase inhibits human airway smooth muscle proliferation via a bilirubin-dependent modulation of erk1/2 phosphorylation. J. Biol. Chem. 2003, 278, 27160–27168. [Google Scholar] [CrossRef] [PubMed]
- Douglas, C.G.; Haldane, J.S.; Haldane, J.B. The laws of combination of hæmoglobin with carbon monoxide and oxygen. J. Physiol. 1912, 44, 275–304. [Google Scholar] [CrossRef]
- Haldane, J.S. A Lecture on the Symptoms, Causes, and Prevention of Anoxaemia (Insufficient Supply of Oxygen to the Tissues), and the Value of Oxygen in its Treatment. Br. Med. J. 1919, 2, 65–71. [Google Scholar] [CrossRef]
- Haldane, J. The supposed oxidation of carbonic oxide in the living body. J. Physiol. 1900, 25, 225–229. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.L. The Pathology of Gas Poisoning, Illustrated by Five Recent Cases. Br. Med. J. 1899, 1, 780–781. [Google Scholar] [CrossRef] [PubMed]
- Haldane, J. Medicolegal contributions of historical interest. The action of carbonic oxide on man. Forensic Sci. 1972, 1, 451–483. [Google Scholar] [CrossRef] [PubMed]
- Llano, A.L.; Raffin, T.A. Management of carbon monoxide poisoning. Chest 1990, 97, 165–169. [Google Scholar] [CrossRef] [PubMed]
- Sendroy, J.; Liu, S.H.; Van Slyke, D.O. The gasometric estimation of the relative affinity constant for carbon monoxide and oxygen in whole blood at 38C. Am. J. Physiol. 1929, 90, 511–512. [Google Scholar]
- Roughton, F.J.W.; Darling, R.C.; Crocker, G.H.; Toth, B.; Jones, J.H.; Agostoni, P.; Swenson, E.R.; Bussotti, M.; Revera, M.; Meriggi, P.; et al. The effect of carbon monoxide on the oxyhemoglobin dissociation curve. Am. J. Physiol. Content 1944, 141, 17–31. [Google Scholar] [CrossRef]
- Sladen, R.N. The oxyhemoglobin dissociation curve. Int. Anesthesiol. Clin. 1981, 19, 39–70. [Google Scholar] [CrossRef]
- Orellano, T.; Dergal, E.; Alijani, M.; Briggs, C.; Vasquez, J.; Goldbaum, L.R.; Absolon, K.B. Studies on the mechanism of carbon monoxide toxicity. J. Surg. Res. 1976, 20, 485–487. [Google Scholar] [CrossRef]
- Goldbaum, L.R.; Ramirez, R.G.; Absalon, K.B. What is the mechanism of carbon monoxide toxicity? Aviat. Space Environ. Med. 1975, 46, 1289–1291. [Google Scholar] [PubMed]
- Alonso, J.R.; Cardellach, F.; López, S.; Casademont, J.; Miró, Ò. Carbon monoxide specifically inhibits cytochrome c oxidase of human mitochondrial respiratory chain. Pharmacol. Toxicol. 2003, 93, 142–146. [Google Scholar] [CrossRef]
- Piantadosi, C.A.; Zhang, J.; Levin, E.D.; Folz, R.J.; Schmechel, D.E. Apoptosis and delayed neuronal damage after carbon monoxide poisoning in the rat. Exp. Neurol. 1997, 147, 103–114. [Google Scholar] [CrossRef] [PubMed]
- Thom, S.R.; Bhopale, V.M.; Han, S.T.; Clark, J.M.; Hardy, K.R. Intravascular neutrophil activation due to carbon monoxide poisoning. Am. J. Respir. Crit. Care Med. 2006, 174, 1239–1248. [Google Scholar] [CrossRef] [PubMed]
- Chin, B.Y.; Jiang, G.; Wegiel, B.; Wang, H.J.; MacDonald, T.; Zhang, X.C.; Gallo, D.; Cszimadia, E.; Bach, F.H.; Lee, P.J.; et al. Hypoxia-inducible factor 1α stabilization by carbon monoxide results in cytoprotective preconditioning. Proc. Natl. Acad. Sci. USA 2007, 104, 5109–5114. [Google Scholar] [CrossRef]
- Thorn, S.R.; Keim, L.W. Carbon monoxide poisoning: A Review epidemiology, pathophysiology, clinical findings, and treatment options including hyperbaric oxygen therapy. J. Toxicol. Clin. Toxicol. 1989, 27, 141–156. [Google Scholar] [CrossRef]
- Cronje, F.J.; Carraway, M.S.; Freiberger, J.J.; Suliman, H.B.; Piantadosi, C.A. Carbon monoxide actuates O2-limited heme degradation in the rat brain. Free Radic. Biol. Med. 2004, 37, 1802–1812. [Google Scholar] [CrossRef]
- Piantadosi, C.A. Toxicity of carbon monoxide: Hemoglobin vs. histotoxic mechanisms. In Carbon Monoxide; Penney, D.G., Ed.; CRC Press: Boca Raton, FL, USA, 1996; pp. 163–186. [Google Scholar]
- Goldbaum, L.R.; Orellano, T.; Dergal, E. Mechanism of the toxic action of carbon monoxide. Ann. Clin. Lab. Sci. 1976, 6, 372–376. [Google Scholar]
- Norkool, D.M.; Kirkpatrick, J.N. Treatment of acute carbon monoxide poisoning with hyperbaric oxygen: A review of 115 cases. Ann. Emerg. Med. 1985, 14, 1168–1171. [Google Scholar] [CrossRef] [PubMed]
- Weaver, L.K. Clinical practice. Carbon monoxide poisoning. N. Engl. J. Med. 2009, 360, 1217–1225. [Google Scholar] [CrossRef] [PubMed]
- Myers, R.A. Carbon monoxide poisoning. J. Emerg. Med. 1984, 1, 245–248. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.D.; Piantadosi, C.A. Recovery of energy metabolism in rat brain after carbon monoxide hypoxia. J. Clin. Investig. 1992, 89, 666–672. [Google Scholar] [CrossRef] [PubMed]
- Rottman, S.J. Carbon monoxide screening in the ED. Am. J. Emerg. Med. 1991, 9, 204–205. [Google Scholar] [CrossRef] [PubMed]
- Hill, B.C. The pathway of CO binding to cytochrome c oxidase Can the gateway be closed? FEBS Lett. 1994, 354, 284–288. [Google Scholar] [CrossRef] [PubMed]
- Chance, B.; Erecinska, M.; Wagner, M. Mitochondrial responses to carbon monoxide toxicity. Ann. N. Y. Acad. Sci. 1970, 174, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Hardy, K.R.; Thom, S.R. Pathophysiology and treatment of carbon monoxide poisoning. J. Toxicol. Clin. Toxicol. 1994, 32, 613–629. [Google Scholar] [CrossRef] [PubMed]
- Thom, S.; Ohnishi, S.; Ischiropoulos, H. Nitric oxide released by platelets inhibits neutrophil b2 integrin function following acute carbon monoxide poisoning. Toxicol. Appl. Pharmacol. 1994, 128, 105–110. [Google Scholar] [CrossRef]
- Florkowski, C.M.; Rossi, M.L.; Carey, M.P.; Poulton, K.; Dickson, G.R.; Ferner, R.E. Rhabdomyolysis and acute renal failure following carbon monoxide poisoning: Two case reports with muscle histopathology and enzyme activities. J. Toxicol. Clin. Toxicol. 1992, 30, 443–454. [Google Scholar] [CrossRef]
- Abdul-Ghaffar, N.U.; Farghaly, M.M.; Swamy, A.S. Acute renal failure, compartment syndrome, and systemic capillary leak syndrome complicating carbon monoxide poisoning. J. Toxicol. Clin. Toxicol. 1996, 34, 713–719. [Google Scholar] [CrossRef] [PubMed]
- Olson, K.R. Carbon monoxide poisoning: Mechanisms, presentation, and controversies in management. J. Emerg. Med. 1984, 1, 233–243. [Google Scholar] [CrossRef] [PubMed]
- DeBias, D.A.; Banerjee, C.M.; Birkhead, N.C.; Greene, C.H.; Scott, S.D.; Harrer, W.V. Effects of carbon monoxide inhalation on ventricular fibrillation. Arch. Environ. Health Int. J. 1976, 31, 42–46. [Google Scholar] [CrossRef] [PubMed]
- Sangalli, B.C.; Bidanset, J.H. A review of carboxymyoglobin formation: A major mechanism of carbon monoxide toxicity. Vet. Hum. Toxicol. 1990, 32, 449–453. [Google Scholar] [PubMed]
- Verma, A.; Hirsch, D.J.; Glatt, C.E.; Ronnett, G.V.; Snyder, S.H. Carbon monoxide: A putative neural messenger. Science 1993, 259, 381–384. [Google Scholar] [CrossRef] [PubMed]
- Coceani, F. Carbon monoxide and dilation of blood vessels. Science 1993, 260, 739. [Google Scholar] [CrossRef] [PubMed]
- Barinaga, M. Carbon monoxide: Killer to brain messenger in one step. Science 1993, 259, 309. [Google Scholar] [CrossRef] [PubMed]
- Fichtner, A.; Eichhorn, L. Kohlenmonoxidintoxikation—Neue Aspekte und aktuelle leitlinienbasierte Empfehlungen [Carbon monoxide intoxication-New aspects and current guideline-based recommendations]. Anaesthesiologie 2022, 71, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Butera, R.; Candura, S.M.; Locatelli, C.; Varango, C.; Li, B.; Manzo, L. Neurological sequelae of carbon monoxide poisoning: Role of hyperbaric oxigen. Indorr. Environ. 1995, 4, 134–139. [Google Scholar]
- Yang, Z.D. Observation of hyperbaric oxigen in 160 patients with later manifestations after acute carbon monoxide poisoning. J. Hyperbar. Med. 1986, 1, 188. [Google Scholar]
- Stewart, R.D.; Peterson, J.E.; Baretta, E.D.; Bachand, R.T.; Hosko, M.J.; Herrmann, A.A. Experimental human exposure to carbon monoxide. Arch. Environ. Health Int. J. 1970, 21, 154–164. [Google Scholar] [CrossRef]
- Stewart, R.D.; Peterson, J.E.; Fisher, T.N.; Hosko, M.J.; Baretta, E.D.; Dodd, H.C.; Herrmann, A.A. Experimental human exposure to high concentrations of carbon monoxide. Arch. Environ. Health Int. J. 1973, 26, 10666210. [Google Scholar] [CrossRef] [PubMed]
- Herman, L.Y. Carbon monoxide poisoning presenting as an isolated seizure. J. Emerg. Med. 1998, 16, 429–432. [Google Scholar] [CrossRef] [PubMed]
- Mori, T.; Nagai, K. Carbon-monoxide poisoning presenting as an afebrile seizure. Pediatr. Neurol. 2000, 22, 330–331. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.S.; Lagasse, J.; Zimmerman, G. Computed tomographic findings after acute carbon monoxide poisoning. Am. J. Emerg. Med. 1994, 12, 448–451. [Google Scholar] [CrossRef]
- Miura, T.; Mitomo, M.; Kawai, R.; Harada, K. CT of the brain in acute carbon monoxide intoxication: Characteristic features and prognosis. AJNR Am. J. Neuroradiol. 1985, 6, 739–742. [Google Scholar] [PubMed]
- Sawada, Y.; Takahashi, M.; Ohashi, N.; Fusamoto, H.; Maemura, K.; Kobayashi, H.; Yoshioka, T.; Sugimoto, T. Computerised tomography as an indication of long-term outcome after acute carbon monoxide poisoning. Lancet 1980, 1, 783–784. [Google Scholar] [PubMed]
- Okeda, R.; Funata, N.; Takano, T.; Miyazaki, Y.; Higashino, F.; Yokoyama, K.; Manabe, M. The pathogenesis of carbon monoxide encephalopathy in the acute phase? Physiological and morphological correlation. Acta Neuropathol. 1981, 54, bf00691327. [Google Scholar] [CrossRef] [PubMed]
- Ginsberg, M.D.; Myers, R.E. Experimental carbon monoxide encephalopathy in the primate I. Physiologic and metabolic aspects. Arch. Neurol. 1974, 30, 202–208. [Google Scholar] [CrossRef]
- Ginsberg, M.D.; Myers, R.E.; McDonagh, B.F. Experimental Carbon Monoxide Encephalopathy in the Primate. II. Clinical aspects, neuropathology, and physiologic correlation. Arch. Neurol. 1974, 30, 209–216. [Google Scholar] [CrossRef]
- Penney, D.G. Hemodynamic response to carbon monoxide. Environ. Health Perspect. 1988, 77, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Penney, D.G. Acute carbon monoxide poisoning: Animal models: A review. Toxicology 1990, 62, 123–160. [Google Scholar] [CrossRef]
- Yanir, Y.; Shupak, A.; Abramovich, A.; Reisner, S.A.; Lorber, A. Cardiogenic shock complicating acute carbon monoxide poisoning despite neurologic and metabolic recovery. Ann. Emerg. Med. 2002, 40, 420–424. [Google Scholar] [CrossRef]
- Atkins, E.H.; Baker, E.L. Exacerbation of coronary artery disease by occupational carbon monoxide exposure: A report of two fatalities and a review of the literature. Am. J. Ind. Med. 1985, 7, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Sheps, D.S.; Herbst, M.C.; Hinderliter, A.L.; Adams, K.F.; Ekelund, L.G.; O’Neil, J.J.; Goldstein, G.M.; Bromberg, P.A.; Dalton, J.L.; Ballenger, M.N.; et al. Production of arrhythmias by elevated carboxyhemoglobin in patients with coronary artery disease. Ann. Intern. Med. 1990, 113, 343–351. [Google Scholar] [CrossRef]
- Wolff, E. Carbon monoxide poisoning with severe myonecrosis and acute renal failure. Am. J. Emerg. Med. 1994, 12, 347–349. [Google Scholar] [CrossRef]
- Herman, G.D.; Shapiro, A.B.; Leikin, J. Myonecrosis in carbon monoxide poisoning. Vet. Hum. Toxicol. 1988, 30, 28–30. [Google Scholar] [PubMed]
- Thom, S.R. Smoke inhalation. Emerg. Med. Clin. N. Am. 1989, 7, 371–387. [Google Scholar] [CrossRef]
- Krantz, T.; Thisted, B.; Strom, J.; Sørensen, M.B. Acute carbon monoxide poisoning. Acta Anaesthesiol. Scand. 1988, 32, 278–282. [Google Scholar] [CrossRef]
- Hosko, M.J. The effect of carbon monoxide on the visual evoked response in man and the spontaneous electroencephalogram. Arch. Environ. Health Int. J. 1970, 21, 174–180. [Google Scholar] [CrossRef]
- Hampson, N.B.; Piantadosi, C.A.; Thom, S.R.; Weaver, L.K. Practice recommendations in the diagnosis, management, and prevention of carbone monoxide poisoning. Am. J. Resp. Crit. Care Med. 2012, 186, 1098–1101. [Google Scholar] [CrossRef] [PubMed]
- Locatelli, C.; Casagranda, I.; Coen, D.; Demattè, P.; Demicheli, V.; Perraro, F.; Pesenti Campagnoni, M.; Porro, F.; Re, G.; Butera, R.; et al. Linee Guida per la Gestione ed il Trattamento del Paziente con Intossicazione Cuta da Monossido di Carbonio; SIMEU: Milano, Italy, 2001. [Google Scholar]
- Longo, L.D.; Hill, E.P. Carbon monoxide uptake and elimination in fetal and maternal sheep. Am. J. Physiol. 1977, 232, H324–H330. [Google Scholar] [CrossRef] [PubMed]
- Norman, C.A.; Halton, D.M. Is carbon monoxide a workplace teratogen? A review and evaluation of the literature. Ann. Occup. Hyg. 1990, 34, 335–347. [Google Scholar] [CrossRef] [PubMed]
- Koren, G.; Sharav, T.; Pastuszak, A.; Garrettson, L.K.; Hill, K.; Samson, I.; Rorem, M.; King, A.; Dolgin, J.E. A multicenter, prospective study of fetal outcome following accidental carbon monoxide poisoning in pregnancy. Reprod. Toxicol. 1991, 5, 397–403. [Google Scholar] [CrossRef] [PubMed]
- Cramer, C.R. Fetal death due to accidental maternal carbon monoxide poisoning. J. Toxicol. Clin. Toxicol. 1982, 19, 297–301. [Google Scholar] [CrossRef] [PubMed]
- Woody, R.C.; Brewster, M.A. Telencephalic dysgenesis associated with presumptive maternal carbon monoxide intoxication in the first trimester of pregnancy. J. Toxicol. Clin. Toxicol. 1990, 28, 467–475. [Google Scholar] [CrossRef] [PubMed]
- Caravati, E.M.; Adams, C.J.; Joyce, S.M.; Schafer, N.C. Fetal toxicity associated with maternal carbon monoxide poisoning. Ann. Emerg. Med. 1988, 17, 714–717. [Google Scholar] [CrossRef] [PubMed]
- Longo, L.D. Carbon monoxide in the pregnant mother and fetus and its exchange across the placenta. Ann. N. Y. Acad. Sci. 1970, 174, 313–341. [Google Scholar] [CrossRef] [PubMed]
- Vreman, H.J.; Mahoney, J.J.; Stevenson, D.K. Carbon monoxide and carboxyhemoglobin. Adv. Pediatr. 1995, 42, 303–334. [Google Scholar] [CrossRef]
- Takeuchi, M.; Abe, Y. Carbon monoxide poisoning of children. Chudoku Kenkyu 2006, 19, 283–284. (In Japanese) [Google Scholar]
- Foster, M.; Goodwin, S.R.; Williams, C.; Loeffler, J. Recurrent acute life-threatening events and lactic acidosis caused by chronic carbon monoxide poisoning in an infant. Pediatrics 1999, 104, e34. [Google Scholar] [CrossRef] [PubMed]
- Zworth, M.; Kareemi, H.; Boroumand, S.; Sikora, L.; Stiell, I.; Yadav, K. Machine learning for the diagnosis of acute coronary syndrome using a 12-lead ECG: A systematic review. CJEM 2023, 25, 818–827. [Google Scholar] [CrossRef] [PubMed]
- Broderick, J.P.; Silva, G.S.; Selim, M.; Kasner, S.E.; Aziz, Y.; Sutherland, J.; Jauch, E.C.; Adeoye, O.M.; Hill, M.D.; Mistry, E.A.; et al. Enhancing Enrollment in Acute Stroke Trials: Current State and Consensus Recommendations. Stroke 2023, 54, 2698–2707. [Google Scholar] [CrossRef] [PubMed]
- Savioli, G.; Ceresa, I.F.; Luzzi, S.; Lucifero, A.G.; Cambiè, G.; Manzoni, F.; Preda, L.; Ricevuti, G.; Bressan, M.A. Mild Head Trauma (MHT) and Antiplatelet Therapy. Reply to Lorenzati et al. Comment on “Savioli et al. Mild Head Trauma: Is Antiplatelet Therapy a Risk Factor for Hemorrhagic Complications?”. Medicina 2021, 57, 889. [Google Scholar] [CrossRef] [PubMed]
- Savioli, G.; Ceresa, I.F.; Ciceri, L.; Sciutti, F.; Belliato, M.; Iotti, G.A.; Luzzi, S.; Del Maestro, M.; Mezzini, G.; Lafe, E.; et al. Mild head trauma in elderly patients: Experience of an emergency department. Heliyon 2020, 6, e04226. [Google Scholar] [CrossRef]
- Savioli, G.; Ceresa, I.; Macedonio, S.; Gerosa, S.; Belliato, M.; Luzzi, S.; Lucifero, A.; Manzoni, F.; Ricevuti, G.; Bressan, M. Major Trauma in Elderly Patients: Worse Mortality and Outcomes in an Italian Trauma Center. J. Emerg. Trauma Shock 2021, 14, 98–103. [Google Scholar] [CrossRef] [PubMed]
- Savioli, G.; Ceresa, I.F.; Giordano, M.; Ferrari, I.; Varesi, A.; Floris, V.; Esposito, C.; Croesi, B.; Ricevuti, G.; Calvi, M.; et al. The Reliability of Anamnestic Data in the Management of Clostridium Tetani Infection in Elderly. Front. Med. 2021, 8, 684594. [Google Scholar] [CrossRef] [PubMed]
- Lapenna, R.; Faralli, M.; Del Zompo, M.R.; Cipriani, L.; Mobaraki, P.D.; Ricci, G. Reliability of an anamnestic questionnaire for the diagnosis of benign paroxysmal positional vertigo in the elderly. Aging Clin. Exp. Res. 2015, 28, 881–888. [Google Scholar] [CrossRef]
- Aboraya, A.; Rankin, E.; France, C.; El-Missiry, A.; John, C. The reliability of psychiatric diagnosis revisited: The clinician’s guide to improve the reliability of psychiatric diagnosis. Psychiatry 2006, 3, 41–50. [Google Scholar]
- Lindner, T.; Slagman, A.; Senkin, A.; Möckel, M.; Searle, J. Medical History of Elderly Patients in the Emergency Setting: Not an Easy Point-of-Care Diagnostic Marker. Emerg. Med. Int. 2015, 2015, 490947. [Google Scholar] [CrossRef]
- Kennedy, M.; Hwang, U.; Han, J.H. Delirium in the emergency department: Moving from tool-based research to system-wide change. J. Am. Geriatr. Soc. 2020, 68, 956–958. [Google Scholar] [CrossRef]
- Carpenter, C.R.; Bassett, E.R.; Fischer, G.M.; Shirshekan, J.; Galvin, J.E.; Morris, J.C. Four sensitive screening tools to detect cognitive dysfunction in geriatric emergency department patients: Brief Alzheimer’s screen, short blessed test, Ottawa 3DY, and the caregiver-completed AD8. Acad. Emerg. Med. 2011, 18, 374–384. [Google Scholar] [CrossRef]
- Savioli, G.; Ceresa, I.; Guarnone, R.; Muzzi, A.; Novelli, V.; Ricevuti, G.; Iotti, G.; Bressan, M.; Oddone, E. Impact of Coronavirus Disease 2019 Pandemic on Crowding: A Call to Action for Effective Solutions to “Access Block”. West J. Emerg. Med. 2021, 22, 860–870. [Google Scholar] [CrossRef]
- Savioli, G.; Ceresa, I.F.; Novelli, V.; Ricevuti, G.; Bressan, M.A.; Oddone, E. How the coronavirus disease 2019 pandemic changed the patterns of healthcare utilization by geriatric patients and the crowding: A call to action for effective solutions to the access block. Intern. Emerg. Med. 2021, 17, 503–514. [Google Scholar] [CrossRef] [PubMed]
- Lauque, D.; Khalemsky, A.; Boudi, Z.; Östlundh, L.; Xu, C.; Alsabri, M.; Onyeji, C.; Cellini, J.; Intas, G.; Soni, K.D.; et al. Length-of-Stay in the Emergency Department and In-Hospital Mortality: A Systematic Review and Meta-Analysis. J. Clin. Med. 2022, 12, 32. [Google Scholar] [CrossRef]
- Wu, L.; Chen, X.; Khalemsky, A.; Li, D.; Zoubeidi, T.; Lauque, D.; Alsabri, M.; Boudi, Z.; Kumar, V.A.; Paxton, J.; et al. The Association between Emergency Department Length of Stay and In-Hospital Mortality in Older Patients Using Machine Learning: An Observational Cohort Study. J. Clin. Med. 2023, 12, 4750. [Google Scholar] [CrossRef] [PubMed]
- Savioli, G.; Ceresa, I.F.; Bressan, M.A.; Piccini, G.B.; Varesi, A.; Novelli, V.; Muzzi, A.; Cutti, S.; Ricevuti, G.; Esposito, C.; et al. Five Level Triage vs. Four Level Triage in a Quaternary Emergency Department: National Analysis on Waiting Time, Validity, and Crowding—The CREONTE (Crowding and RE-Organization National TriagE) Study Group. Medicina 2023, 59, 781. [Google Scholar] [CrossRef] [PubMed]
- Savioli, G.; Ceresa, I.F.; Maggioni, P.; Lava, M.; Ricevuti, G.; Manzoni, F.; Oddone, E.; Bressan, M.A. Impact of ED Organization with a Holding Area and a Dedicated Team on the Adherence to International Guidelines for Patients with Acute Pulmonary Embolism: Experience of an Emergency Department Organized in Areas of Intensity of Care. Medicines 2020, 7, 60. [Google Scholar] [CrossRef]
- Savioli, G.; Ceresa, I.F.; Manzoni, F.; Ricevuti, G.; Bressan, M.A.; Oddone, E. Role of a Brief Intensive Observation Area with a Dedicated Team of Doctors in the Management of Acute Heart Failure Patients: A Retrospective Observational Study. Medicina 2020, 56, 251. [Google Scholar] [CrossRef]
- Crocker, P.J.; Walker, J.S. Pediatric carbon monoxide toxicity. J. Emerg. Med. 1985, 3, 443–448. [Google Scholar] [CrossRef]
- Strope, G.L.; Watkins, C.G. Chronic carbon monoxide poisoning as a major contributing factor in the sudden infant death syndrome. Am. J. Dis. Child. 1986, 140, 619. [Google Scholar] [CrossRef] [PubMed]
- Wright, J. Chronic and occult carbon monoxide poisoning: We don’t know what we’re missing. Emerg. Med. J. 2002, 19, 386–390. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, G.J.; Glaser, G.H. Neurologic manifestations of chronic carbon monoxide poisoning. N. Engl. J. Med. 1959, 261, 1217–1220. [Google Scholar] [CrossRef] [PubMed]
- Khan, K.; Sharief, N. Chronic carbon monoxide poisoning in children. Acta Paediatr. 1995, 84, 742. [Google Scholar] [CrossRef] [PubMed]
- Myers, R.A.M.; DeFazio, A.; Kelly, M.P. Chronic carbon monoxide exposure: A clinical syndrome detected by neuropsychological tests. J. Clin. Psychol. 1998, 54, 555–567. [Google Scholar] [CrossRef]
- Knobeloch, L.; Jackson, R. Recognition of chronic carbon monoxide poisoning. WMJ 1999, 98, 26–29. [Google Scholar] [PubMed]
- Seger, D.; Welch, L. Carbon monoxide controversies: Neuropsychological tessting, mechanism of toxicity, and hyperbaric oxyge. Ann. Emerg. Med. 1994, 24, 242–248. [Google Scholar] [CrossRef] [PubMed]
- Min, S.K. A brain syndrome associated with delayed neuropsychiatric sequelae following acute carbon monoxide intoxication. Acta Psychiatr. Scand. 1986, 73, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Thom, S.R.; Taber, R.L.; I Mendiguren, I.; Clark, J.M.; Hardy, K.R.; Fisher, A.B. Delayed neuropsychologic sequelae after carbon monoxide poisoning: Prevention by treatment with hyperbaric oxygen. Ann. Emerg. Med. 1995, 25, 474–480. [Google Scholar] [CrossRef]
- Choi, I.S. Delayed neurologic sequelae in carbon monoxide intoxication. Arch. Neurol. 1983, 40, 433–435. [Google Scholar] [CrossRef]
- Smith, J.S.; Brandon, S. Morbility from acute carbon monoxide poisoning at three-year follow-up. Br. Med. 1973, 1, 318–321. [Google Scholar] [CrossRef]
- Mathieu, D.; Nolf, M.; Durocher, A.; Saulnier, F.; Frimat, P.; Furon, D.; Wattel, F. Acute carbon monoxide poisoning risk of late sequelae and treatment by hyperbaric oxygen. J. Toxicol. Clin. Toxicol. 1985, 23, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.C.; Chen, T.H.; Ho, C.H.; Chen, Y.C.; Hsu, C.C.; Lin, H.J.; Wang, J.J.; Chang, C.P.; Guo, H.R. Increased risk of congestive heart failure following carbon monoxide poisoning. Circ. Heart Fail. 2021, 14, e007267. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.C.; Ho, C.H.; Chen, Y.C.; Lin, H.J.; Hsu, C.C.; Wang, J.J.; Su, S.B.; Guo, H.R. Risk of myocardial infarction after carbon monoxide poisoning: A nationwide population-based cohort study. Cardiovasc. Toxicol. 2019, 19, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.C.; Ho, C.H.; Chen, Y.C.; Hsu, C.C.; Lin, H.J.; Wang, J.J.; Su, S.B.; Guo, H.R. Effects of hyperbaric oxygen therapy on acute myocardial infarction following carbon monoxide poisoning. Cardiovasc. Toxicol. 2020, 20, 291–300. [Google Scholar] [CrossRef]
- Huang, C.C.; Ho, C.H.; Chen, Y.C.; Hsu, C.C.; Lin, H.J.; Wang, J.J.; Su, S.B.; Guo, H.R. Association between carbon monoxide poisoning and adrenal insufficiency: A nationwide cohort study. Sci. Rep. 2022, 12, 16219. [Google Scholar] [CrossRef]
- Huang, C.C.; Chung, M.H.; Weng, S.F.; Chien, C.C.; Lin, S.J.; Lin, H.J.; Guo, H.R.; Su, S.B.; Hsu, C.C.; Juan, C.W. Long-Term prognosis of patients with carbon monoxide poisoning: A nationwide cohort study. PLoS ONE 2014, 9, e105503. [Google Scholar] [CrossRef]
- Huang, C.C.; Ho, C.H.; Chen, Y.C.; Lin, H.J.; Hsu, C.C.; Wang, J.J.; Su, S.B.; Guo, H.R. Hyperbaric oxygen therapy is associated with lower short- and long-term mortality in patients with carbon monoxide poisoning. Chest 2017, 152, 943–953. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.C.; Ho, C.H.; Chen, Y.C.; Lin, H.J.; Hsu, C.C.; Wang, J.J.; Su, S.B.; Guo, H.R. Increased risk for diabetes mellitus in patients with carbon monoxide poisoning. Oncotarget 2017, 8, 63680–63690. [Google Scholar] [CrossRef]
- Nieman, L.K. Patient Education: Adrenal Insufciency (Addison’s Disease) (Beyond the Basics). 2019. Available online: https://medilib.ir/uptodate/show/15599 (accessed on 15 December 2023).
- Bornstein, S.R. Predisposing Factors for Adrenal Insufficiency. N. Engl. J. Med. 2009, 360, 2328–2339. [Google Scholar] [CrossRef]
- Inoue, O. Clinical description of dementia. Kangogaku Zasshi 1985, 49, 1173–1176. [Google Scholar] [PubMed]
- Inoue, S. Various types of dementia. (3). Subdural hematoma, hypothyroidism, anterior pituitary hypofunction, carbon monoxide poisoning, and pseudodementia. Kangogaku Zasshi 1986, 50, 574–577. [Google Scholar]
- Huang, C.C.; Ho, C.H.; Chen, Y.C.; Hsu, C.C.; Lin, H.J.; Su, S.B.; Wang, J.J.; Guo, H.R. Increased risk for hypothyroidism associated with carbon monoxide poisoning: A nationwide population-based cohort study. Sci. Rep. 2019, 9, 16512. [Google Scholar] [CrossRef] [PubMed]
- Didier, K.; Bolko, L.; Giusti, D.; Toquet, S.; Robbins, A.; Antonicelli, F.; Servettaz, A. Autoantibodies Associated With Connective Tissue Diseases: What Meaning for Clinicians? Front. Immunol. 2018, 9, 541. [Google Scholar] [CrossRef] [PubMed]
- Kumagai, S.; Jikimoto, T.; Saegusa, J. Pathological roles of oxidative stress in autoimmune diseases. Rinsho Byori 2003, 51, 126–132. [Google Scholar] [PubMed]
- Huang, C.C.; Ho, C.H.; Chen, Y.C.; Hsu, C.C.; Lin, H.J.; Wang, J.J.; Guo, H.R. Autoimmune Connective Tissue Disease Following Carbon Monoxide Poisoning: A Nationwide Population-Based Cohort Study. Clin. Epidemiol. 2020, 12, 1287–1298. [Google Scholar] [CrossRef] [PubMed]
- Pagano, G.; Castello, G.; Pallardó, F.V. Sjøgren’s syndrome-associated oxidative stress and mitochondrial dysfunction: Prospects for chemoprevention trials. Free. Radic. Res. 2013, 47, 71–73. [Google Scholar] [CrossRef] [PubMed]
- Perl, A. Oxidative stress in the pathology and treatment of systemic lupus erythematosus. Nat. Rev. Rheumatol. 2013, 9, 674–686. [Google Scholar] [CrossRef] [PubMed]
- Cooper, G.S.; Dooley, M.A.; Treadwell, E.L.; St Clair, E.W.; Gilkeson, G.S. Risk factors for development of systemic lupus erythematosus: Allergies, infections, and family history. J. Clin. Epidemiol. 2002, 55, 982–989. [Google Scholar] [CrossRef] [PubMed]
- Van Hal, T.W.; van Bon, L.; Radstake, T.R.D.J. A system out of breath: How hypoxia possibly contributes to the pathogenesis of systemic sclerosis. Int. J. Rheumatol. 2011, 2011, 824972. [Google Scholar] [CrossRef]
- Hampson, N.B.; Mathieu, D.; Piantadosi, C.A.; Thom, S.R.; Weaver, L.K. Carbon monoxide poisoning: Interpretation of randomized clinical trials and unresolved treatment issues. Undersea Hyperb. Med. 2001, 28, 157–164. [Google Scholar]
- Mastrangelo, G.; Mapp, C. Ossido di carboniop. In Medicina del Lavoro; Crepet, M., Ed.; UTET: Torino, Italy, 1979. [Google Scholar]
- Chiappino, G.; Tomasini, M. Gas tossici. In Medicina del Lavoro; Chiappino, G., Tomasini, M., Eds.; Cortina: Milano, Italy, 1994. [Google Scholar]
- Mennoia, V.; Gazzaruso, C.; Geroldi, D.; Cadura, S.M. Fenotipi di apolipoproteina(a) come indicatori di suscettiblità genetica per rischio cardiovascolare in medicina del lavoro. G. Ital. Med. Lav. Erg. 1999, 21, 206–217. [Google Scholar]
- Barret, L.; Danel, V.; Faure, J. Carbon monnoxide poisoning, a diagnosis frequently overlooked. J. Toxicol. Clin. Toxicol. 1985, 23, 309–313. [Google Scholar] [CrossRef] [PubMed]
- Ernst, A.; Zibrak, J.D. Carbon monoxide poisoning. N. Engl. J. Med. 1998, 339, 1603–1608. [Google Scholar] [CrossRef] [PubMed]
- Weaver, L.K.; Howe, S.; Hopkins, R.; Chan, K.J. Carboxyhemoglobin half-life in carbon monoxide-poisoned patients treated with 100% oxygen at atmospheric pressure. Chest 2000, 117, 801–808. [Google Scholar] [CrossRef]
- Weaver, L.K.; Valentine, K.J.; Hopkins, R.O. Carbon monoxide poisoning: Risk factors for cognitive sequelae and the role of hyperbaric oxygen. Am. J. Respir. Crit. Care Med. 2007, 176, 491–497. [Google Scholar] [CrossRef] [PubMed]
- Touger, M.; Gallagher, E.; Tyrell, J. Relationship between venous and arterial carboxyhemoglobin levels in patients with suspected carbon monoxide poisoning. Ann. Emerg. Med. 1995, 25, 481–483. [Google Scholar] [CrossRef] [PubMed]
- Candura, S.M.; Fonte, R.; Finozzi, E.; Guarnone, F.; Taglione, L.; Manzo, L.; Biscaldi, G. Inquinamento indoor: Presentazione di due casi clinici di ossicarbonismo occulto. G. Ital. Med. Lav. 1993, 15, 67–70. [Google Scholar]
- Winter, P.M.; Miller, J.N. Carbon monoxide poisoning. JAMA 1976, 236, 1502. [Google Scholar] [CrossRef]
- Pace, N.; Strajman, E.; Walker, E.L. Acceleration of Carbon Monoxide Elimination in Man by High Pressure Oxygen. Science 1950, 111, 652–654. [Google Scholar] [CrossRef]
- Jay, G.D.; McKindley, D.S. Alterations in pharmacokinetics of carboxyhemoglobin produced by oxygen under pressure. Undersea Hyperb. Med. 1997, 24, 165–173. [Google Scholar]
- Araki, R.; Nashimoto, I.; Takano, T. The effect of hyperbaric oxygen on cerebral hemoglobin oxygenation and dissociation rate of carboxyhemoglobin in anesthetized rats: Spectroscopic approach. Adv. Exp. Med. Biol. 1988, 222, 375–381. [Google Scholar] [CrossRef] [PubMed]
- Smith, G. The treatment of carbon monoxide poisoning with oxygen at two atmospheres absolute. Ann. Occup. Hyg. 1962, 5, 259–263. [Google Scholar] [CrossRef] [PubMed]
- Loyning, Y. Treatment with oxygen under pressure in carbon monoxide poisoning. Tidsskr. Nor. Laegeforen. 1961, 81, 1207–1208. [Google Scholar] [PubMed]
- Thom, S.R. Antagonism of carbon monoxide-mediated brain lipid peroxidation by hyperbaric oxygen. Toxicol. Appl. Pharmacol. 1990, 105, 340–344. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.D.; Piantadosi, C.A. Reversal of carbon monoxide-cytochrome c oxidase binding by hyperbaric oxygen in vivo. Adv. Exp. Med. Biol. 1989, 248, 747–754. [Google Scholar] [CrossRef] [PubMed]
- Thom, S. Functional inhibition of leukocyte b2 integrins by hyperbaric oxygen in carbon monoxide-mediated brain injury in rats. Toxicol. Appl. Pharmacol. 1993, 123, 248–256. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Tyssebotn, I. Normobaric and hyperbaric oxygen treatment of acute carbon monoxide poisoning in rats. Undersea Hyperb. Med. 1997, 24, 107–116. [Google Scholar] [PubMed]
- Gesell, L.B.; Trott, A. De novo cataract development following a standard course of hyperbaric oxygen therapy. Undersea Hyperb. Med. 2008, 34, 389–392. [Google Scholar]
- Gesell, L.B. Hyperbaric Oxygen 2009: Indications and Results: The Hyperbaric Oxygen Therapy Committee Report; Undersea and Hyperbaric Medical Society: Durham, NC, USA, 2008. [Google Scholar]
- Weaver, L.K.; Hopkins, R.O.; Chan, K.J.; Churchill, S.; Elliott, C.G.; Clemmer, T.P.; Orme, J.F., Jr.; Thomas, F.O.; Morris, A.H. Hyperbaric oxygen for acute carbon monoxide poisoning. N. Engl. J. Med. 2002, 347, 1057–1067. [Google Scholar] [CrossRef]
- Ducassé, J.L.; Celsis, P.; Marc-Vergnes, J.P. Non-comatose patients with acute carbon monoxide poisoning: Hyperbaric or normobaric oxygenation? Undersea Hyperb. Med. 1995, 22, 9–15. [Google Scholar]
- Raphael, J.C.; Elkharrat, D.; Jars-Guincestre, M.C.; Chastang, C.; Vercken, J.B.; Chasles, V.; Gajdos, P. Trial of normobaric and hyperbaric oxygen for acute carbon monoxide intoxication. Lancet 1989, 334, 414–419. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| CAS 630-08-0 EC: 006-001-00-2 Other names: Carbon monoxide, carbonic oxide | Molecular formula: CO |
| Physical state at 25 °C: Gas (odorless and tasteless) Melting point: −205 °C Boiling temperature: −191 °C | Molecular weight: 28.01 g/mol Vapor pressure: Not applicable |
| Relative vapor density (air = 1): 0.97 | Solubility in water: 2.3 mL/100 mL at 20 °C |
| Self-ignition temperature: 605 °C Flash point: −101.6 °C Explosive limit: 12.5–74.2 vol (%) | Conversion factors: - 1 mL/m3 = 1.83 mg/m3 - 1 ppm = 1.14 mg/m3 - 1 mg/m3 = 0.87 ppm |
| Industrial Sector | Processing |
|---|---|
| Fuel |
- Treatment of coke oven gas - Coke extinguishing - Production and distribution of illuminating gas - Gasification of solid fuels |
| Siliceous products | - Use of ovens |
| Chemistry and petrochemistry |
- Use of ovens - Production of CO and its mixtures - Production of calcium carbide (CaC2), acetylene (C2H2), hydrogen cyanide (HCN), and organic compounds |
| Metallurgy and engineering |
- Use of ovens - Welding and cutting with a blowtorch - Repair, maintenance, and use of engines |
| EFFECT | MECHANISM |
|---|---|
HEMOGLOBIN
|
|
TISSUES
|
|
| SEVERITY CLASS | SIGNS AND SYMPTOMS |
|---|---|
| ASYMPTOMATIC (grade 1) | Absent (with positive COHb values) |
| MILD (grade 2) | Headache, dizziness, nausea, vomiting |
| AVERAGE (grade 3) | Mental confusion, slow thinking, blurred vision, asthenia, ataxia, abnormal behavior, shallow breathing, exertional dyspnea, tachypnea, tachycardia, abnormal psychometric test |
| SERIOUS (grade 4) | Drowsiness, sensory blunting, coma, convulsions, syncope, disorientation, brain CT changes, hypotension, chest pain, palpitations, arrhythmias, ECG ischemic signs, pulmonary edema, myonecrosis, skin bullae, lactic acidosis |
| INDICATIONS FOR HYPERBARIC THERAPY |
|---|
| Syncope |
| Coma |
| Convulsions |
| Altered mental state |
| Cerebellar signs |
| Carboxyhemoglobin > 25% |
| Fetal distress (in pregnancy) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Savioli, G.; Gri, N.; Ceresa, I.F.; Piccioni, A.; Zanza, C.; Longhitano, Y.; Ricevuti, G.; Daccò, M.; Esposito, C.; Candura, S.M. Carbon Monoxide Poisoning: From Occupational Health to Emergency Medicine. J. Clin. Med. 2024, 13, 2466. https://doi.org/10.3390/jcm13092466
Savioli G, Gri N, Ceresa IF, Piccioni A, Zanza C, Longhitano Y, Ricevuti G, Daccò M, Esposito C, Candura SM. Carbon Monoxide Poisoning: From Occupational Health to Emergency Medicine. Journal of Clinical Medicine. 2024; 13(9):2466. https://doi.org/10.3390/jcm13092466
Chicago/Turabian StyleSavioli, Gabriele, Nicole Gri, Iride Francesca Ceresa, Andrea Piccioni, Christian Zanza, Yaroslava Longhitano, Giovanni Ricevuti, Maurizio Daccò, Ciro Esposito, and Stefano M. Candura. 2024. "Carbon Monoxide Poisoning: From Occupational Health to Emergency Medicine" Journal of Clinical Medicine 13, no. 9: 2466. https://doi.org/10.3390/jcm13092466