

Increased Expression of α-Hemoglobin Stabilizing Protein (AHSP) mRNA in Erythroid Precursor Cells Isolated from β-Thalassemia Patients Treated with Sirolimus (Rapamycin)
 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Culture and Treatment of Human Erythroid Precursor Cells (ErPCs)
2.2. RT-qPCR Analysis
2.3. Western Blotting
2.4. Statistical Analysis
3. Results
3.1. Production of AHSP in β-Thalassemic Erythroid Precursor Cells (ErPCs): Comparison with ErPCs from Healthy Subjects
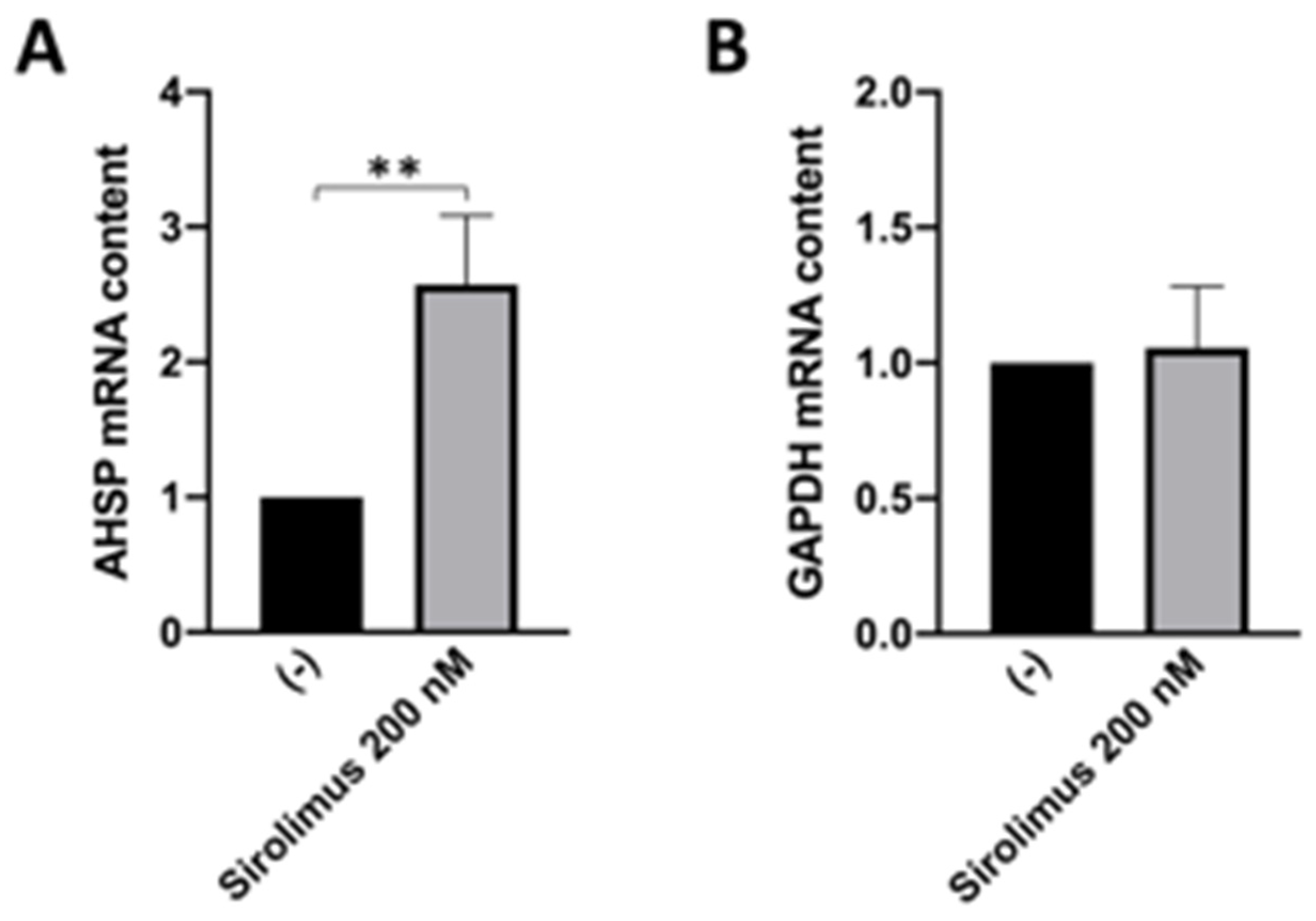
3.2. Sirolimus (Rapamycin) Induces Increase in AHSP mRNA Expression in ErPCs from β-Thalassemia Patients
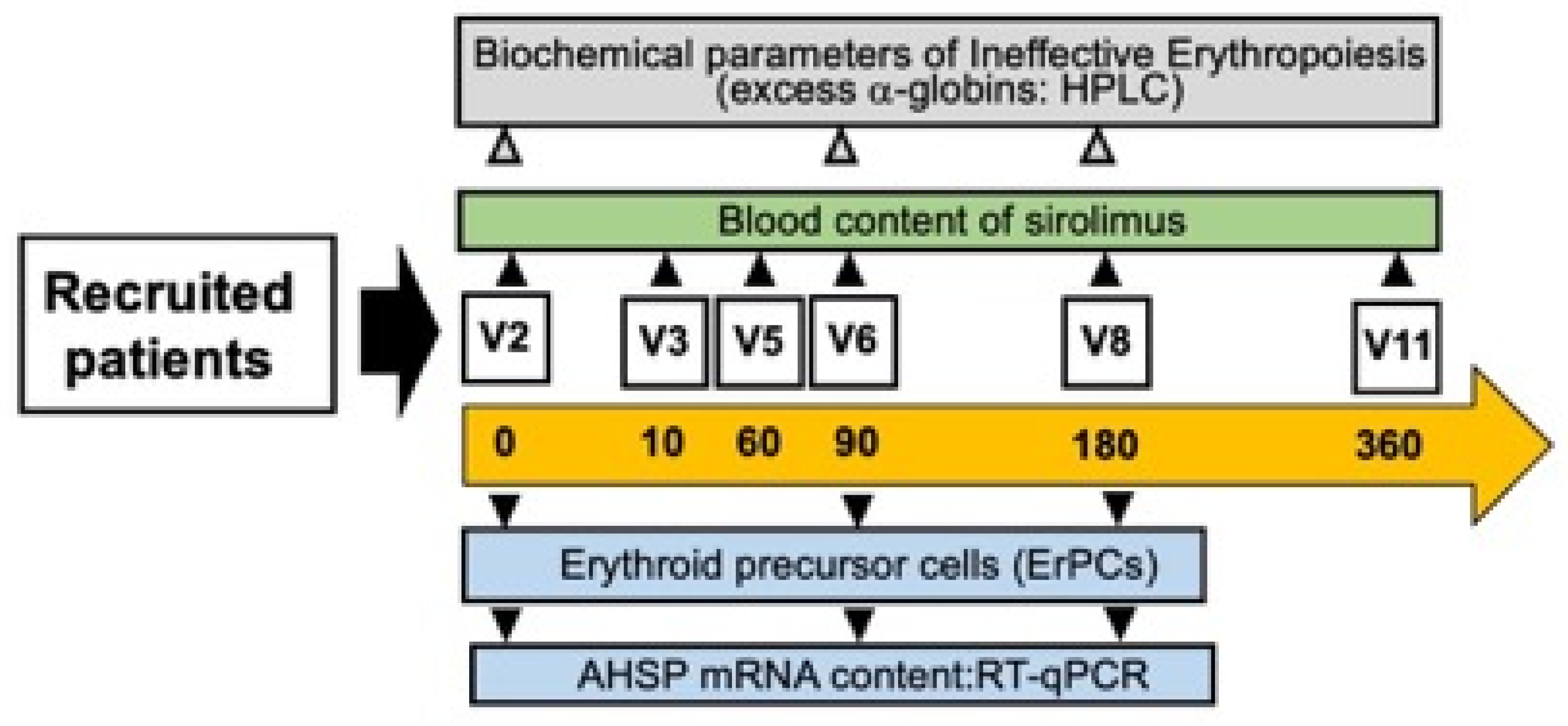
3.3. Selected Activities of the Sirthalaclin NCT03877809 Clinical Trial
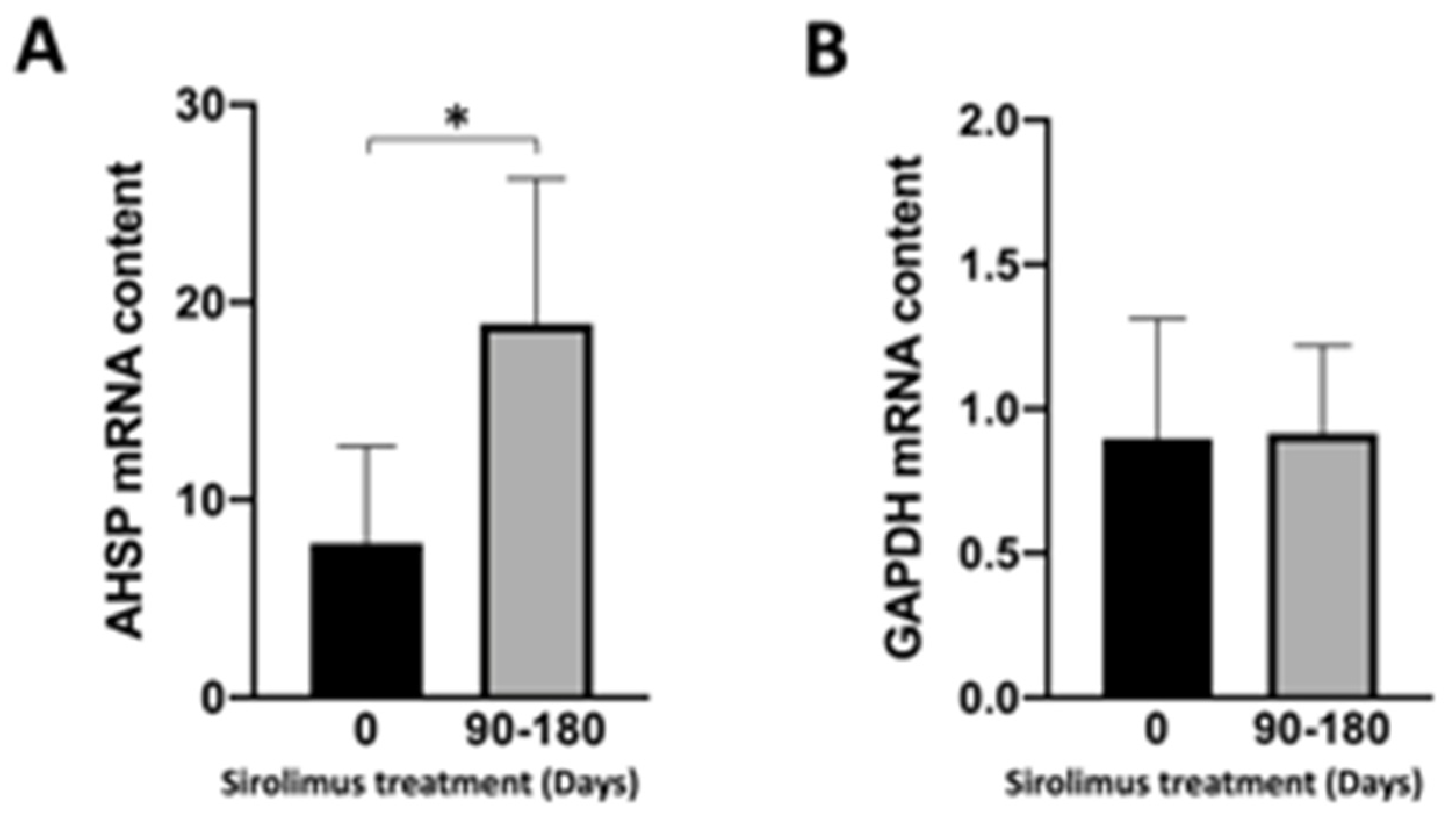
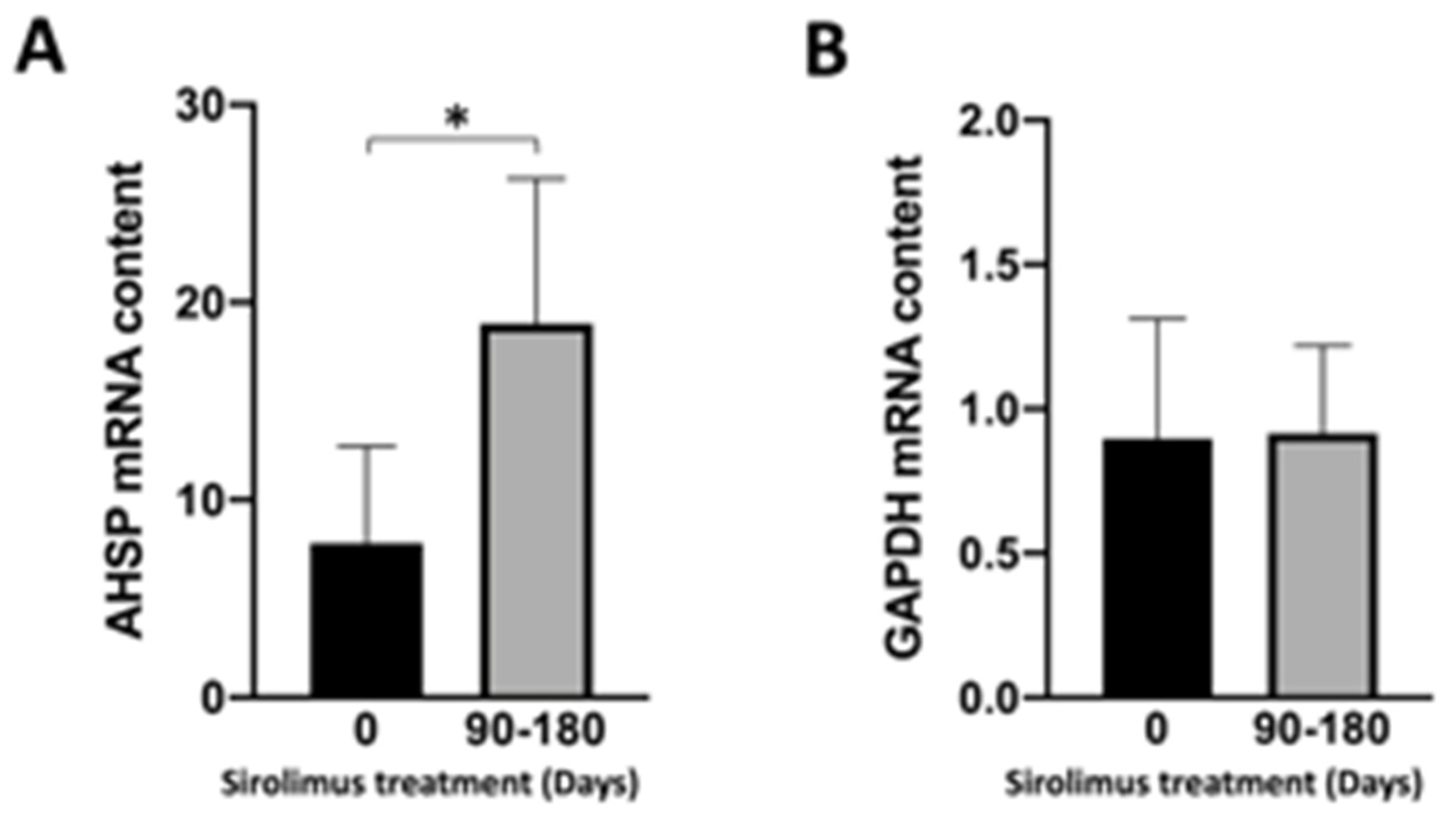
3.4. Sirolimus (Rapamycin) Treatment of β-Thalassemia Patients Participating to the Sirthalaclin NCT03877809 Trial Induces an Increase in AHSP mRNA Production in ErPCs from Sirolimus-Treated β-Thalassemia Patients
4. Discussion
4.1. Strength of the Study
4.2. Limitations and Drawbacks of the Study
4.3. Future Research Efforts and Perspectives
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Higgs, D.R.; Engel, J.D.; Stamatoyannopoulos, G. Thalassaemia. Lancet 2012, 379, 373–383. [Google Scholar] [CrossRef] [PubMed]
- Thein, S.L. Molecular basis of β thalassemia and potential therapeutic targets. Blood Cells Mol. Dis. 2018, 70, 54–65. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Sagar, C.; Sharma, D.; Kishor, P. β-globin genes: Mutation hot-spots in the global thalassemia belt. Hemoglobin 2015, 39, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Rao, E.; Kumar Chandraker, S.; Misha Singh, M.; Kumar, R. Global distribution of β-thalassemia mutations: An update. Gene 2023, 896, 148022. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.Y.; Orkin, S.H.; Sankaran, V.G. Fetal Hemoglobin Regulation in Beta-Thalassemia. Hematol. Oncol. Clin. N. Am. 2023, 37, 301–312. [Google Scholar] [CrossRef]
- Pavan, A.R.; Lopes, J.R.; Dos Santos, J.L. The state of the art of fetal hemoglobin-inducing agents. Expert Opin. Drug Discov. 2022, 17, 1279–1293. [Google Scholar] [CrossRef]
- Vasseur, C.; Pissard, S.; Domingues-Hamdi, E.; Marden, M.C.; Galactéros, F.; Baudin-Creuza, V. Evaluation of the free α-hemoglobin pool in red blood cells: A new test providing a scale of β-thalassemia severity. Am. J. Hematol. 2011, 86, 199–202. [Google Scholar] [CrossRef]
- Mettananda, S.; Higgs, D.R. Molecular Basis and Genetic Modifiers of Thalassemia. Hematol. Oncol. Clin. N. Am. 2018, 32, 177–191. [Google Scholar] [CrossRef] [PubMed]
- Arlet, J.B.; Dussiot, M.; Moura, I.C.; Hermine, O.; Courtois, G. Novel players in β-thalassemia dyserythropoiesis and new therapeutic strategies. Curr. Opin. Hematol. 2016, 23, 181–188. [Google Scholar] [CrossRef]
- Mettananda, S.; Gibbons, R.J.; Higgs, D.R. α-Globin as a molecular target in the treatment of β-thalassemia. Blood 2015, 125, 3694–3701. [Google Scholar] [CrossRef]
- Nienhuis, A.W.; Nathan, D.G. Pathophysiology and Clinical Manifestations of the β-Thalassemias. Cold Spring Harb. Perspect Med. 2012, 2, a011726. [Google Scholar] [CrossRef]
- Origa, R.; Galanello, R. Pathophysiology of beta thalassaemia. Pediatr. Endocrinol. Rev. 2011, 8, 263–270. [Google Scholar]
- Shaeffer, J.R.; Kania, M.A. Degradation of monoubiquitinated alpha-globin by 26S proteasomes. Biochemistry 1995, 34, 4015–4021. [Google Scholar] [CrossRef] [PubMed]
- Wickramasinghe, S.; Lee, M. Evidence that the ubiquitin proteolytic pathway is involved in the degradation of precipitated globin chains in thalassaemia. Br. J. Haematol. 1998, 101, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Shaeffer, J.R.; Cohen, R.E. Ubiquitin aldehyde increases adenosine triphosphate–dependent proteolysis of hemoglobin α-subunits in β-thalassemic hemolysates. Blood 1997, 90, 1300–1308. [Google Scholar] [CrossRef]
- Braverman, A.S.; Lester, D. Evidence for increased proteolysis in intact beta thalassemia erythroid cells. Hemoglobin. 1981, 5, 549–564. [Google Scholar] [CrossRef] [PubMed]
- Loukopoulos, D.; Karoulias, A.; Fessas, P. Proteolysis in thalassemia: Studies with protease inhibitors. Ann. N. Y. Acad. Sci. 1980, 344, 323–335. [Google Scholar] [CrossRef]
- Khandros, E.; Thom, C.S.; D’Souza, J.; Weiss, M.J. Integrated protein quality-control pathways regulate free α-globin in murine β-thalassemia. Blood. 2012, 119, 5265–5275. [Google Scholar] [CrossRef] [PubMed]
- Lechauve, C.; Keith, J.; Khandros, E.; Fowler, S.; Mayberry, K.; Freiwan, A.; Thom, C.S.; Delbini, P.; Romero, E.B.; Zhang, J.; et al. The autophagy-activating kinase ULK1 mediates clearance of free α-globin in β-thalassemia. Sci. Transl. Med. 2019, 11, eaav4881. [Google Scholar] [CrossRef]
- Zurlo, M.; Zuccato, C.; Cosenza, L.C.; Gasparello, J.; Gamberini, M.R.; Stievano, A.; Fortini, M.; Prosdocimi, M.; Finotti, A.; Gambari, R. Decrease in α-Globin and Increase in the Autophagy-Activating Kinase ULK1 mRNA in Erythroid Precursors from β-Thalassemia Patients Treated with Sirolimus. Int. J. Mol. Sci. 2023, 24, 15049. [Google Scholar] [CrossRef]
- Keith, J.; Christakopoulos, G.E.; Fernandez, A.G.; Yao, Y.; Zhang, J.; Mayberry, K.; Telange, R.; Sweileh, R.B.A.; Dudley, M.; Westbrook, C.; et al. Loss of miR-144/451 alleviates β-thalassemia by stimulating ULK1-mediated autophagy of free α-globin. Blood 2023, 142, 918–932. [Google Scholar] [CrossRef] [PubMed]
- Kihm, A.J.; Kong, Y.; Hong, W.; Russell, J.E.; Rouda, S.; Adachi, K.; Simon, M.C.; Blobel, G.A.; Weiss, M.J. An abundant erythroid protein that stabilizes free alpha-haemoglobin. Nature 2002, 417, 758–763. [Google Scholar] [CrossRef] [PubMed]
- Viprakasit, V.; Tanphaichitr, V.S.; Chinchang, W.; Sangkla, P.; Weiss, M.J.; Higgs, D.R. Evaluation of alpha hemoglobin stabilizing protein (AHSP) as a genetic modifier in patients with beta thalassemia. Blood 2004, 103, 3296–3299. [Google Scholar] [CrossRef] [PubMed]
- Kong, Y.; Zhou, S.; Kihm, A.J.; Katein, A.M.; Yu, X.; Gell, D.A.; Mackay, J.P.; Adachi, K.; Foster-Brown, L.; Louden, C.S.; et al. Loss of alpha-hemoglobin-stabilizing protein impairs erythropoiesis and exacerbates beta-thalassemia. J. Clin. Investig. 2004, 114, 1457–1466. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Gell, D.A.; Zhou, S.; Gu, L.; Kong, Y.; Li, J.; Hu, M.; Yan, N.; Lee, C.; Rich, A.M.; et al. Molecular mechanism of AHSP-mediated stabilization of alpha-hemoglobin. Cell 2004, 119, 629–640. [Google Scholar] [CrossRef] [PubMed]
- Weiss, M.J.; Zhou, S.; Feng, L.; Gell, D.A.; Mackay, J.P.; Shi, Y.; Gow, A.J. Role of alpha-hemoglobin-stabilizing protein in normal erythropoiesis and beta-thalassemia. Ann. N. Y. Acad. Sci. 2005, 1054, 103–117. [Google Scholar] [CrossRef] [PubMed]
- Che Yaacob, N.S.; Islam, M.A.; Alsaleh, H.; Ibrahim, I.K.; Hassan, R. Alpha-hemoglobin-stabilizing protein (AHSP): A modulatory factor in β-thalassemia. Int. J. Hematol. 2020, 111, 352–359. [Google Scholar] [CrossRef] [PubMed]
- Voon, H.P.; Vadolas, J. Controlling alpha-globin: A review of alpha-globin expression and its impact on beta-thalassemia. Haematologica 2008, 93, 1868–1876. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, H.M.; Shoeib, A.A.; Abd El Ghany, S.M.; Reda, M.M.; Ragab, I.A. Study of alpha hemoglobin stabilizing protein expression in patients with β thalassemia and sickle cell anemia and its impact on clinical severity. Blood Cells Mol. Dis. 2015, 55, 358–362. [Google Scholar] [CrossRef]
- Sagar, C.S.; Kumar, R.; Sharma, D.C.; Kishor, P. Alpha hemoglobin stabilizing protein: Its causal relationship with the severity of beta thalassemia. Blood Cells Mol. Dis. 2015, 55, 104–107. [Google Scholar] [CrossRef]
- Ranjbaran, R.; Okhovat, M.A.; Mobarhanfard, A.; Aboualizadeh, F.; Abbasi, M.; Moezzi, L.; Golafshan, H.A.; Behzad-Behbahani, A.; Bagheri, M.; Sharifzadeh, S. Relationship between AHSP gene expression, β/α globin mRNA ratio, and clinical severity of the β-thalassemia patients. Ann. Clin. Lab. Sci. 2014, 44, 189–193. [Google Scholar] [PubMed]
- Turbpaiboon, C.; Wilairat, P. Alpha-hemoglobin stabilizing protein: Molecular function and clinical correlation. Front. Biosci. 2010, 15, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Scheps, K.G.; Varela, V.; Targovnik, H.M. The Chaperones Involved in Hemoglobin Synthesis Take the Spotlight: Analysis of AHSP in the Argentinean Population and Review of the Literature. Hemoglobin 2018, 42, 310–314. [Google Scholar] [CrossRef] [PubMed]
- dos Santos, C.O.; Costa, F.F. AHSP and beta-thalassemia: A possible genetic modifier. Hematology 2005, 10, 157–161. [Google Scholar] [CrossRef] [PubMed]
- Fibach, E.; Bianchi, N.; Borgatti, M.; Zuccato, C.; Finotti, A.; Lampronti, I.; Prus, E.; Mischiati, C.; Gambari, R. Effects of rapamycin on accumulation of alpha-, beta- and gamma-globin mRNAs in erythroid precursor cells from beta-thalassaemia patients. Eur. J. Haematol. 2006, 77, 437–441. [Google Scholar] [CrossRef] [PubMed]
- Pecoraro, A.; Troia, A.; Calzolari, R.; Scazzone, C.; Rigano, P.; Martorana, A.; Sacco, M.; Maggio, A.; Di Marzo, R. Efficacy of Rapamycin as Inducer of Hb F in Primary Erythroid Cultures from Sickle Cell Disease and β-Thalassemia Patients. Hemoglobin 2015, 39, 225–229. [Google Scholar] [CrossRef] [PubMed]
- Khaibullina, A.; Almeida, L.E.; Wang, L.; Kamimura, S.; Wong, E.C.; Nouraie, M.; Maric, I.; Albani, S.; Finkel, J.; Quezado, Z.M. Rapamycin increases fetal hemoglobin and ameliorates the nociception phenotype in sickle cell mice. Blood Cells Mol. Dis. 2015, 55, 363–372. [Google Scholar] [CrossRef]
- Wang, J.; Tran, J.; Wang, H.; Guo, C.; Harro, D.; Campbell, A.D.; Eitzman, D.T. mTOR Inhibition improves anaemia and reduces organ damage in a murine model of sickle cell disease. Br. J. Haematol. 2016, 174, 461–469. [Google Scholar] [CrossRef]
- Gambari, R.; Zuccato, C.; Cosenza, L.C.; Zurlo, M.; Gasparello, J.; Finotti, A.; Gamberini, M.R.; Prosdocimi, M. The Long Scientific Journey of Sirolimus (Rapamycin): From the Soil of Easter Island (Rapa Nui) to Applied Research and Clinical Trials on β-Thalassemia and Other Hemoglobinopathies. Biology 2023, 12, 1202. [Google Scholar] [CrossRef]
- Gaudre, N.; Cougoul, P.; Bartolucci, P.; Dörr, G.; Bura-Riviere, A.; Kamar, N.; Del Bello, A. Improved Fetal Hemoglobin with mTOR Inhibitor-Based Immunosuppression in a Kidney Transplant Recipient with Sickle Cell Disease. Am. J. Transplant. 2017, 17, 2212–2214. [Google Scholar] [CrossRef]
- Al-Khatti, A.A.; Alkhunaizi, A.M. Additive effect of sirolimus and hydroxycarbamide on fetal haemoglobin level in kidney transplant patients with sickle cell disease. Br. J. Haematol. 2019, 185, 959–961. [Google Scholar] [CrossRef] [PubMed]
- Gamberini, M.R.; Prosdocimi, M.; Gambari, R. Sirolimus for Treatment of β-Thalassemia: From Pre-Clinical Studies to the Design of Clinical Trials. Health Educ. Public Health 2021, 4, 425–435. [Google Scholar]
- Zuccato, C.; Cosenza, L.C.; Zurlo, M.; Gasparello, J.; Papi, C.; D’Aversa, E.; Breveglieri, G.; Lampronti, I.; Finotti, A.; Borgatti, M.; et al. Expression of γ-globin genes in β-thalassemia patients treated with sirolimus: Results from a pilot clinical trial (Sirthalaclin). Ther. Adv. Hematol. 2022, 13, 20406207221100648. [Google Scholar] [CrossRef] [PubMed]
- Fibach, E.; Manor, D.; Oppenheim, A.; Rachmilewitz, E.A. Proliferation and maturation of human erythroid progenitors in liquid culture. Blood 1989, 73, 100–103. [Google Scholar] [CrossRef] [PubMed]
- Fibach, E.; Prus, E. Differentiation of human erythroid cells in culture. Curr. Protoc. Immunol. 2005, 69, 22F. 7.1–22F. 7.10. [Google Scholar] [CrossRef] [PubMed]
- Finotti, A.; Bianchi, N.; Fabbri, E.; Borgatti, M.; Breveglieri, G.; Gasparello, J.; Gambari, R. Erythroid induction of K562 cells treated with mithramycin is associated with inhibition of raptor gene transcription and mammalian target of rapamycin complex 1 (mTORC1) functions. Pharmacol. Res. 2015, 91, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Zurlo, M.; Nicoli, F.; Proietto, D.; Dallan, B.; Zuccato, C.; Cosenza, L.C.; Gasparello, J.; Papi, C.; d’Aversa, E.; Borgatti, M.; et al. Effects of Sirolimus treatment on patients with β-Thalassemia: Lymphocyte immunophenotype and biological activity of memory CD4+ and CD8+ T cells. J. Cell. Mol. Med. 2023, 27, 353–364. [Google Scholar] [CrossRef]
- Wang, Q.; Luo, M.; Xiang, B.; Chen, S.; Ji, Y. The efficacy and safety of pharmacological treatments for lymphangioleiomyomatosis. Respir Res. 2020, 21, 55. [Google Scholar] [CrossRef]
- Liu, N.; Li, P.; Zang, S.; Liu, Q.; Ma, D.; Sun, X.; Ji, C. Novel agent nitidine chloride induces erythroid differentiation and apoptosis in CML cells through c-Myc-miRNAs axis. PLoS ONE 2015, 10, e0116880. [Google Scholar] [CrossRef]
- Han, G.; Cao, C.; Yang, X.; Zhao, G.W.; Hu, X.J.; Yu, D.L.; Yang, R.F.; Yang, K.; Zhang, Y.Y.; Wang, W.T.; et al. Nrf2 expands the intracellular pool of the chaperone AHSP in a cellular model of β-thalassemia. Redox Biol. 2022, 50, 102239. [Google Scholar] [CrossRef]
- Shah, A.; Varma, M.; Bhandari, R. Exploring sulforaphane as neurotherapeutic: Targeting Nrf2-Keap & Nf-Kb pathway crosstalk in ASD. Metab. Brain Dis. 2023, 39, 373–385. [Google Scholar] [CrossRef] [PubMed]
- Houghton, C.A.; Fassett, R.G.; Coombes, J.S. Sulforaphane and Other Nutrigenomic Nrf2 Activators: Can the Clinician’s Expectation Be Matched by the Reality? Oxid. Med. Cell Longev. 2016, 2016, 7857186. [Google Scholar] [CrossRef] [PubMed]
- Foong, W.C.; Loh, C.K.; Ho, J.J.; Lau, D.S. Foetal haemoglobin inducers for reducing blood transfusion in non-transfusion-dependent beta-thalassaemias. Cochrane Database Syst Rev. 2023, 1, CD013767. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primers and Probes | Sequences |
|---|---|
| primer forward AHSP | 5′-GAGACATATACAGCCTGTTAGACC-3′ |
| primer reverse AHSP | 5′-GAGGATCATTGAAGACCTGCT-3′ |
| probe AHSP | 5′-FAM-ATGAGATCCTTATTGGCCTTAAGAAGAGCC-BFQ-3′ |
| primer forward RPL13A | 5′-GGCAATTTCTACAGAAACAAGTTG-3′ |
| primer reverse RPL13A | 5′-GTTTTGTGGGGCAGCATACC-3′ |
| probe RPL13A | 5′-HEX-CGCACGGTCCGCCAGAAGAT-BFQ-3′ |
| primer forward β-actin | 5′-ACAGAGCCTCGCCTTTG-3′ |
| primer reverse β-actin | 5′-ACGATGGAGGGGAAGACG-3′ |
| probe β-actin | 5′-Cy5-CCTTGCACATGCCGGAGCC-BRQ-3′ |
| primer forward GAPDH | 5′-ACATCGCTCAGACACCATG-3′ |
| primer reverse GAPDH | 5′-TGTAGTTGAGGTCAATGAAGGG-3′ |
| probe GAPDH | 5′-FAM-AAGGTCGGAGTCAACGGATTTGGTC-BFQ-3′ |
| Target | Primary Antibody | Cat.n. | Secondary Antibody | Cat.n. |
|---|---|---|---|---|
| AHSP | Rabbit anti-AHSP (ABclonal, Woburn, MA, USA) | A6465 | Mouse Anti-rabbit IgG HRP (Cell Signalling Technology, Danvers, MA, USA | 7074 |
| GAPDH | Mouse anti-GAPDH (Thermo Fisher, Waltham, MA, USA) | MA1-16783 | Goat Anti-mouse IgG HRP (Thermo Fisher, Waltham, MA, USA) | 32430 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zurlo, M.; Zuccato, C.; Cosenza, L.C.; Gamberini, M.R.; Finotti, A.; Gambari, R. Increased Expression of α-Hemoglobin Stabilizing Protein (AHSP) mRNA in Erythroid Precursor Cells Isolated from β-Thalassemia Patients Treated with Sirolimus (Rapamycin). J. Clin. Med. 2024, 13, 2479. https://doi.org/10.3390/jcm13092479
Zurlo M, Zuccato C, Cosenza LC, Gamberini MR, Finotti A, Gambari R. Increased Expression of α-Hemoglobin Stabilizing Protein (AHSP) mRNA in Erythroid Precursor Cells Isolated from β-Thalassemia Patients Treated with Sirolimus (Rapamycin). Journal of Clinical Medicine. 2024; 13(9):2479. https://doi.org/10.3390/jcm13092479
Chicago/Turabian StyleZurlo, Matteo, Cristina Zuccato, Lucia Carmela Cosenza, Maria Rita Gamberini, Alessia Finotti, and Roberto Gambari. 2024. "Increased Expression of α-Hemoglobin Stabilizing Protein (AHSP) mRNA in Erythroid Precursor Cells Isolated from β-Thalassemia Patients Treated with Sirolimus (Rapamycin)" Journal of Clinical Medicine 13, no. 9: 2479. https://doi.org/10.3390/jcm13092479