Preimplantation Genetic Diagnosis: Prenatal Testing for Embryos Finally Achieving Its Potential

Abstract
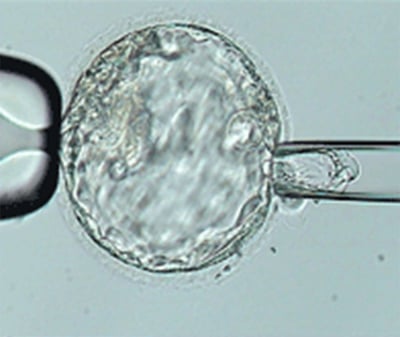
:
1. Introduction
2. Utilization of PGD

{kind=link}
{kind=link}
{kind=link}
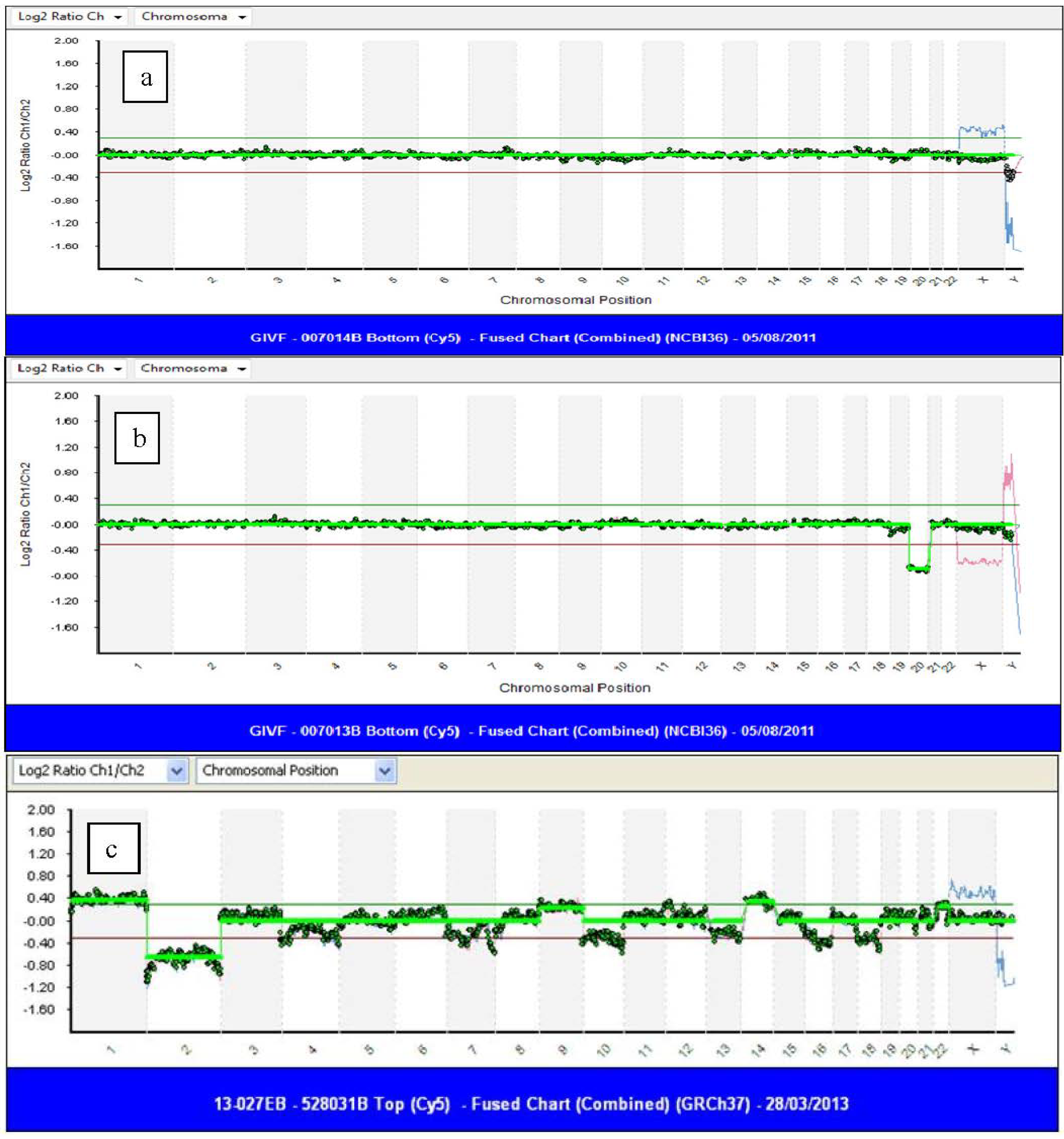{kind=link}
| Indication | Cycles | Percentage |
|---|---|---|
| Single gene disorders | 11,084 | 21% |
| Aneuploidy screening | 30,033 | 58% |
| Inherited chromosome abnormalities | 8104 | 16% |
| Sexing for X-linked disease | 1603 | 3% |
| Non-medical (social) sexing | 765 | 2% |
| Indication for PGD | Percent of Clinics Offering This Type of PGD |
|---|---|
| Aneuploidy testing | 93% |
| Single gene disorders | 82% |
| Structural chromosome rearrangements | 67% |
| Fetal sex for X-linked disease | 58% |
| Non-medical sex selection | 42% |
| Avoid adult onset disorder | 28% |
| HLA * typing with disease testing | 24% |
| HLA typing without disease testing | 6% |
| Selection for a disability | 3% |
3. Tissue Biopsy for PGD
3.1. Polar Body Biopsy
3.2. Cleavage Stage (Blastomere) Biopsy
3.3. Blastocyst (Trophectoderm) Biopsy
4. Technique of Genetic Analysis in PGD
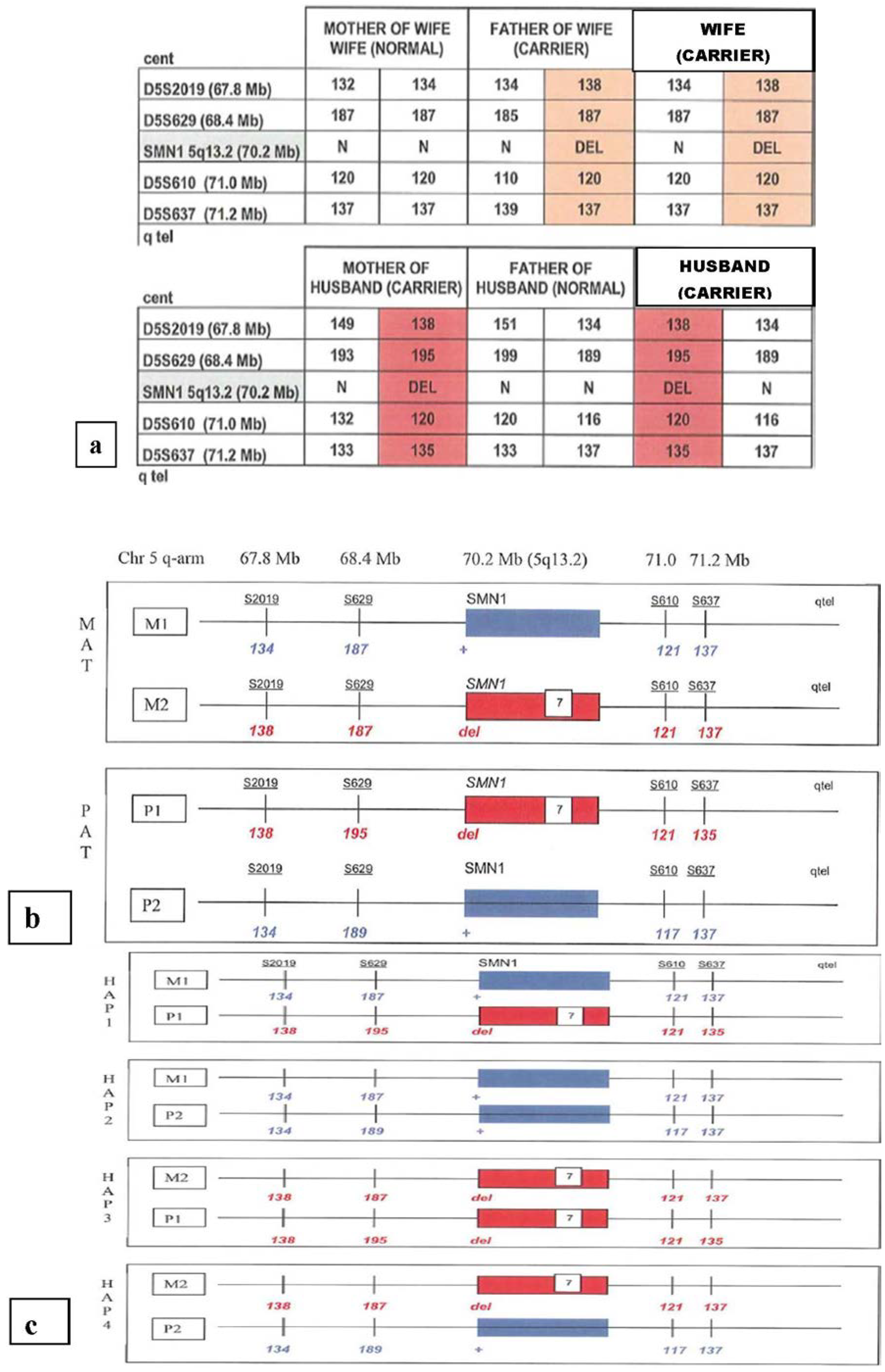
4.1. Single Gene Disorders

4.2. Age-Related Chromosome Aneuploidy

4.3. Structural Chromosome Rearrangements
5. Next Generation Sequencing and PGD of the Future
6. Conclusions
Conflicts of Interest
References
- Edwards, R.G.; Gardner, R.L. Sexing of live rabbit blastocysts. Nature 1967, 214, 576–577. [Google Scholar] [CrossRef]
- Steptoe, P.C.; Edwards, R.G. Birth after the reimplantation of a human embryo. Lancet 1978, 2. [Google Scholar] [CrossRef]
- Monk, M.H.; Handyside, A.H. Sexing of preimplantation embryos by measurement of X-linked gene dosage in a single blastomere. J. Reprod. Fert. 1988, 82, 365–368. [Google Scholar] [CrossRef]
- Handyside, A.H.; Kontogianni, E.H.; Hardy, K.; Winston, R.M. Pregnancies from biopsied human preimplantation embryos sexed by Y-specific DNA amplification. Nature 1990, 344, 768–770. [Google Scholar] [CrossRef]
- Handyside, A.H.; Lesko, J.G.; Tarin, J.J.; Winston, R.M.; Hughes, M.R. Birth of a normal girl after in vitro fertilization and preimplantation diagnostic testing for cystic fibrosis. N. Engl. J. Med. 1992, 327, 905–909. [Google Scholar] [CrossRef]
- Verlinsky, Y.; Ginsberg, N.; Lifchez, A.; Valle, J.; Moise, J.; Strom, C.M. Analysis of the first polar body: Preconception genetic diagnosis. Hum. Reprod. 1990, 5, 826–829. [Google Scholar]
- Griffin, D.K.; Handyside, A.H.; Penketh, R.J.; Winston, R.M.; Delhanty, J.D. Fluorescent in-situ hybridization to interphase nuclei of human preimplantation embryos with X and Y chromosome specific probes. Hum. Reprod. 1991, 6, 101–105. [Google Scholar]
- Grifo, J.A.; Boyle, A.; Tang, Y.X.; Ward, D.C. Preimplantation genetic diagnosis. In situ hybridization as a tool for analysis. Arch. Pathol. Lab. Med. 1992, 116, 393–397. [Google Scholar]
- Munne, S.; Lee, A.; Rosenwaks, Z.; Grifo, J.; Cohen, J. Diagnosis of major chromosome aneuploidies in human preimplantation embryos. Hum. Reprod. 1993, 8, 2185–2191. [Google Scholar]
- Traeger-Synodinos, J.C.; Goossens, E.V. Data from ESHRE PGD Consortium. Hum. Reprod. 2013, 28, S1. [Google Scholar]
- Baruch, S.; Kaufman, D.; Hudson, K.L. Genetic testing of embryos: Practices and perspectives of US in vitro fertilization clinics. Fertil. Steril. 2008, 89, 1053–1058. [Google Scholar] [CrossRef]
- Verlinsky, Y.; Rechitsky, S.; Schoolcraft, W.; Strom, C.; Kuliev, A. Preimplantation diagnosis for Fanconi anemia combined with HLA matching. JAMA 2001, 285, 3130–3133. [Google Scholar] [CrossRef]
- Verlinsky, Y.; Rechitsky, S.; Sharapova, T.; Morris, R.; Taranissi, M.; Kuliev, A. Preimplantation HLA testing. JAMA 2004, 291, 2079–2085. [Google Scholar] [CrossRef]
- Baruch, S. Preimplantation genetic diagnosis and parental preferences: Beyond deadly disease. Houst. J. Health Law Policy 2008, 8, 245–270. [Google Scholar]
- Baruch, S.; Kaufman, D.J.; Hudson, K.L. Preimplantation genetic screening: A survey of in vitro fertilization clinics. Genet. Med. 2008, 10, 685–690. [Google Scholar] [CrossRef]
- Stern, H.; Wiley, R.; Matken, R.; Karabinus, D.; Blauer, K. MicroSort™ babies: 1994–2002 preliminary postnatal follow-up results. Fertil. Steril. 2002, 78, 133. [Google Scholar]
- Evans, M.I.; Rosner, M.; Andriole, S.; Alkalay, A.; Gebb, J.; Britt, D.W. Evolution of gender options in multiple pregnancy management. Prenat. Diagn. 2013, 33, 935–939. [Google Scholar]
- Knoppers, B.M.; Isasi, R.M. Regulatory approaches to reproductive genetic testing. Hum. Reprod. 2004, 19, 2695–2701. [Google Scholar] [CrossRef]
- Gianaroli, L.; Crivello, A.M.; Stanghellini, I.; Ferraretti, A.P.; Tabanelli, C.; Magli, M.C. Reiterative changes in the Italian regulation on IVF: The effect on PGD patients’ reproductive decisions. Reprod. Biomed. Online 2014, 28, 125–132. [Google Scholar] [CrossRef]
- Corveleyn, A.; Zika, E.; Morris, M.; Dequeker, E.; Davies, L.J.; Sermon, K.; Antiñolo, G.; Schmutzler, A.; Vanecek, J.; Palau, F.; et al. Preimplantation Genetic Diagnosis in Europe; Office for Official Publications of the European Communities: Luxembourg, Luxembourg, 2007. [Google Scholar]
- Corveleyn, A.; Morris, M.A.; Dequeker, E.; Sermon, K.; Davies, J.L.; Antinolo, G.; Schmutzler, A.; Vanecek, J.; Nagels, N.; Zika, E.; et al. Provision and quality assurance of preimplantation genetic diagnosis in Europe. Eur. J. Hum. Genet. 2008, 16, 290–299. [Google Scholar] [CrossRef] [Green Version]
- Scott, K.L.; Hong, K.H.; Scott, R.T. Selecting the optimal time to perform biopsy for preimplantation genetic testing. Fertil. Steril. 2013, 100, 608–614. [Google Scholar] [CrossRef]
- Montag, M.; Koster, M.; Strowitzki, T.; Toth, B. Poar body biopsy. Fertil. Steril. 2013, 100, 603–607. [Google Scholar] [CrossRef]
- Kuliev, A.; Rechitsky, S. Polar body-based preimplantation genetic diagnosis for Mendelian disorders. Mol. Hum. Reprod. 2011, 17, 275–285. [Google Scholar] [CrossRef]
- Kuliev, A.; Zlatopolsky, Z.; Kirillova, I.; Spivakova, J.; Cieslak Janzen, J. Meiosis errors in over 20,000 oocytes studied in the practice of preimplantation aneuploidy testing. Reprod. Biomed. Online 2010, 22, 2–8. [Google Scholar]
- Verlinsky, Y.; Cieslak, J.; Ivakhnenko, V.; Evsikov, S.; Wolf, G.; White, M.; Lifchez, A.; Kaplan, B.; Moise, J.; Valle, J.; et al. Prevention of age-related aneuploidies by polar body testing of oocytes. J. Assist. Reprod. Genet. 1999, 16, 165–169. [Google Scholar] [CrossRef]
- Handyside, A.H.; Montag, M.; Magli, M.C.; Repping, S.; Harper, J.; Schmutzler, A.; Vesela, K.; Gianaroli, L.; Geraedts, J. Multiple meiotic errors caused by predivision of chromatids in women of advanced maternal age undergoing in vitro fertilisation. Eur. J. Hum. Genet. 2012, 20, 742–747. [Google Scholar] [CrossRef]
- Magli, M.C.; Montag, M.; Koster, M.; Muzi, L.; Geraedts, J.; Collins, J.; Goossens, V.; Handyside, A.H.; Harper, J.; Repping, S.; et al. Polar body array CGH for prediction of the status of the corresponding oocyte. Part II: Technical aspects. Hum. Reprod. 2011, 26, 3181–3185. [Google Scholar] [CrossRef]
- Geraedts, J.; Montag, M.; Magli, M.C.; Repping, S.; Handyside, A.; Staessen, C.; Harper, J.; Schmutzler, A.; Collins, J.; Goossens, V.; et al. Polar body array CGH for prediction of the status of the corresponding oocyte. Part I: Clinical results. Hum. Reprod. 2011, 26, 3173–3180. [Google Scholar]
- Scriven, P.N.; Ogilvie, C.M.; Khalaf, Y. Embryo selection in IVF: Is polar body array comparative genomic hybridization accurate enough? Hum. Reprod. 2012, 27, 951–953. [Google Scholar] [CrossRef]
- Staessen, C.; Platteau, P.; van Assche, E.; Michiels, A.; Tournaye, H.; Camus, M.; Devroey, P.; Liebaers, I.; van Steirteghem, A. Comparison of blastocyst transfer with or without preimplantation genetic diagnosis for aneuploidy screening in couples with advanced maternal age: A prospective randomized controlled trial. Hum. Reprod. 2004, 19, 2849–2858. [Google Scholar] [CrossRef]
- Dumoulin, J.C.; Bras, M.; Coonen, E.; Dreesen, J.; Geraedts, J.P.; Evers, J.L. Effect of Ca2+/Mg2+-free medium on the biopsy procedure for preimplantation genetic diagnosis and further development of human embryos. Hum. Reprod. 1998, 13, 2880–2883. [Google Scholar] [CrossRef]
- Goossens, V.; de Rycke, M.; de Vos, A.; Staessen, C.; Michiels, A.; Verpoest, W.; van Steirteghem, A.; Bertrand, C.; Liebaers, I.; Devroey, P.; et al. Diagnostic efficiency, embryonic development and clinical outcome after the biopsy of one or two blastomeres for preimplantation genetic diagnosis. Hum. Reprod. 2008, 23, 481–492. [Google Scholar] [CrossRef]
- Cohen, J.; Wells, D.; Munne, S. Removal of 2 cells from cleavage stage embryos is likely to reduce the efficacy of chromosomal tests that are used to enhance implantation rates. Fertil. Steril. 2007, 87, 496–503. [Google Scholar] [CrossRef]
- Van Echten-Arends, J.; Mastenbroek, S.; Sikkema-Raddatz, B.; Korevaar, J.C.; Heineman, M.J.; van der Veen, F.; Repping, S. Chromosomal mosaicism in human preimplantation embryos: A systematic review. Hum. Reprod. Updat. 2011, 17, 620–627. [Google Scholar] [CrossRef]
- Bielanska, M.; Tan, S.L.; Ao, A. Chromosomal mosaicism throughout human preimplantation development in vitro: Incidence, type, and relevance to embryo outcome. Hum. Reprod. 2002, 17, 413–419. [Google Scholar] [CrossRef]
- Bielanska, M.; Tan, S.L.; Ao, A. High rate of mixoploidy among human blastocysts cultured in vitro. Fertil. Steril. 2002, 78, 1248–1253. [Google Scholar] [CrossRef]
- Northrop, L.E.; Treff, N.R.; Levy, B.; Scott, R.T., Jr. SNP microarray-based 24 chromosome aneuploidy screening demonstrates that cleavage-stage FISH poorly predicts aneuploidy in embryos that develop to morphologically normal blastocysts. Mol. Hum. Reprod. 2010, 16, 590–600. [Google Scholar] [CrossRef]
- Hardy, K.; Handyside, A.H.; Winston, R.M. The human blastocyst: Cell number, death and allocation during late preimplantation development in vitro. Development 1989, 107, 597–604. [Google Scholar]
- Edgar, D.H.; Bourne, H.; Speirs, A.L.; McBain, J.C. A quantitative analysis of the impact of cryopreservation on the implantation potential of human early cleavage stage embryos. Hum. Reprod. 2000, 15, 175–179. [Google Scholar] [CrossRef]
- McArthur, S.J.; Leigh, D.; Marshall, J.T.; de Boer, K.A.; Jansen, R.P. Pregnancies and live births after trophectoderm biopsy and preimplantation genetic testing of human blastocysts. Fertil. Steril. 2005, 84, 1628–1636. [Google Scholar] [CrossRef]
- McArthur, S.J.; Leigh, D.; Marshall, J.T.; Gee, A.J.; de Boer, K.A.; Jansen, R.P. Blastocyst trophectoderm biopsy and preimplantation genetic diagnosis for familial monogenic disorders and chromosomal translocations. Prenat. Diagn. 2008, 28, 434–442. [Google Scholar] [CrossRef]
- Scott, R.T., Jr.; Upham, K.M.; Forman, E.J.; Zhao, T.; Treff, N.R. Cleavage-stage biopsy significantly impairs human embryonic implantation potential while blastocyst biopsy does not: A randomized and paired clinical trial. Fertil. Steril. 2013, 100, 624–630. [Google Scholar]
- Holding, C.; Monk, M. Diagnosis of beta-thalassaemia by DNA amplification in single blastomeres from mouse preimplantation embryos. Lancet 1989, 2, 532–535. [Google Scholar] [CrossRef]
- Rechitsky, S.; Strom, C.; Verlinsky, O.; Amet, T.; Ivakhnenko, V.; Kukharenko, V.; Kuliev, A.; Verlinsky, Y. Allele dropout in polar bodies and blastomeres. J. Assist. Reprod. Genet. 1998, 15, 253–257. [Google Scholar] [CrossRef]
- Wells, D.; Sherlock, J.K. Strategies for preimplantation genetic diagnosis of single gene disorders by DNA amplification. Prenat. Diagn. 1998, 18, 1389–1401. [Google Scholar] [CrossRef]
- Zhang, C.; Zhang, C.; Chen, S.; Yin, X.; Pan, X.; Lin, G.; Tan, Y.; Tan, K.; Xu, Z.; Hu, P.; et al. A single cell level based method for copy number variation analysis by low coverage massively parallel sequencing. PLoS One 2013, 8, e54236. [Google Scholar]
- Wells, D.; Delhanty, J.D. Comprehensive chromosomal analysis of human preimplantation embryos using whole genome amplification and single cell comparative genomic hybridization. Mol. Hum. Reprod. 2000, 6, 1055–1062. [Google Scholar] [CrossRef]
- Treff, N.R.; Su, J.; Tao, X.; Levy, B.; Scott, R.T., Jr. Accurate single cell 24 chromosome aneuploidy screening using whole genome amplification and single nucleotide polymorphism microarrays. Fertil. Steril. 2010, 94, 2017–2021. [Google Scholar] [CrossRef]
- Handyside, A.H.; Robinson, M.D.; Simpson, R.J.; Omar, M.B.; Shaw, M.A.; Grudzinskas, J.G.; Rutherford, A. Isothermal whole genome amplification from single and small numbers of cells: A new era for preimplantation genetic diagnosis of inherited disease. Mol. Hum. Reprod. 2004, 10, 767–772. [Google Scholar] [CrossRef]
- Hellani, A.; Coskun, S.; Benkhalifa, M.; Tbakhi, A.; Sakati, N.; Al-Odaib, A.; Ozand, P. Multiple displacement amplification on single cell and possible PGD applications. Mol. Hum. Reprod. 2004, 10, 847–852. [Google Scholar] [CrossRef]
- Renwick, P.; Trussler, J.; Lashwood, A.; Braude, P.; Ogilvie, C.M. Preimplantation genetic haplotyping: 127 Diagnostic cycles demonstrating a robust, efficient alternative to direct mutation testing on single cells. Reprod. Biomed. Online 2010, 20, 470–476. [Google Scholar] [CrossRef]
- Renwick, P.J.; Trussler, J.; Ostad-Saffari, E.; Fassihi, H.; Black, C.; Braude, P.; Ogilvie, C.M.; Abbs, S. Proof of principle and first cases using preimplantation genetic haplotyping—A paradigm shift for embryo diagnosis. Reprod. Biomed. Online 2006, 13, 110–119. [Google Scholar] [CrossRef]
- Zong, C.; Lu, S.; Chapman, A.R.; Xie, X.S. Genome-wide detection of single-nucleotide and copy-number variations of a single human cell. Science 2012, 338, 1622–1626. [Google Scholar] [CrossRef]
- Tran, K.D.; Mariani, B.D.; Shrestha, S.; Sands, M.C.; Novik, V.; Stern, H.J. Successful application of simultaneous chromosomal microarray analysis (CMA) and genetic disease diagnosis from day 5 trophectoderm embryo biopsies with day 6 transfer. Fertil. Steril. 2012, 98, S135. [Google Scholar]
- Rabinowitz, M.; Ryan, A.; Gemelos, G.; Hill, M.; Baner, J.; Cinnioglu, C.; Banjevic, M.; Potter, D.; Petrov, D.A.; Demko, Z. Origins and rates of aneuploidy in human blastomeres. Fertil. Steril. 2012, 97, 395–401. [Google Scholar] [CrossRef]
- Delhanty, J.D.; Harper, J.C.; Ao, A.; Handyside, A.H.; Winston, R.M. Multicolour FISH detects frequent chromosomal mosaicism and chaotic division in normal preimplantation embryos from fertile patients. Hum. Genet. 1997, 99, 755–760. [Google Scholar] [CrossRef]
- Hodes-Wertz, B.; Grifo, J.; Ghadir, S.; Kaplan, B.; Laskin, C.A.; Glassner, M.; Munne, S. Idiopathic recurrent miscarriage is caused mostly by aneuploid embryos. Fertil. Steril. 2012, 98, 675–680. [Google Scholar] [CrossRef]
- Menasha, J.; Levy, B.; Hirschhorn, K.; Kardon, N.B. Incidence and spectrum of chromosome abnormalities in spontaneous abortions: New insights from a 12-year study. Genet. Med. 2005, 7, 251–263. [Google Scholar] [CrossRef]
- Verlinsky, Y.; Cieslak, J.; Freidine, M.; Ivakhnenko, V.; Wolf, G.; Kovalinskaya, L.; White, M.; Lifchez, A.; Kaplan, B.; Moise, J.; et al. Pregnancies following pre-conception diagnosis of common aneuploidies by fluorescent in-situ hybridization. Hum. Reprod. 1995, 10, 1923–1927. [Google Scholar]
- Bielanska, M.; Jin, S.; Bernier, M.; Tan, S.L.; Ao, A. Diploid-aneuploid mosaicism in human embryos cultured to the blastocyst stage. Fertil. Steril. 2005, 84, 336–342. [Google Scholar] [CrossRef]
- Wells, D.; Alfarawati, S.; Fragouli, E. Use of comprehensive chromosomal screening for embryo assessment: Microarrays and CGH. Mol. Hum. Reprod. 2008, 14, 703–710. [Google Scholar] [CrossRef]
- Voullaire, L.; Slater, H.; Williamson, R.; Wilton, L. Chromosome analysis of blastomeres from human embryos by using comparative genomic hybridization. Hum. Genet. 2000, 106, 210–217. [Google Scholar] [CrossRef]
- Gianaroli, L.; Magli, M.C.; Ferraretti, A.P.; Munne, S. Preimplantation diagnosis for aneuploidies in patients undergoing in vitro fertilization with a poor prognosis: Identification of the categories for which it should be proposed. Fertil. Steril. 1999, 72, 837–844. [Google Scholar] [CrossRef]
- Munne, S.; Magli, C.; Cohen, J.; Morton, P.; Sadowy, S.; Gianaroli, L.; Tucker, M.; Marquez, C.; Sable, D.; Ferraretti, A.P.; et al. Positive outcome after preimplantation diagnosis of aneuploidy in human embryos. Hum. Reprod. 1999, 14, 2191–2199. [Google Scholar] [CrossRef]
- Munne, S.; Sandalinas, M.; Escudero, T.; Velilla, E.; Walmsley, R.; Sadowy, S.; Cohen, J.; Sable, D. Improved implantation after preimplantation genetic diagnosis of aneuploidy. Reprod. Biomed. Online 2003, 7, 91–97. [Google Scholar] [CrossRef]
- Platteau, P.; Staessen, C.; Michiels, A.; van Steirteghem, A.; Liebaers, I.; Devroey, P. Preimplantation genetic diagnosis for aneuploidy screening in women older than 37 years. Fertil. Steril. 2005, 84, 319–324. [Google Scholar] [CrossRef]
- Platteau, P.; Staessen, C.; Michiels, A.; van Steirteghem, A.; Liebaers, I.; Devroey, P. Preimplantation genetic diagnosis for aneuploidy screening in patients with unexplained recurrent miscarriages. Fertil. Steril. 2005, 83, 393–397. [Google Scholar] [CrossRef]
- Fragouli, E.; Wells, D. Aneuploidy screening for embryo selection. Semin. Reprod. Med. 2012, 30, 289–301. [Google Scholar] [CrossRef]
- Mastenbroek, S.; Twisk, M.; van Echten-Arends, J.; Sikkema-Raddatz, B.; Korevaar, J.C.; Verhoeve, H.R.; Vogel, N.E.; Arts, E.G.; de Vries, J.W.; Bossuyt, P.M.; et al. In vitro fertilization with preimplantation genetic screening. N. Engl. J. Med. 2007, 357, 9–17. [Google Scholar] [CrossRef]
- Munne, S.; Gianaroli, L.; Tur-Kaspa, I.; Magli, C.; Sandalinas, M.; Grifo, J.; Cram, D.; Kahraman, S.; Verlinsky, Y.; Simpson, J.L. Substandard application of preimplantation genetic screening may interfere with its clinical success. Fertil. Steril. 2007, 88, 781–784. [Google Scholar] [CrossRef]
- Cohen, J.; Grifo, J.A. Multicentre trial of preimplantation genetic screening reported in the New England Journal of Medicine: An in-depth look at the findings. Reprod. Biomed. Online 2007, 15, 365–366. [Google Scholar] [CrossRef]
- Munne, S.; Wells, D.; Cohen, J. Technology requirements for preimplantation genetic diagnosis to improve assisted reproduction outcomes. Fertil. Steril. 2010, 94, 408–430. [Google Scholar] [CrossRef]
- Simpson, J.L.; Bombard, A.T. Chromosome Abnormalities in Spontaneous Abortion: Frequency, Pathology and Genetic Counseling; Blackwell: London, UK, 1987. [Google Scholar]
- Magli, M.C.; Jones, G.M.; Gras, L.; Gianaroli, L.; Korman, I.; Trounson, A.O. Chromosome mosaicism in day 3 aneuploid embryos that develop to morphologically normal blastocysts in vitro. Hum. Reprod. 2000, 15, 1781–1786. [Google Scholar] [CrossRef]
- Kuo, H.C.; Ogilvie, C.M.; Handyside, A.H. Chromosomal mosaicism in cleavage-stage human embryos and the accuracy of single-cell genetic analysis. J. Assist. Reprod. Genet. 1998, 15, 276–280. [Google Scholar] [CrossRef]
- Harper, J.; Sermon, K.; Geraedts, J.; Vesela, K.; Harton, G.; Thornhill, A.; Pehlivan, T.; Fiorentino, F.; SenGupta, S.; de Die-Smulders, C.; et al. What next for preimplantation genetic screening? Hum. Reprod. 2008, 23, 478–480. [Google Scholar] [CrossRef]
- DeUgarte, C.M.; Li, M.; Surrey, M.; Danzer, H.; Hill, D.; DeCherney, A.H. Accuracy of FISH analysis in predicting chromosomal status in patients undergoing preimplantation genetic diagnosis. Fertil. Steril. 2008, 90, 1049–1054. [Google Scholar] [CrossRef]
- Magli, M.C.; Gianaroli, L.; Fortini, D.; Ferraretti, A.P.; Munne, S. Impact of blastomere biopsy and cryopreservation techniques on human embryo viability. Hum. Reprod. 1999, 14, 770–773. [Google Scholar] [CrossRef]
- Kuwayama, M. Highly efficient vitrification for cryopreservation of human oocytes and embryos: The Cryotop method. Theriogenology 2007, 67, 73–80. [Google Scholar] [CrossRef]
- Fragouli, E.; Alfarawati, S.; Goodall, N.N.; Sanchez-Garcia, J.F.; Colls, P.; Wells, D. The cytogenetics of polar bodies: Insights into female meiosis and the diagnosis of aneuploidy. Mol. Hum. Reprod. 2011, 17, 286–295. [Google Scholar] [CrossRef]
- Wells, D.; Escudero, T.; Levy, B.; Hirschhorn, K.; Delhanty, J.D.; Munne, S. First clinical application of comparative genomic hybridization and polar body testing for preimplantation genetic diagnosis of aneuploidy. Fertil. Steril. 2002, 78, 543–549. [Google Scholar] [CrossRef]
- Fragouli, E.; Escalona, A.; Gutierrez-Mateo, C.; Tormasi, S.; Alfarawati, S.; Sepulveda, S.; Noriega, L.; Garcia, J.; Wells, D.; Munne, S. Comparative genomic hybridization of oocytes and first polar bodies from young donors. Reprod. Biomed. Online 2009, 19, 228–237. [Google Scholar] [CrossRef]
- Fragouli, E.; Katz-Jaffe, M.; Alfarawati, S.; Stevens, J.; Colls, P.; Goodall, N.N.; Tormasi, S.; Gutierrez-Mateo, C.; Prates, R.; Schoolcraft, W.B.; et al. Comprehensive chromosome screening of polar bodies and blastocysts from couples experiencing repeated implantation failure. Fertil. Steril. 2010, 94, 875–887. [Google Scholar] [CrossRef]
- Fragouli, E.; Alfarawati, S.; Daphnis, D.D.; Goodall, N.N.; Mania, A.; Griffiths, T.; Gordon, A.; Wells, D. Cytogenetic analysis of human blastocysts with the use of FISH, CGH and aCGH: Scientific data and technical evaluation. Hum. Reprod. 2011, 26, 480–490. [Google Scholar] [CrossRef]
- Fragouli, E.; Lenzi, M.; Ross, R.; Katz-Jaffe, M.; Schoolcraft, W.B.; Wells, D. Comprehensive molecular cytogenetic analysis of the human blastocyst stage. Hum. Reprod. 2008, 23, 2596–2608. [Google Scholar] [CrossRef]
- Gutierrez-Mateo, C.; Colls, P.; Sanchez-Garcia, J.; Escudero, T.; Prates, R.; Ketterson, K.; Wells, D.; Munne, S. Validation of microarray comparative genomic hybridization for comprehensive chromosome analysis of embryos. Fertil. Steril. 2011, 95, 953–958. [Google Scholar] [CrossRef]
- Treff, N.R.; Levy, B.; Su, J.; Northrop, L.E.; Tao, X.; Scott, R.T., Jr. SNP microarray-based 24 chromosome aneuploidy screening is significantly more consistent than FISH. Mol. Hum. Reprod. 2010, 16, 583–589. [Google Scholar] [CrossRef]
- Mamas, T.; Gordon, A.; Brown, A.; Harper, J.; Sengupta, S. Detection of aneuploidy by array comparative genomic hybridization using cell lines to mimic a mosaic trophectoderm biopsy. Fertil. Steril. 2012, 97, 943–947. [Google Scholar] [CrossRef]
- Novik, V.; Morris, E.B.; Tran, K.D.; Stern, H.J.; Mariani, B.D.; Stanley, W.S. Clinical validation for mosaicism detected in trophectoderm biopsy samples analyzed by chromosomal microarrays. Fertil. Steril. 2012, 98, S136. [Google Scholar]
- Schoolcraft, W.B.; Fragouli, E.; Stevens, J.; Munne, S.; Katz-Jaffe, M.G.; Wells, D. Clinical application of comprehensive chromosomal screening at the blastocyst stage. Fertil. Steril. 2010, 94, 1700–1706. [Google Scholar] [CrossRef]
- Schoolcraft, W.B.; Treff, N.R.; Stevens, J.M.; Ferry, K.; Katz-Jaffe, M.; Scott, R.T., Jr. Live birth outcome with trophectoderm biopsy, blastocyst vitrification, and single-nucleotide polymorphism microarray-based comprehensive chromosome screening in infertile patients. Fertil. Steril. 2011, 96, 638–640. [Google Scholar] [CrossRef]
- Treff, N.R.; Tao, X.; Ferry, K.M.; Su, J.; Taylor, D.; Scott, R.T., Jr. Development and validation of an accurate quantitative real-time polymerase chain reaction-based assay for human blastocyst comprehensive chromosomal aneuploidy screening. Fertil. Steril. 2012, 97, 819–824. [Google Scholar] [CrossRef]
- Handyside, A.H. 24-Chromosome copy number analysis: A comparison of available technologies. Fertil. Steril. 2013, 100, 595–602. [Google Scholar] [CrossRef]
- Scott, R.T., Jr.; Ferry, K.; Su, J.; Tao, X.; Scott, K.; Treff, N.R. Comprehensive chromosome screening is highly predictive of the reproductive potential of human embryos: A prospective, blinded, nonselection study. Fertil. Steril. 2012, 97, 870–875. [Google Scholar] [CrossRef]
- Harton, G.L.; Munne, S.; Surrey, M.; Grifo, J.; Kaplan, B.; McCulloh, D.H.; Griffin, D.K.; Wells, D. Diminished effect of maternal age on implantation after preimplantation genetic diagnosis with array comparative genomic hybridization. Fertil. Steril. 2013, 100, 1695–1703. [Google Scholar] [CrossRef]
- Scott, R.T., Jr.; Upham, K.M.; Forman, E.J.; Hong, K.H.; Scott, K.L.; Taylor, D.; Tao, X.; Treff, N.R. Blastocyst biopsy with comprehensive chromosome screening and fresh embryo transfer significantly increases in vitro fertilization implantation and delivery rates: A randomized controlled trial. Fertil. Steril. 2013, 100, 697–703. [Google Scholar] [CrossRef]
- ASRM Practice Committee. Multiple gestation associated with infertility therapy: An American Society for Reproductive Medicine Practice Committee opinion. Fertil. Steril. 2012, 97, 825–834. [Google Scholar] [CrossRef]
- Yang, Z.; Liu, J.; Collins, G.S.; Salem, S.A.; Liu, X.; Lyle, S.S.; Peck, A.C.; Sills, E.S.; Salem, R.D. Selection of single blastocysts for fresh transfer via standard morphology assessment alone and with array CGH for good prognosis IVF patients: Results from a randomized pilot study. Mol. Cytogenet. 2012, 5. [Google Scholar] [CrossRef]
- Forman, E.J.; Hong, K.H.; Ferry, K.M.; Tao, X.; Taylor, D.; Levy, B.; Treff, N.R.; Scott, R.T., Jr. In vitro fertilization with single euploid blastocyst transfer: A randomized controlled trial. Fertil. Steril. 2013, 100, 100–107. [Google Scholar] [CrossRef]
- Van Dyke, D.L.; Weiss, L.; Roberson, J.R.; Babu, V.R. The frequency and mutation rate of balanced autosomal rearrangements in man estimated from prenatal genetic studies for advanced maternal age. Am. J. Hum. Genet. 1983, 35, 301–308. [Google Scholar]
- Scriven, P.N.; Handyside, A.H.; Ogilvie, C.M. Chromosome translocations: Segregation modes and strategies for preimplantation genetic diagnosis. Prenat. Diagn. 1998, 18, 1437–1449. [Google Scholar] [CrossRef]
- Fiorentino, F.; Kokkali, G.; Biricik, A.; Stavrou, D.; Ismailoglu, B.; de Palma, R.; Arizzi, L.; Harton, G.; Sessa, M.; Pantos, K. Polymerase chain reaction-based detection of chromosomal imbalances on embryos: The evolution of preimplantation genetic diagnosis for chromosomal translocations. Fertil. Steril. 2010, 94, 2001–2011. [Google Scholar] [CrossRef]
- Alfarawati, S.; Fragouli, E.; Colls, P.; Wells, D. First births after preimplantation genetic diagnosis of structural chromosome abnormalities using comparative genomic hybridization and microarray analysis. Hum. Reprod. 2011, 26, 1560–1574. [Google Scholar] [CrossRef]
- Fiorentino, F.; Spizzichino, L.; Bono, S.; Biricik, A.; Kokkali, G.; Rienzi, L.; Ubaldi, F.M.; Iammarrone, E.; Gordon, A.; Pantos, K. PGD for reciprocal and Robertsonian translocations using array comparative genomic hybridization. Hum. Reprod. 2011, 26, 1925–1935. [Google Scholar] [CrossRef]
- Treff, N.R.; Tao, X.; Schillings, W.J.; Bergh, P.A.; Scott, R.T., Jr.; Levy, B. Use of single nucleotide polymorphism microarrays to distinguish between balanced and normal chromosomes in embryos from a translocation carrier. Fertil. Steril. 2011, 96, 58–65. [Google Scholar] [CrossRef]
- Scriven, P.N.; Flinter, F.A.; Khalaf, Y.; Lashwood, A.; Mackie Ogilvie, C. Benefits and drawbacks of preimplantation genetic diagnosis (PGD) for reciprocal translocations: Lessons from a prospective cohort study. Eur. J. Hum. Genet. 2013, 21, 1035–1041. [Google Scholar] [CrossRef]
- Mertes, F.; Elsharawy, A.; Sauer, S.; van Helvoort, J.M.; van der Zaag, P.J.; Franke, A.; Nilsson, M.; Lehrach, H.; Brookes, A.J. Targeted enrichment of genomic DNA regions for next-generation sequencing. Brief. Funct. Genomics 2011, 10, 374–386. [Google Scholar] [CrossRef]
- Treff, N.R.; Fedick, A.; Tao, X.; Devkota, B.; Taylor, D.; Scott, R.T., Jr. Evaluation of targeted next-generation sequencing-based preimplantation genetic diagnosis of monogenic disease. Fertil. Steril. 2013, 99, 1377–1384. [Google Scholar] [CrossRef]
- Fan, H.C.; Blumenfeld, Y.J.; Chitkara, U.; Hudgins, L.; Quake, S.R. Noninvasive diagnosis of fetal aneuploidy by shotgun sequencing DNA from maternal blood. Proc. Natl. Acad. Sci. USA 2008, 105, 16266–16271. [Google Scholar] [CrossRef]
- Yin, X.; Tan, K.; Vajta, G.; Jiang, H.; Tan, Y.; Zhang, C.; Chen, F.; Chen, S.; Zhang, C.; Pan, X.; et al. Massively parallel sequencing for chromosomal abnormality testing in trophectoderm cells of human blastocysts. Biol. Reprod. 2013, 88. [Google Scholar] [CrossRef]
- Li, J.; Yin, X.Y.; Tan, K.; Tan, Y.Q.; Chen, F.; Zhang, L.E.I.; Lin, G.; Jiang, H.; Wang, W. Clinical Application of Massively Parallel Sequencing on Chromosome Abnormality Detection of Human Blastocysts. In Proceedings of the 2013 ESHRE Annual Conference, London, UK, 7–10 July 2013.
- Wells, D.; Kaur, K.; Grifo, J.; Anderson, S.; Taylor, J.; Fragouli, E.; Munne, S. A Novel Embryo Screening Technique Provides New Insight into Embryo Biology and Yeilds the First Pregnancies Following Genome Sequencing. In Proceedings of the 2013 ESHRE Annual Conference, London, UK, 7–10 July 2013.
- Hens, K.; Dondorp, W.; Handyside, A.H.; Harper, J.; Newson, A.J.; Pennings, G.; Rehmann-Sutter, C.; de Wert, G. Dynamics and ethics of comprehensive preimplantation genetic testing: A review of the challenges. Hum. Reprod. Updat. 2013, 19, 366–375. [Google Scholar] [CrossRef]
- Hens, K.; Dondorp, W.J.; Geraedts, J.P.; de Wert, G.M. Comprehensive embryo testing. Experts’ opinions regarding future directions: An expert panel study on comprehensive embryo testing. Hum. Reprod. 2013, 28, 1418–1425. [Google Scholar] [CrossRef]
- Harper, J.C.; Geraedts, J.; Borry, P.; Cornel, M.C.; Dondorp, W.; Gianaroli, L.; Harton, G.; Milachich, T.; Kaariainen, H.; Liebaers, I.; et al. Current issues in medically assisted reproduction and genetics in Europe: Research, clinical practice, ethics, legal issues and policy. European Society of Human Genetics and European Society of Human Reproduction and Embryology. Eur. J. Hum. Genet. 2013, 21, 1–21. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Stern, H.J. Preimplantation Genetic Diagnosis: Prenatal Testing for Embryos Finally Achieving Its Potential. J. Clin. Med. 2014, 3, 280-309. https://doi.org/10.3390/jcm3010280
Stern HJ. Preimplantation Genetic Diagnosis: Prenatal Testing for Embryos Finally Achieving Its Potential. Journal of Clinical Medicine. 2014; 3(1):280-309. https://doi.org/10.3390/jcm3010280
Chicago/Turabian StyleStern, Harvey J. 2014. "Preimplantation Genetic Diagnosis: Prenatal Testing for Embryos Finally Achieving Its Potential" Journal of Clinical Medicine 3, no. 1: 280-309. https://doi.org/10.3390/jcm3010280
APA StyleStern, H. J. (2014). Preimplantation Genetic Diagnosis: Prenatal Testing for Embryos Finally Achieving Its Potential. Journal of Clinical Medicine, 3(1), 280-309. https://doi.org/10.3390/jcm3010280