The Roles of Vitamin A in the Regulation of Carbohydrate, Lipid, and Protein Metabolism

Abstract
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
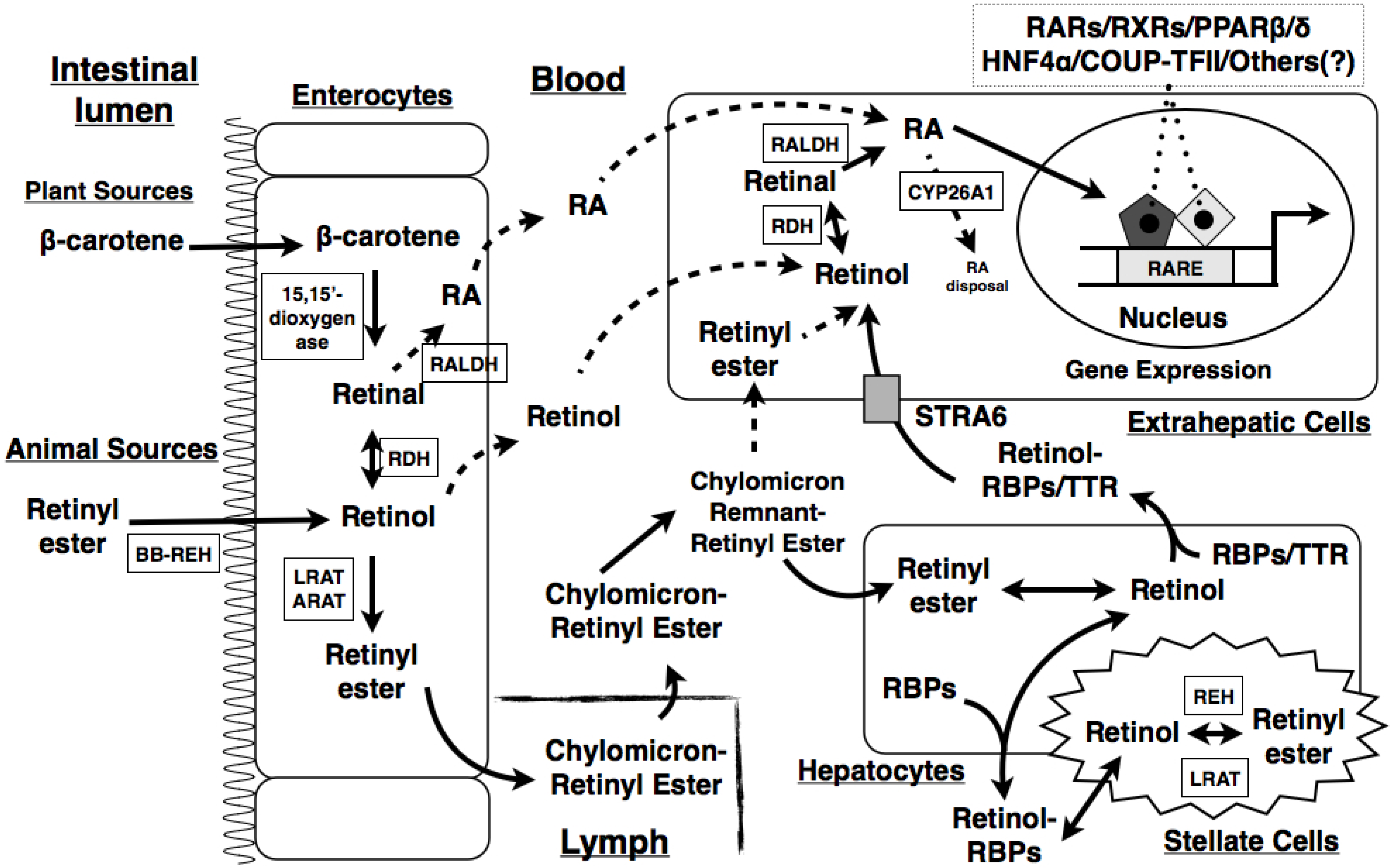
2. Overview of VA and Its Metabolism

3. VA and Growth, Appetite, Taste and Olfaction
4. VA and Plasma Parameters: Clinical Evidence
5. VA and Metabolism in the Liver
5.1. VA and Hepatic Carbohydrate Metabolism
5.2. VA and Hepatic Protein Metabolism
5.3. VA and Hepatic Lipid Metabolism
5.4. VA and Mitochondrial Functions in Hepatocytes
6. VA and Islets of Langerhans in the Pancreas
7. VA and Metabolism in Adipose Tissues
7.1. VA and Metabolism in White Adipose Tissue

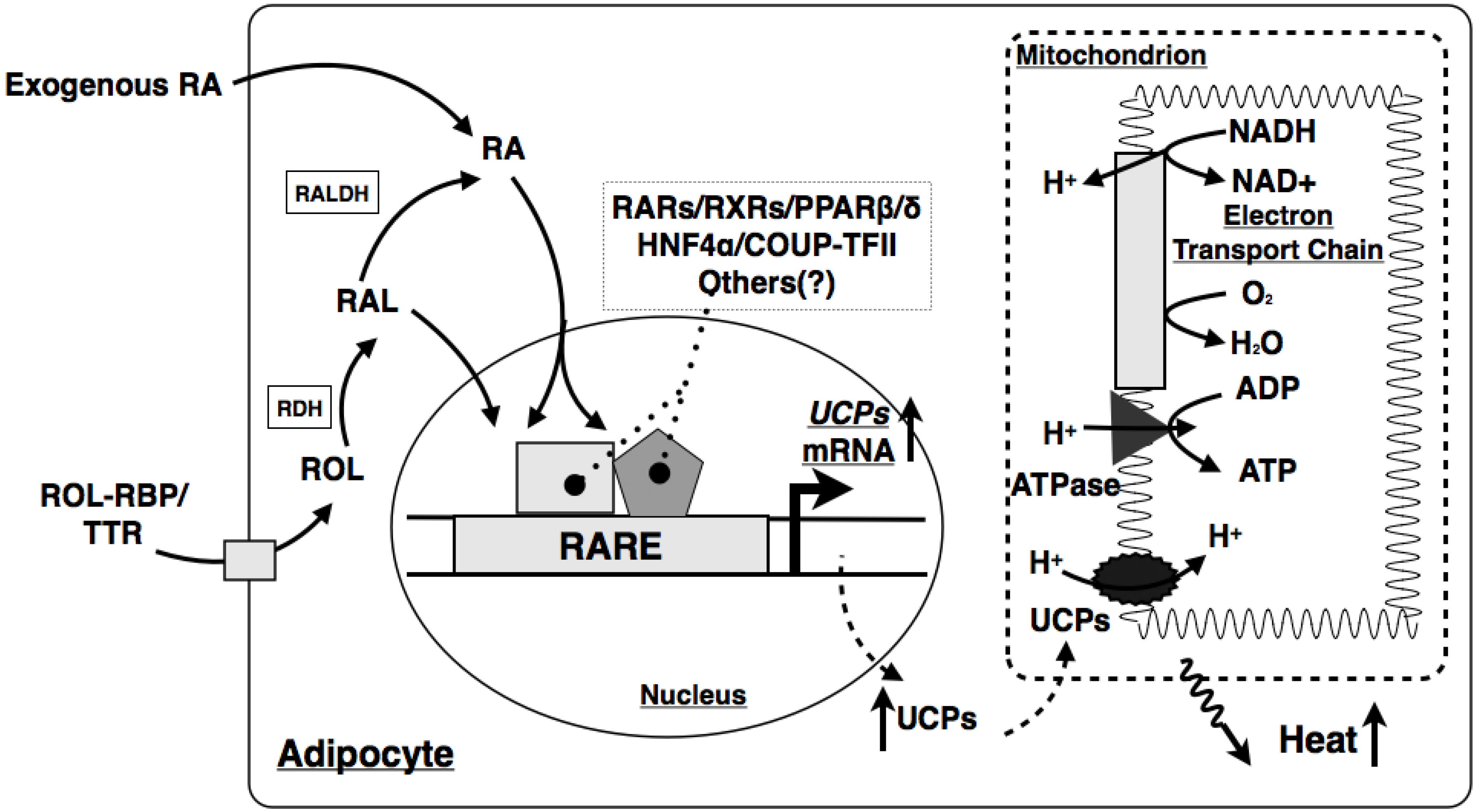
7.2. VA and Metabolism in Brown Adipose Tissue
8. VA and Metabolism in the Skeletal Muscle
9. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- WHO. Consultation, Obesity: Preventing and Managing the Global Epidemic. In World Health Organization Technical Report Series; WHO: Geneva, Switzerland, 2000. [Google Scholar]
- WHO. Burden: Mortality, Morbidity and Risk Factors. In Global Status Report on Noncommunicable Diseases 2010—Description of the Global Burden of NCDs, Their Risk Factors and Determinants; World Health Organization: Geneva, Switzerland, 2011; pp. 1–23. [Google Scholar]
- Jensen, M.D.; Ryan, D.H.; Apovian, C.M.; Loria, C.M.; Ard, J.D.; Millen, B.E.; Comuzzie, A.G.; Nonas, C.A.; Donato, K.A.; Pi-Sunyer, F.X.; et al. 2013 AHA/ACC/TOS Guideline for the Management of Overweight and Obesity in Adults: A Report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and The Obesity Society. J. Am. Coll. Cardiol. 2013. [Google Scholar] [CrossRef]
- Zhao, S.; Li, R.; Li, Y.; Chen, W.; Zhang, Y.; Chen, G. Roles of vitamin A status and retinoids in glucose and fatty acid metabolism. Biochem. Cell Biol. 2012, 90, 142–152. [Google Scholar] [CrossRef]
- Chen, G. Roles of Vitamin A Metabolism in the Development of Hepatic Insulin Resistance. ISRN Hepatol. 2013, 2013, 1–21. [Google Scholar]
- Fall, T.; Ingelsson, E. Genome-wide association studies of obesity and metabolic syndrome. Mol. Cell. Endocrinol. 2014, 382, 740–757. [Google Scholar] [CrossRef]
- Kopelman, P. Symposium 1: Overnutrition: Consequences and solutions. Foresight Report: The obesity challenge ahead. Proc. Nutr. Soc. 2010, 69, 80–85. [Google Scholar] [CrossRef]
- Key, T.J.; Spencer, E.A.; Reeves, G.K. Symposium 1: Overnutrition: Consequences and solutions. Obesity and cancer risk. Proc. Nutr. Soc. 2010, 69, 86–90. [Google Scholar] [CrossRef]
- White, M.F. Insulin signaling in health and disease. Science 2003, 302, 1710–1711. [Google Scholar] [CrossRef]
- Kasuga, M.; Karlsson, F.A.; Kahn, C.R. Insulin stimulates the phosphorylation of the 95,000-dalton subunit of its own receptor. Science 1982, 215, 185–187. [Google Scholar]
- Petruzzelli, L.M.; Ganguly, S.; Smith, C.J.; Cobb, M.H.; Rubin, C.S.; Rosen, O.M. Insulin activates a tyrosine-specific protein kinase in extracts of 3T3-L1 adipocytes and human placenta. Proc. Natl. Acad. Sci. USA 1982, 79, 6792–6796. [Google Scholar] [CrossRef]
- McKern, N.M.; Lawrence, M.C.; Streltsov, V.A.; Lou, M.-Z.; Adams, T.E.; Lovrecz, G.O.; Elleman, T.C.; Richards, K.M.; Bentley, J.D.; Pilling, P.A.; et al. Structure of the insulin receptor ectodomain reveals a folded-over conformation. Nature 2006, 443, 218–221. [Google Scholar] [CrossRef]
- Cohen, P. The twentieth century struggle to decipher insulin signalling. Nat. Rev. Mol. Cell Biol. 2006, 7, 867–873. [Google Scholar] [CrossRef]
- Taniguchi, C.M.; Emanuelli, B.; Kahn, C.R. Critical nodes in signalling pathways: Insights into insulin action. Nat. Rev. Mol. Cell Biol. 2006, 7, 85–96. [Google Scholar] [CrossRef]
- Brown, M.S.; Goldstein, J.L. Selective versus total insulin resistance: A pathogenic paradox. Cell Metab. 2008, 7, 95–96. [Google Scholar] [CrossRef]
- Johnson, A.M.F.; Olefsky, J.M. The Origins and Drivers of Insulin Resistance. Cell 2013, 152, 673–684. [Google Scholar] [CrossRef]
- IARC. IARC Handbooks of Cancer Prevention; IARC Scientific Publications: Lyon, France, 1999. [Google Scholar]
- Chambon, P. The retinoid signaling pathway: Molecular and genetic analyses. Semin. Cell Biol. 1994, 5, 115–125. [Google Scholar] [CrossRef]
- Lefebvre, P.; Martin, P.J.; Flajollet, S.; Dedieu, S.; Billaut, X.; Lefebvre, B. Transcriptional activities of retinoic acid receptors. Vitam. Horm. 2005, 70, 199–264. [Google Scholar] [CrossRef]
- Moore, T. Vitamin A and carotene: The absence of the liver oil vitamin A from carotene. VI. The conversion of carotene to vitamin A in vivo. Biochem. J. 1930, 24, 692–702. [Google Scholar]
- Moore, T. Vitamin A; Elsevier Publishing Company: Amsterdam, The Netherlands, 1957. [Google Scholar]
- Rigtrup, K.M.; Ong, D.E. A retinyl ester hydrolase activity intrinsic to the brush border membrane of rat small intestine. Biochemistry 1992, 31, 2920–2926. [Google Scholar] [CrossRef]
- Rigtrup, K.M.; Kakkad, B.; Ong, D.E. Purification and partial characterization of a retinyl ester hydrolase from the brush border of rat small intestine mucosa: Probable identity with brush border phospholipase B. Biochemistry 1994, 33, 2661–2666. [Google Scholar] [CrossRef]
- Erdman, J.W.; Bierer, T.L.; Gugger, E.T. Absorption and transport of carotenoids. Ann. N. Y. Acad. Sci. 1993, 691, 76–85. [Google Scholar] [CrossRef]
- Parker, R.S. Absorption, metabolism, and transport of carotenoids. FASEB J. 1996, 10, 542–551. [Google Scholar]
- Dew, S.E.; Ong, D.E. Specificity of the retinol transporter of the rat small intestine brush border. Biochemistry 1994, 33, 12340–12345. [Google Scholar] [CrossRef]
- Goodman, D.S.; Huang, A.S. Biosynthesis of vitamin A with rat intestinal enzymes. Science 1965, 149, 879–880. [Google Scholar]
- Wang, X.D.; Russell, R.M.; Liu, C.; Stickel, F.; Smith, D.E.; Krinsky, N.I. β-Oxidation in rabbit liver in vitro and in the perfused ferret liver contributes to retinoic acid biosynthesis from β-apocarotenoic acids. J. Biol. Chem. 1996, 271, 26490–26498. [Google Scholar] [CrossRef]
- Kiefer, C.; Hessel, S.; Lampert, J.M.; Vogt, K.; Lederer, M.O.; Breithaupt, D.E.; Lintig, J. Identification and characterization of a mammalian enzyme catalyzing the asymmetric oxidative cleavage of provitamin A. J. Biol. Chem. 2001, 276, 14110–14116. [Google Scholar]
- Harrison, E.H. Mechanisms of digestion and absorption of dietary vitamin A. Annu. Rev. Nutr. 2005, 25, 87–103. [Google Scholar] [CrossRef]
- MacDonald, P.N.; Ong, D.E. Evidence for a lecithin-retinol acyltransferase activity in the rat small intestine. J. Biol. Chem. 1988, 263, 12478–12482. [Google Scholar]
- Helgerud, P.; Petersen, L.B.; Norum, K.R. Acyl CoA: Retinol acyltransferase in rat small intestine: Its activity and some properties of the enzymic reaction. J. Lipid Res. 1982, 23, 609–618. [Google Scholar]
- Ross, A.C.; Li, N.-Q. Retinol combined with retinoic acid increases retinol uptake and esterification in the lungs of young adult rats when delivered by the intramuscular as well as oral routes. J. Nutr. 2007, 137, 2371–2376. [Google Scholar]
- Blomhoff, R.; Green, M.H.; Berg, T.; Norum, K.R. Transport and storage of vitamin A. Science 1990, 250, 399–404. [Google Scholar] [CrossRef]
- Kawaguchi, R.; Yu, J.; Honda, J.; Hu, J.; Whitelegge, J.; Ping, P.; Wiita, P.; Bok, D.; Sun, H. A membrane receptor for retinol binding protein mediates cellular uptake of vitamin A. Science 2007, 315, 820–825. [Google Scholar] [CrossRef]
- Chassaing, N.; Golzio, C.; Odent, S.; Lequeux, L.; Vigouroux, A.; Martinovic-Bouriel, J.; Tiziano, F.D.; Masini, L.; Piro, F.; Maragliano, G.; et al. Phenotypic spectrum of STRA6 mutations: From Matthew-Wood syndrome to non-lethal anophthalmia. Hum. Mutat. 2009, 30, E673–E681. [Google Scholar] [CrossRef]
- Golzio, C.; Martinovic-Bouriel, J.; Thomas, S.; Mougou-Zrelli, S.; Grattagliano-Bessieres, B.; Bonniere, M.; Delahaye, S.; Munnich, A.; Encha-Razavi, F.; Lyonnet, S.; et al. Matthew-Wood syndrome is caused by truncating mutations in the retinol-binding protein receptor gene STRA6. Am. J. Hum. Genet. 2007, 80, 1179–1187. [Google Scholar] [CrossRef] [Green Version]
- Berry, D.C.; Jacobs, H.; Marwarha, G.; Gely-Pernot, A.; O’Byrne, S.M.; DeSantis, D.; Klopfenstein, M.; Feret, B.; Dennefeld, C.; Blaner, W.S.; et al. The STRA6 receptor is essential for retinol-binding protein-induced insulin resistance but not for maintaining vitamin A homeostasis in tissues other than the eye. J. Biol. Chem. 2013, 288, 24528–24539. [Google Scholar] [CrossRef]
- Ruiz, A.; Mark, M.; Jacobs, H.; Klopfenstein, M.; Hu, J.; Lloyd, M.; Habib, S.; Tosha, C.; Radu, R.A.; Ghyselinck, N.B.; et al. Retinoid content, visual responses, and ocular morphology are compromised in the retinas of mice lacking the retinol-binding protein receptor, STRA6. Investig. Ophthalmol. Vis. Sci. 2012, 53, 3027–3039. [Google Scholar] [CrossRef]
- Terra, R.; Wang, X.; Hu, Y.; Charpentier, T.; Lamarre, A.; Zhong, M.; Sun, H.; Mao, J.; Qi, S.; Luo, H.; et al. To investigate the necessity of STRA6 upregulation in T cells during T cell immune responses. PLoS One 2013, 8, e82808. [Google Scholar] [CrossRef]
- Napoli, J.L. Physiological insights into all-trans-retinoic acid biosynthesis. Biochim. Biophys. Acta 2012, 1821, 152–167. [Google Scholar]
- Wolf, G. Retinoic acid homeostasis: Retinoic acid regulates liver retinol esterification as well as its own catabolic oxidation in liver. Nutr. Rev. 2001, 59, 391–394. [Google Scholar] [CrossRef]
- Akawi, Z.; Napoli, J.L. Rat liver cytosolic retinal dehydrogenase: Comparison of 13-cis-, 9-cis-, and all-trans-retinal as substrates and effects of cellular retinoid-binding proteins and retinoic acid on activity. Biochemistry 1994, 33, 1938–1943. [Google Scholar] [CrossRef]
- Matt, N.; Dupé, V.; Garnier, J.-M.; Dennefeld, C.; Chambon, P.; Mark, M.; Ghyselinck, N.B. Retinoic acid-dependent eye morphogenesis is orchestrated by neural crest cells. Development 2005, 132, 4789–4800. [Google Scholar] [CrossRef]
- Ziouzenkova, O.; Orasanu, G.; Sharlach, M.; Akiyama, T.E.; Berger, J.P.; Viereck, J.; Hamilton, J.A.; Tang, G.; Dolnikowski, G.G.; Vogel, S.; et al. Retinaldehyde represses adipogenesis and diet-induced obesity. Nat. Med. 2007, 13, 695–702. [Google Scholar] [CrossRef]
- Reichert, B.; Yasmeen, R.; Jeyakumar, S.M.; Yang, F.; Thomou, T.; Alder, H.; Duester, G.; Maiseyeu, A.; Mihai, G.; Harrison, E.H.; et al. Concerted action of aldehyde dehydrogenases influences depot-specific fat formation. Mol. Endocrinol. 2011, 25, 799–809. [Google Scholar] [CrossRef]
- Zhai, Y.; Sperkova, Z.; Napoli, J.L. Cellular expression of retinal dehydrogenase types 1 and 2: Effects of vitamin A status on testis mRNA. J. Cell. Physiol. 2001, 186, 220–232. [Google Scholar] [CrossRef]
- Lin, M.; Zhang, M.; Abraham, M.; Smith, S.M.; Napoli, J.L. Mouse retinal dehydrogenase 4 (RALDH4), molecular cloning, cellular expression, and activity in 9-cis-retinoic acid biosynthesis in intact cells. J. Biol. Chem. 2003, 278, 9856–9861. [Google Scholar] [CrossRef]
- Sima, A.; Parisotto, M.; Mader, S.; Bhat, P.V. Kinetic characterization of recombinant mouse retinal dehydrogenase types 3 and 4 for retinal substrates. Biochim. Biophys. Acta 2009, 1790, 1660–1664. [Google Scholar]
- Roberts, A.B.; Nichols, M.D.; Newton, D.L.; Sporn, M.B. In vitro metabolism of retinoic acid in hamster intestine and liver. J. Biol. Chem. 1979, 254, 6296–6302. [Google Scholar]
- Fujii, H.; Sato, T.; Kaneko, S.; Gotoh, O.; Fujii-Kuriyama, Y.; Osawa, K.; Kato, S.; Hamada, H. Metabolic inactivation of retinoic acid by a novel P450 differentially expressed in developing mouse embryos. EMBO J. 1997, 16, 4163–4173. [Google Scholar]
- Wang, Y.; Zolfaghari, R.; Ross, A.C. Cloning of rat cytochrome P450RAI (CYP26) cDNA and regulation of its gene expression by all-trans-retinoic acid in vivo. Arch. Biochem. Biophys. 2002, 401, 235–243. [Google Scholar] [CrossRef]
- Ray, W.J.; Bain, G.; Yao, M.; Gottlieb, D.I. CYP26, a novel mammalian cytochrome P450, is induced by retinoic acid and defines a new family. J. Biol. Chem. 1997, 272, 18702–18708. [Google Scholar]
- Abu-Abed, S.; Dollé, P.; Metzger, D.; Beckett, B.; Chambon, P.; Petkovich, M. The retinoic acid-metabolizing enzyme, CYP26A1, is essential for normal hindbrain patterning, vertebral identity, and development of posterior structures. Genes Dev. 2001, 15, 226–240. [Google Scholar] [CrossRef]
- Mangelsdorf, D.J.; Thummel, C.; Beato, M.; Herrlich, P.; Schütz, G.; Umesono, K.; Blumberg, B.; Kastner, P.; Mark, M.; Chambon, P.; et al. The nuclear receptor superfamily: The second decade. Cell 1995, 83, 835–839. [Google Scholar] [CrossRef]
- Lehmann, J.M.; Dawson, M.I.; Hobbs, P.D.; Husmann, M.; Pfahl, M. Identification of retinoids with nuclear receptor subtype-selective activities. Cancer Res. 1991, 51, 4804–4809. [Google Scholar]
- Berry, D.C.; Noy, N. Is PPAR β/δ a Retinoid Receptor? PPAR Res. 2007, 2007. [Google Scholar] [CrossRef]
- Mayer, J.; Krehl, W.A. Influence of vitamin A deficiency on the gross efficiency of growth of rats. Yale J. Biol. Med. 1948, 20, 403–405. [Google Scholar]
- Sampson, M.M.; Korenchevsky, V. The influence of vitamin A deficiency on male rats in paired feeding experiments. Biochem. J. 1932, 26, 1322–1339. [Google Scholar]
- Zhang, Y.; Li, R.; Li, Y.; Chen, W.; Zhao, S.; Chen, G. Vitamin A status affects obesity development and hepatic expression of key genes for fuel metabolism in Zucker fatty rats. Biochem. Cell Biol. 2012, 90, 548–557. [Google Scholar] [CrossRef]
- Rogers, W.E.; Bieri, J.G. Vitamin A deficiency in the rat prior to weaning. Proc. Soc. Exp. Biol. Med. 1969, 132, 622–624. [Google Scholar] [CrossRef]
- Anzano, M.A.; Lamb, A.J.; Olson, J.A. Growth, appetite, sequence of pathological signs and survival following the induction of rapid, synchronous vitamin A deficiency in the rat. J. Nutr. 1979, 109, 1419–1431. [Google Scholar]
- McCarthy, P.T.; Cerecedo, L.R. Vitamin A deficiency in the mouse. J. Nutr. 1952, 46, 361–376. [Google Scholar]
- Sagazio, A.; Piantedosi, R.; Alba, M.; Blaner, W.S.; Salvatori, R. Vitamin A deficiency does not influence longitudinal growth in mice. Nutrition 2007, 23, 483–488. [Google Scholar] [CrossRef]
- Friedman, J.M.; Halaas, J.L. Leptin and the regulation of body weight in mammals. Nature 1998, 395, 763–770. [Google Scholar] [CrossRef]
- Kumar, M.V.; Sunvold, G.D.; Scarpace, P.J. Dietary vitamin A supplementation in rats: Suppression of leptin and induction of UCP1 mRNA. J. Lipid Res. 1999, 40, 824–829. [Google Scholar]
- Hollung, K.; Rise, C.P.; Drevon, C.A.; Reseland, J.E. Tissue-specific regulation of leptin expression and secretion by all-trans retinoic acid. J. Cell. Biochem. 2004, 92, 307–315. [Google Scholar] [CrossRef]
- Felipe, F.; Mercader, J.; Ribot, J.; Palou, A.; Bonet, M.L. Effects of retinoic acid administration and dietary vitamin A supplementation on leptin expression in mice: Lack of correlation with changes of adipose tissue mass and food intake. Biochim. Biophys. Acta 2005, 1740, 258–265. [Google Scholar]
- Bernard, R.A.; Halpern, B.P. Taste changes in vitamin A deficiency. J. Gen. Physiol. 1968, 52, 444–464. [Google Scholar] [CrossRef]
- Reifen, R.; Agami, O.; Weiser, H.; Biesalski, H.; Naim, M. Impaired responses to sweet taste in vitamin A-deficient rats. Metab. Clin. Exp. 1998, 47, 1–2. [Google Scholar]
- Garrett-Laster, M.; Russell, R.M.; Jacques, P.F. Impairment of taste and olfaction in patients with cirrhosis: The role of vitamin A. Hum. Nutr. Clin. Nutr. 1984, 38, 203–214. [Google Scholar]
- Moore, T. Vitamin A and carotene: The vitamin A reserve of the adult human being in health and disease. Biochem. J. 1937, 31, 155–164. [Google Scholar]
- Boeck, W.C.; Yater, W.M. Xanthemia and xanthosis (carotinemia): Clinical study. J. Lab. Clin. Med. 1929, 14, 1129–1143. [Google Scholar]
- Rabinowitch, I.M. Carotinemia and diabetes: II. The relationship between the sugar, cholesterol and carotin contents of blood plasma. Arch. Intern. Med. 1930, 45, 586–592. [Google Scholar] [CrossRef]
- Mosenthal, H.O.; Loughlin, W.C. Vitamins A, B and C in diabetic children. Arch. Intern. Med. 1944, 73, 391–396. [Google Scholar] [CrossRef]
- Basu, T.K.; Tze, W.J.; Leichter, J. Serum vitamin A and retinol-binding protein in patients with insulin-dependent diabetes mellitus. Am. J. Clin. Nutr. 1989, 50, 329–331. [Google Scholar]
- Graham, T.E.; Yang, Q.; Blüher, M.; Hammarstedt, A.; Ciaraldi, T.P.; Henry, R.R.; Wason, C.J.; Oberbach, A.; Jansson, P.-A.; Smith, U.; et al. Retinol-binding protein 4 and insulin resistance in lean, obese, and diabetic subjects. N. Engl. J. Med. 2006, 354, 2552–2563. [Google Scholar] [CrossRef]
- Cho, Y.M.; Youn, B.-S.; Lee, H.; Lee, N.; Min, S.-S.; Kwak, S.H.; Lee, H.K.; Park, K.S. Plasma retinol-binding protein-4 concentrations are elevated in human subjects with impaired glucose tolerance and type 2 diabetes. Diabetes Care 2006, 29, 2457–2461. [Google Scholar]
- Wolf, G. Serum retinol-binding protein: A link between obesity, insulin resistance, and type 2 diabetes. Nutr. Rev. 2007, 65, 251–256. [Google Scholar] [CrossRef]
- Craig, R.L.; Chu, W.S.; Elbein, S.C. Retinol binding protein 4 as a candidate gene for type 2 diabetes and prediabetic intermediate traits. Mol. Genet. Metab. 2007, 90, 338–344. [Google Scholar] [CrossRef]
- Kovacs, P.; Geyer, M.; Berndt, J.; Klöting, N.; Graham, T.E.; Böttcher, Y.; Enigk, B.; Tönjes, A.; Schleinitz, D.; Schön, M.R.; et al. Effects of genetic variation in the human retinol binding protein-4 gene (RBP4) on insulin resistance and fat depot-specific mRNA expression. Diabetes 2007, 56, 3095–3100. [Google Scholar] [CrossRef]
- Janke, J.; Engeli, S.; Boschmann, M.; Adams, F.; Böhnke, J.; Luft, F.C.; Sharma, A.M.; Jordan, J. Retinol-binding protein 4 in human obesity. Diabetes 2006, 55, 2805–2810. [Google Scholar] [CrossRef]
- Ulgen, F.; Herder, C.; Kühn, M.C.; Willenberg, H.S.; Schott, M.; Scherbaum, W.A.; Schinner, S. Association of serum levels of retinol-binding protein 4 with male sex but not with insulin resistance in obese patients. Arch. Physiol. Biochem. 2010, 116, 57–62. [Google Scholar] [CrossRef]
- Kotnik, P.; Fischer-Posovszky, P.; Wabitsch, M. RBP4: A controversial adipokine. Eur. J. Endocrinol. 2011, 165, 703–711. [Google Scholar] [CrossRef]
- Yang, Q.; Graham, T.E.; Mody, N.; Preitner, F.; Peroni, O.D.; Zabolotny, J.M.; Kotani, K.; Quadro, L.; Kahn, B.B. Serum retinol binding protein 4 contributes to insulin resistance in obesity and type 2 diabetes. Nature 2005, 436, 356–362. [Google Scholar] [CrossRef]
- Motani, A.; Wang, Z.; Conn, M.; Siegler, K.; Zhang, Y.; Liu, Q.; Johnstone, S.; Xu, H.; Thibault, S.; Wang, Y.; et al. Identification and characterization of a non-retinoid ligand for retinol-binding protein 4 which lowers serum retinol-binding protein 4 levels in vivo. J. Biol. Chem. 2009, 284, 7673–7680. [Google Scholar] [CrossRef]
- Bershad, S.; Rubinstein, A.; Paterniti, J.R.; Le, N.A.; Poliak, S.C.; Heller, B.; Ginsberg, H.N.; Fleischmajer, R.; Brown, W.V. Changes in plasma lipids and lipoproteins during isotretinoin therapy for acne. N. Engl. J. Med. 1985, 313, 981–985. [Google Scholar] [CrossRef]
- Tallman, M.S.; Kwaan, H.C. Reassessing the hemostatic disorder associated with acute promyelocytic leukemia. Blood 1992, 79, 543–553. [Google Scholar]
- Miller, V.A.; Rigas, J.R.; Muindi, J.R.; Tong, W.P.; Venkatraman, E.; Kris, M.G.; Warrell, R.P. Modulation of all-trans retinoic acid pharmacokinetics by liarozole. Cancer Chemother. Pharmacol. 1994, 34, 522–526. [Google Scholar] [CrossRef]
- Vu-Dac, N.; Gervois, P.; Torra, I.P.; Fruchart, J.C.; Kosykh, V.; Kooistra, T.; Princen, H.M.; Dallongeville, J.; Staels, B. Retinoids increase human apo C-III expression at the transcriptional level via the retinoid X receptor. Contribution to the hypertriglyceridemic action of retinoids. J. Clin. Investig. 1998, 102, 625–632. [Google Scholar] [CrossRef]
- Shachter, N.S. Apolipoproteins C-I and C-III as important modulators of lipoprotein metabolism. Curr. Opin. Lipidol. 2001, 12, 297–304. [Google Scholar] [CrossRef]
- Preitner, F.; Mody, N.; Graham, T.E.; Peroni, O.D.; Kahn, B.B. Long-term Fenretinide treatment prevents high-fat diet-induced obesity, insulin resistance, and hepatic steatosis. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E1420–E1429. [Google Scholar] [CrossRef]
- Mcilroy, G.D.; Delibegovic, M.; Owen, C.; Stoney, P.N.; Shearer, K.D.; McCaffery, P.J.; Mody, N. Fenretinide treatment prevents diet-induced obesity in association with major alterations in retinoid homeostatic gene expression in adipose, liver, and hypothalamus. Diabetes 2013, 62, 825–836. [Google Scholar] [CrossRef]
- Wolf, G.; Lane, M.D.; Johnson, B.C. Studies on the function of vitamin A in metabolism. J. Biol. Chem. 1957, 225, 995–1008. [Google Scholar]
- Johnson, B.C.; Wolf, G. The function of vitamin A in carbohydrate metabolism. Its role in adrenocorticoid production. Vitam. Horm. 1960, 18, 457–483. [Google Scholar] [CrossRef]
- Ray, A.; Sadhu, D.P. Carbohydrate metabolism in hypervitaminosis A. Nature 1959, 184. [Google Scholar] [CrossRef]
- Singh, M.; Singh, V.N.; Venkitasubramanian, T.A. Early effects of feeding excess vitamin A: Hepatic glycogen, blood lactic acid, plasma NEFA and glucose tolerance in rats. Life Sci. 1968, 7, 239–247. [Google Scholar] [CrossRef]
- Singh, M.; Singh, V.N.; Venkitasubramanian, T.A. Early effects of excessive retinol intake on hepatic glycogen metabolism. Arch. Biochem. Biophys. 1976, 173, 93–99. [Google Scholar] [CrossRef]
- Shankar, S.; Creek, K.E.; de Luca, L.M. The effect of the progression of vitamin A deficiency on glucose, galactose and mannose incorporation into sugar phosphates and sugar nucleotides in hamster liver. J. Nutr. 1990, 120, 361–374. [Google Scholar]
- Singh, V.N.; Singh, M.; Dileepan, K.N. Early effects of vitamin A toxicity on hepatic glycolysis in rat. J. Nutr. 1978, 108, 1959–1962. [Google Scholar]
- Chen, G.; Zhang, Y.; Lu, D.; Li, N.-Q.; Ross, A.C. Retinoids synergize with insulin to induce hepatic Gck expression. Biochem. J. 2009, 419, 645–653. [Google Scholar]
- Dileepan, K.N.; Singh, V.N.; Ramachandran, C.K. Decreased hepatic gluconeogenesis in vitamin A-deficient rats. Proc. Soc. Exp. Biol. Med. 1981, 167, 248–253. [Google Scholar]
- Singh, M.; Ningh, V.N.; Venkitasubramanian, T.A. Role of adrenals in the vitamin A-mediated increase in the activities of gluconeogenic enzymes of rat liver. Life Sci. 1975, 17, 859–865. [Google Scholar] [CrossRef]
- Dileepan, K.N.; Singh, V.N.; Ramachandran, C.K. Early effects of hypervitaminosis A on gluconeogenic activity and amino acid metabolizing enzymes of rat liver. J. Nutr. 1977, 107, 1809–1815. [Google Scholar]
- Singh, M.; Singh, V.N.; Venkitasubramanian, T.A. Early effects of excessive retinol intake on gluconeogenesis. Involvement of adrenals in the increased activities on Gluconeogenic Enzymes of rat. Arch. Biochem. Biophys. 1976, 173, 82–92. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, R.; Chen, W.; Li, Y.; Chen, G. Retinoids induced Pck1 expression and attenuated insulin-mediated suppression of its expression via activation of retinoic acid receptor in primary rat hepatocytes. Mol. Cell. Biochem. 2011, 355, 1–8. [Google Scholar] [CrossRef]
- Shin, D.-J.; Odom, D.P.; Scribner, K.B.; Ghoshal, S.; McGrane, M.M. Retinoid regulation of the phosphoenolpyruvate carboxykinase gene in liver. Mol. Cell. Endocrinol. 2002, 195, 39–54. [Google Scholar] [CrossRef]
- Scribner, K.B.; Odom, D.P.; McGrane, M.M. Nuclear receptor binding to the retinoic acid response elements of the phosphoenolpyruvate carboxykinase gene in vivo: Effects of vitamin A deficiency. J. Nutr. Biochem. 2007, 18, 206–214. [Google Scholar] [CrossRef]
- Sampson, M.M.; Dennison, M.; Korenchevsky, V. The absorption of nitrogen and of fat from the alimentary canal of rats kept on a vitamin A-deficient diet. Biochem. J. 1932, 26, 1315–1321. [Google Scholar]
- Brown, E.F.; Morgan, A.F. The effect of vitamin A deficiency upon the nitrogen metabolism of the rat. J. Nutr. 1948, 35, 425–438. [Google Scholar]
- Esteban-Pretel, G.; Marín, M.P.; Cabezuelo, F.; Moreno, V.; Renau-Piqueras, J.; Timoneda, J.; Barber, T. Vitamin A deficiency increases protein catabolism and induces urea cycle enzymes in rats. J. Nutr. 2010, 140, 792–798. [Google Scholar] [CrossRef]
- John, A.; Sivakumar, B. Effect of vitamin A deficiency on nitrogen balance and hepatic urea cycle enzymes and intermediates in rats. J. Nutr. 1989, 119, 29–35. [Google Scholar]
- Rao, B.S.N. Effect of vitamin A deficiency on the incorporation of 14C-Leucine into tissue proteins of rats. Nature 1966, 210, 306–307. [Google Scholar]
- Tryfiates, G.P.; Krause, R.F. Effect of vitamin A deficiency on the protein synthetic activity of rat liver ribosomes. Proc. Soc. Exp. Biol. Med. 1971, 136, 946–948. [Google Scholar] [CrossRef]
- Nerurkar, M.K.; Sahasrabudhe, M.B. Metabolism of calcium, phosphorus and nitrogen in hypervitaminosis A in young rats. Biochem. J. 1956, 63, 344–349. [Google Scholar]
- Gerber, L.E.; Erdman, J.W. Changes in lipid metabolism during retinoid administration. J. Am. Acad. Dermatol. 1982, 6, 664–674. [Google Scholar] [CrossRef]
- Ellis, C.N.; Kang, S.; Vinik, A.I.; Grekin, R.C.; Cunningham, W.J.; Voorhees, J.J. Glucose and insulin responses are improved in patients with psoriasis during therapy with etretinate. Arch. Dermatol. 1987, 123, 471–475. [Google Scholar] [CrossRef]
- Yehya, A.; Baer, J.T.; Smiley, W.; Dollar, A.; Sperling, L. Hypervitaminosis A altering the lipid profile in a hypercholesterolemic patient. J. Clin. Lipidol. 2009, 3, 205–207. [Google Scholar] [CrossRef]
- Ellis, J.K.; Russell, R.M.; Makrauer, F.L.; Schaefer, E.J. Increased risk for vitamin A toxicity in severe hypertriglyceridemia. Ann. Intern. Med. 1986, 105, 877–879. [Google Scholar] [CrossRef]
- Lettinga, K.D.; Gutter, W.; van Noorden, C.J.; Schellens, J.P.; Frederiks, W.M. Early effects of high doses of retinol (vitamin A) on the in situ cellular metabolism in rat liver. Liver 1996, 16, 1–11. [Google Scholar] [CrossRef]
- Misra, U.K. Hypervitaminosis A and tissue fatty acids. Can. J. Biochem. 1965, 43, 1885–1886. [Google Scholar] [CrossRef]
- Singh, V.N.; Singh, M.; Venkitasubramanian, T.A. Early effects of feeding excess vitamin A: Mechanism of fatty liver production in rats. J. Lipid Res. 1969, 10, 395–401. [Google Scholar]
- Ramachandran, C.K.; Dileepan, K.N.; Singh, V.; Venkitasubramanian, T.A. Effect of excess and deficiency of vitamin A on the utilization of FFA by liver and skeletal muscle. Environ. Physiol. Biochem. 1975, 5, 208–214. [Google Scholar]
- Singh, M.; Singh, V.N. Fatty liver in hypervitaminosis A: Synthesis and release of hepatic triglycerides. Am. J. Physiol. 1978, 234, E511–E514. [Google Scholar]
- Oliveros, L.B.; Domeniconi, M.A.; Vega, V.A.; Gatica, L.V.; Brigada, A.M.; Gimenez, M.S. Vitamin A deficiency modifies lipid metabolism in rat liver. Br. J. Nutr. 2007, 97, 263–272. [Google Scholar] [CrossRef]
- Khanna, A.; Reddy, T.S. Effect of undernutrition and vitamin A deficiency on the phospholipid composition of rat tissues at 21 days of age—I. Liver, spleen and kidney. Int. J. Vitam. Nutr. Res. 1983, 53, 3–8. [Google Scholar]
- Green, B.; Lowe, J.S.; Morton, R.A. The effect of vitamin A deficiency on the cholesterol levels of the plasma and liver of the rat. Biochem. J. 1955, 61, 447–453. [Google Scholar]
- Wiss, O.; Wiss, V. Alterations of the lipid metabolism of rat liver as early symptoms of vitamin A deficiency. Int. J. Vitam. Nutr. Res. 1980, 50, 233–237. [Google Scholar]
- Green, H.N. Fat metabolism in vitamin A deficiency: The utilisation of fat and the desaturation of fat in the liver. Biochem. J. 1934, 28, 25–30. [Google Scholar]
- Kang, H.W.; Bhimidi, G.R.; Odom, D.P.; Brun, P.-J.; Fernandez, M.-L.; McGrane, M.M. Altered lipid catabolism in the vitamin: A deficient liver. Mol. Cell. Endocrinol. 2007, 271, 18–27. [Google Scholar] [CrossRef]
- Bonet, M.L.; Ribot, J.; Palou, A. Lipid metabolism in mammalian tissues and its control by retinoic acid. Biochim. Biophys. Acta 2012, 1821, 177–189. [Google Scholar]
- Horton, J.D.; Goldstein, J.L.; Brown, M.S. SREBPs: Activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Investig. 2002, 109, 1125–1131. [Google Scholar] [CrossRef]
- Chen, G.; Liang, G.; Ou, J.; Goldstein, J.L.; Brown, M.S. Central role for liver X receptor in insulin-mediated activation of Srebp-1c transcription and stimulation of fatty acid synthesis in liver. Proc. Natl. Acad. Sci. USA 2004, 101, 11245–11250. [Google Scholar] [CrossRef]
- Li, R.; Chen, W.; Li, Y.; Zhang, Y.; Chen, G. Retinoids synergized with insulin to induce Srebp-1c expression and activated its promoter via the two liver X receptor binding sites that mediate insulin action. Biochem. Biophys. Res. Commun. 2011, 406, 268–272. [Google Scholar] [CrossRef]
- Deluca, H.F.; Manatt, M.R.; Madsen, N.; Olson, E.B. Action of vitamin A on liver homogenate oxidation of tricarboxylic acid cycle intermediates. J. Nutr. 1963, 81, 383–386. [Google Scholar]
- Seward, C.R.; Vaughan, G.; Hove, E.L. Respiratory activities of hypo- and hypervitaminotic A rat liver homogenates. Proc. Soc. Exp. Biol. Med. 1964, 117, 477–480. [Google Scholar] [CrossRef]
- Seward, C.R.; Vaughan, G.; Hove, E.L. Effect of vitamin A deficiency or excess on the oxidative phosphorylation by rat liver mitochondria. J. Biol. Chem. 1966, 241, 1229–1232. [Google Scholar]
- Brissova, M.; Fowler, M.J.; Nicholson, W.E.; Chu, A.; Hirshberg, B.; Harlan, D.M.; Powers, A.C. Assessment of human pancreatic islet architecture and composition by laser scanning confocal microscopy. J. Histochem. Cytochem. 2005, 53, 1087–1097. [Google Scholar] [CrossRef]
- Andralojc, K.M.; Mercalli, A.; Nowak, K.W.; Albarello, L.; Calcagno, R.; Luzi, L.; Bonifacio, E.; Doglioni, C.; Piemonti, L. Ghrelin-producing epsilon cells in the developing and adult human pancreas. Diabetologia 2009, 52, 486–493. [Google Scholar] [CrossRef]
- Unger, R.H.; Cherrington, A.D. Glucagonocentric restructuring of diabetes: A pathophysiologic and therapeutic makeover. J. Clin. Investig. 2012, 122, 4–12. [Google Scholar] [CrossRef]
- Gylfe, E.; Gilon, P. Glucose regulation of glucagon secretion. Diabetes Res. Clin. Pract. 2013, 103, 1–10. [Google Scholar] [CrossRef]
- Jensen, M.V.; Joseph, J.W.; Ronnebaum, S.M.; Burgess, S.C.; Sherry, A.D.; Newgard, C.B. Metabolic cycling in control of glucose-stimulated insulin secretion. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E1287–E1297. [Google Scholar] [CrossRef]
- Chertow, B.S.; Driscoll, H.K.; Blaner, W.S.; Meda, P.; Cordle, M.B.; Matthews, K.A. Effects of vitamin A deficiency and repletion on rat glucagon secretion. Pancreas 1994, 9, 475–484. [Google Scholar] [CrossRef]
- Chertow, B.S.; Driscoll, H.K.; Primerano, D.A.; Cordle, M.B.; Matthews, K.A. Retinoic acid receptor transcripts and effects of retinol and retinoic acid on glucagon secretion from rat islets and glucagon-secreting cell lines. Metab. Clin. Exp. 1996, 45, 300–305. [Google Scholar] [CrossRef]
- Matthews, K.A.; Rhoten, W.B.; Driscoll, H.K.; Chertow, B.S. Vitamin A deficiency impairs fetal islet development and causes subsequent glucose intolerance in adult rats. J. Nutr. 2004, 134, 1958–1963. [Google Scholar]
- Chertow, B.S.; Blaner, W.S.; Baranetsky, N.G.; Sivitz, W.I.; Cordle, M.B.; Thompson, D.; Meda, P. Effects of vitamin A deficiency and repletion on rat insulin secretion in vivo and in vitro from isolated islets. J. Clin. Investig. 1987, 79, 163–169. [Google Scholar] [CrossRef]
- Chertow, B.S.; Baker, G.R. The effects of vitamin A on insulin release and glucose oxidation in isolated rat islets. Endocrinology 1978, 103, 1562–1572. [Google Scholar] [CrossRef]
- Chertow, B.S.; Goking, N.Q.; Driscoll, H.K.; Primerano, D.A.; Matthews, K.A. Effects of all-trans-retinoic acid (ATRA) and retinoic acid receptor (RAR) expression on secretion, growth, and apoptosis of insulin-secreting RINm5F cells. Pancreas 1997, 15, 122–131. [Google Scholar] [CrossRef]
- Driscoll, H.K.; Adkins, C.D.; Chertow, T.E.; Cordle, M.B.; Matthews, K.A.; Chertow, B.S. Vitamin A stimulation of insulin secretion: Effects on transglutaminase mRNA and activity using rat islets and insulin-secreting cells. Pancreas 1997, 15, 69–77. [Google Scholar] [CrossRef]
- Cabrera-Valladares, G.; German, M.S.; Matschinsky, F.M.; Wang, J.; Fernandez-Mejia, C. Effect of retinoic acid on glucokinase activity and gene expression and on insulin secretion in primary cultures of pancreatic islets. Endocrinology 1999, 140, 3091–3096. [Google Scholar]
- Kane, M.A.; Folias, A.E.; Pingitore, A.; Perri, M.; Obrochta, K.M.; Krois, C.R.; Cione, E.; Ryu, J.Y.; Napoli, J.L. Identification of 9-cis-retinoic acid as a pancreas-specific autacoid that attenuates glucose-stimulated insulin secretion. Proc. Natl. Acad. Sci. USA 2010, 107, 21884–21889. [Google Scholar] [CrossRef]
- Kane, M.A.; Folias, A.E.; Pingitore, A.; Perri, M.; Krois, C.R.; Ryu, J.Y.; Cione, E.; Napoli, J.L. CrbpI modulates glucose homeostasis and pancreas 9-cis-retinoic acid concentrations. Mol. Cell. Biol. 2011, 31, 3277–3285. [Google Scholar] [CrossRef]
- Ramachandran, C.K.; Dileepan, K.N.; Singh, V.N.; Venkitasubramanian, T.A. Metabolic potential of the adipose tissue of rats during hyper- and hypovitaminosis A. Proc. Soc. Exp. Biol. Med. 1986, 182, 73–78. [Google Scholar]
- Ribot, J.; Felipe, F.; Bonet, M.L.; Palou, A. Changes of Adiposity in Response to Vitamin A Status Correlate with Changes of PPARγ2 Expression. Obesity 2001, 9, 500–509. [Google Scholar] [CrossRef]
- De Souza Valente da Silva, L.; Valeria da Veiga, G.; Ramalho, R.A. Association of serum concentrations of retinol and carotenoids with overweight in children and adolescents. Nutrition 2007, 23, 392–397. [Google Scholar] [CrossRef]
- Zulet, M.A.; Puchau, B.; Hermsdorff, H.H.M.; Navarro, C.; Martínez, J.A. Vitamin A intake is inversely related with adiposity in healthy young adults. J. Nutr. Sci. Vitaminol. 2008, 54, 347–352. [Google Scholar] [CrossRef]
- Berry, D.C.; Noy, N. All-trans-retinoic acid represses obesity and insulin resistance by activating both peroxisome proliferation-activated receptor β/δ and retinoic acid receptor. Mol. Cell. Biol. 2009, 29, 3286–3296. [Google Scholar] [CrossRef]
- Berry, D.C.; DeSantis, D.; Soltanian, H.; Croniger, C.M.; Noy, N. Retinoic acid upregulates preadipocyte genes to block adipogenesis and suppress diet-induced obesity. Diabetes 2012, 61, 1112–1121. [Google Scholar] [CrossRef]
- Mercader, J.; Ribot, J.; Murano, I.; Felipe, F.; Cinti, S.; Bonet, M.L.; Palou, A. Remodeling of white adipose tissue after retinoic acid administration in mice. Endocrinology 2006, 147, 5325–5332. [Google Scholar] [CrossRef]
- Murholm, M.; Isidor, M.S.; Basse, A.L.; Winther, S.; Sørensen, C.; Skovgaard-Petersen, J.; Nielsen, M.M.; Hansen, A.S.; Quistorff, B.; Hansen, J.B. Retinoic acid has different effects on UCP1 expression in mouse and human adipocytes. BMC Cell Biol. 2013, 14. [Google Scholar] [CrossRef]
- Schug, T.T.; Berry, D.C.; Shaw, N.S.; Travis, S.N.; Noy, N. Opposing effects of retinoic acid on cell growth result from alternate activation of two different nuclear receptors. Cell 2007, 129, 723–733. [Google Scholar] [CrossRef]
- Shaw, N.; Elholm, M.; Noy, N. Retinoic acid is a high affinity selective ligand for the peroxisome proliferator-activated receptor β/δ. J. Biol. Chem. 2003, 278, 41589–41592. [Google Scholar] [CrossRef]
- Kiefer, F.W.; Vernochet, C.; O’Brien, P.; Spoerl, S.; Brown, J.D.; Nallamshetty, S.; Zeyda, M.; Stulnig, T.M.; Cohen, D.E.; Kahn, C.R.; et al. Retinaldehyde dehydrogenase 1 regulates a thermogenic program in white adipose tissue. Nat. Med. 2012, 18, 918–925. [Google Scholar] [CrossRef]
- Dimaculangan, D.D.; Chawla, A.; Boak, A.; Kagan, H.M.; Lazar, M.A. Retinoic acid prevents downregulation of ras recision gene/lysyl oxidase early in adipocyte differentiation. Differentiation 1994, 58, 47–52. [Google Scholar] [CrossRef]
- Berry, D.C.; Soltanian, H.; Noy, N. Repression of cellular retinoic acid-binding protein II during adipocyte differentiation. J. Biol. Chem. 2010, 285, 15324–15332. [Google Scholar] [CrossRef]
- Xue, J.C.; Schwarz, E.J.; Chawla, A.; Lazar, M.A. Distinct stages in adipogenesis revealed by retinoid inhibition of differentiation after induction of PPARγ. Mol. Cell. Biol. 1996, 16, 1567–1575. [Google Scholar]
- Safonova, I.; Darimont, C.; Amri, E.Z.; Grimaldi, P.; Ailhaud, G.; Reichert, U.; Shroot, B. Retinoids are positive effectors of adipose cell differentiation. Mol. Cell. Endocrinol. 1994, 104, 201–211. [Google Scholar] [CrossRef]
- Cannon, B.; Nedergaard, J. Brown adipose tissue: Function and physiological significance. Physiol. Rev. 2004, 84, 277–359. [Google Scholar] [CrossRef]
- Timmons, J.A.; Wennmalm, K.; Larsson, O.; Walden, T.B.; Lassmann, T.; Petrovic, N.; Hamilton, D.L.; Gimeno, R.E.; Wahlestedt, C.; Baar, K.; et al. Myogenic gene expression signature establishes that brown and white adipocytes originate from distinct cell lineages. Proc. Natl. Acad. Sci. USA 2007, 104, 4401–4406. [Google Scholar] [CrossRef]
- Seale, P.; Kajimura, S.; Yang, W.; Chin, S.; Rohas, L.M.; Uldry, M.; Tavernier, G.; Langin, D.; Spiegelman, B.M. Transcriptional control of brown fat determination by PRDM16. Cell Metab. 2007, 6, 38–54. [Google Scholar] [CrossRef]
- Wolf, G. Brown adipose tissue: The molecular mechanism of its formation. Nutr. Rev. 2009, 67, 167–171. [Google Scholar] [CrossRef]
- Petrovic, N.; Walden, T.B.; Shabalina, I.G.; Timmons, J.A.; Cannon, B.; Nedergaard, J. Chronic peroxisome proliferator-activated receptor gamma (PPARγ) activation of epididymally derived white adipocyte cultures reveals a population of thermogenically competent, UCP1-containing adipocytes molecularly distinct from classic brown adipocytes. J. Biol. Chem. 2010, 285, 7153–7164. [Google Scholar]
- Wu, J.; Boström, P.; Sparks, L.M.; Ye, L.; Choi, J.H.; Giang, A.-H.; Khandekar, M.; Virtanen, K.A.; Nuutila, P.; Schaart, G.; et al. Beige adipocytes are a distinct type of thermogenic fat cell in mouse and human. Cell 2012, 150, 366–376. [Google Scholar] [CrossRef]
- Cypess, A.M.; White, A.P.; Vernochet, C.; Schulz, T.J.; Xue, R.; Sass, C.A.; Huang, T.L.; Roberts-Toler, C.; Weiner, L.S.; Sze, C.; et al. Anatomical localization, gene expression profiling and functional characterization of adult human neck brown fat. Nat. Med. 2013, 19, 635–639. [Google Scholar] [CrossRef]
- Lidell, M.E.; Betz, M.J.; Dahlqvist Leinhard, O.; Heglind, M.; Elander, L.; Slawik, M.; Mussack, T.; Nilsson, D.; Romu, T.; Nuutila, P.; et al. Evidence for two types of brown adipose tissue in humans. Nat. Med. 2013, 19, 631–634. [Google Scholar] [CrossRef]
- Nedergaard, J.; Cannon, B. How brown is brown fat? It depends where you look. Nat. Med. 2013, 19, 540–541. [Google Scholar] [CrossRef]
- Bonet, M.L.; Oliver, J.; Picó, C.; Felipe, F.; Ribot, J.; Cinti, S.; Palou, A. Opposite effects of feeding a vitamin A-deficient diet and retinoic acid treatment on brown adipose tissue uncoupling protein 1 (UCP1), UCP2 and leptin expression. J. Endocrinol. 2000, 166, 511–517. [Google Scholar] [CrossRef]
- Puigserver, P.; Vázquez, F.; Bonet, M.L.; Picó, C.; Palou, A. In vitro and in vivo induction of brown adipocyte uncoupling protein (thermogenin) by retinoic acid. Biochem. J. 1996, 317, 827–833. [Google Scholar]
- Alvarez, R.; de Andrés, J.; Yubero, P.; Viñas, O.; Mampel, T.; Iglesias, R.; Giralt, M.; Villarroya, F. A novel regulatory pathway of brown fat thermogenesis. Retinoic acid is a transcriptional activator of the mitochondrial uncoupling protein gene. J. Biol. Chem. 1995, 270, 5666–5673. [Google Scholar] [CrossRef]
- Larose, M.; Cassard-Doulcier, A.M.; Fleury, C.; Serra, F.; Champigny, O.; Bouillaud, F.; Ricquier, D. Essential cis-acting elements in rat uncoupling protein gene are in an enhancer containing a complex retinoic acid response domain. J. Biol. Chem. 1996, 271, 31533–31542. [Google Scholar] [CrossRef]
- Ribot, J.; Felipe, F.; Bonet, M.L.; Palou, A. Retinoic acid administration and vitamin A status modulate retinoid X receptor alpha and retinoic acid receptor alpha levels in mouse brown adipose tissue. Mol. Cell. Biochem. 2004, 266, 25–30. [Google Scholar] [CrossRef]
- Pedersen, B.K. Muscles and their myokines. J. Exp. Biol. 2011, 214, 337–346. [Google Scholar] [CrossRef]
- Sundeen, G.; Richards, J.F.; Bragg, D.B. The effect of vitamin A deficiency on some postmortem parameters of avian muscle. Poult. Sci. 1980, 59, 2225–2236. [Google Scholar] [CrossRef]
- Lee, Y.M.; Lee, J.O.; Jung, J.-H.; Kim, J.H.; Park, S.-H.; Park, J.M.; Kim, E.-K.; Suh, P.-G.; Kim, H.S. Retinoic acid leads to cytoskeletal rearrangement through AMPK-Rac1 and stimulates glucose uptake through AMPK-p38 MAPK in skeletal muscle cells. J. Biol. Chem. 2008, 283, 33969–33974. [Google Scholar] [CrossRef]
- Narbonne, J.F.; Daubeze, M.; Bonmort, F. Protein metabolism in vitamin A deficient rats. II. Protein synthesis in striated muscle. Ann. Nutr. Aliment 1978, 32, 59–75. [Google Scholar]
- Hillgartner, F.B.; Morin, D.; Hansen, R.J. Effect of excessive vitamin A intake on muscle protein turnover in the rat. Biochem. J. 1982, 202, 499–508. [Google Scholar]
- Amengual, J.; Ribot, J.; Bonet, M.L.; Palou, A. Retinoic acid treatment increases lipid oxidation capacity in skeletal muscle of mice. Obesity 2008, 16, 585–591. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Chen, W.; Chen, G. The Roles of Vitamin A in the Regulation of Carbohydrate, Lipid, and Protein Metabolism. J. Clin. Med. 2014, 3, 453-479. https://doi.org/10.3390/jcm3020453
Chen W, Chen G. The Roles of Vitamin A in the Regulation of Carbohydrate, Lipid, and Protein Metabolism. Journal of Clinical Medicine. 2014; 3(2):453-479. https://doi.org/10.3390/jcm3020453
Chicago/Turabian StyleChen, Wei, and Guoxun Chen. 2014. "The Roles of Vitamin A in the Regulation of Carbohydrate, Lipid, and Protein Metabolism" Journal of Clinical Medicine 3, no. 2: 453-479. https://doi.org/10.3390/jcm3020453
APA StyleChen, W., & Chen, G. (2014). The Roles of Vitamin A in the Regulation of Carbohydrate, Lipid, and Protein Metabolism. Journal of Clinical Medicine, 3(2), 453-479. https://doi.org/10.3390/jcm3020453