
Repurposing Treatments to Enhance Innate Immunity. Can Statins Improve Neutrophil Functions and Clinical Outcomes in COPD?

 ,
,
Abstract
:1. Introduction
1.1. Inflammation, the Neutrophil, and COPD
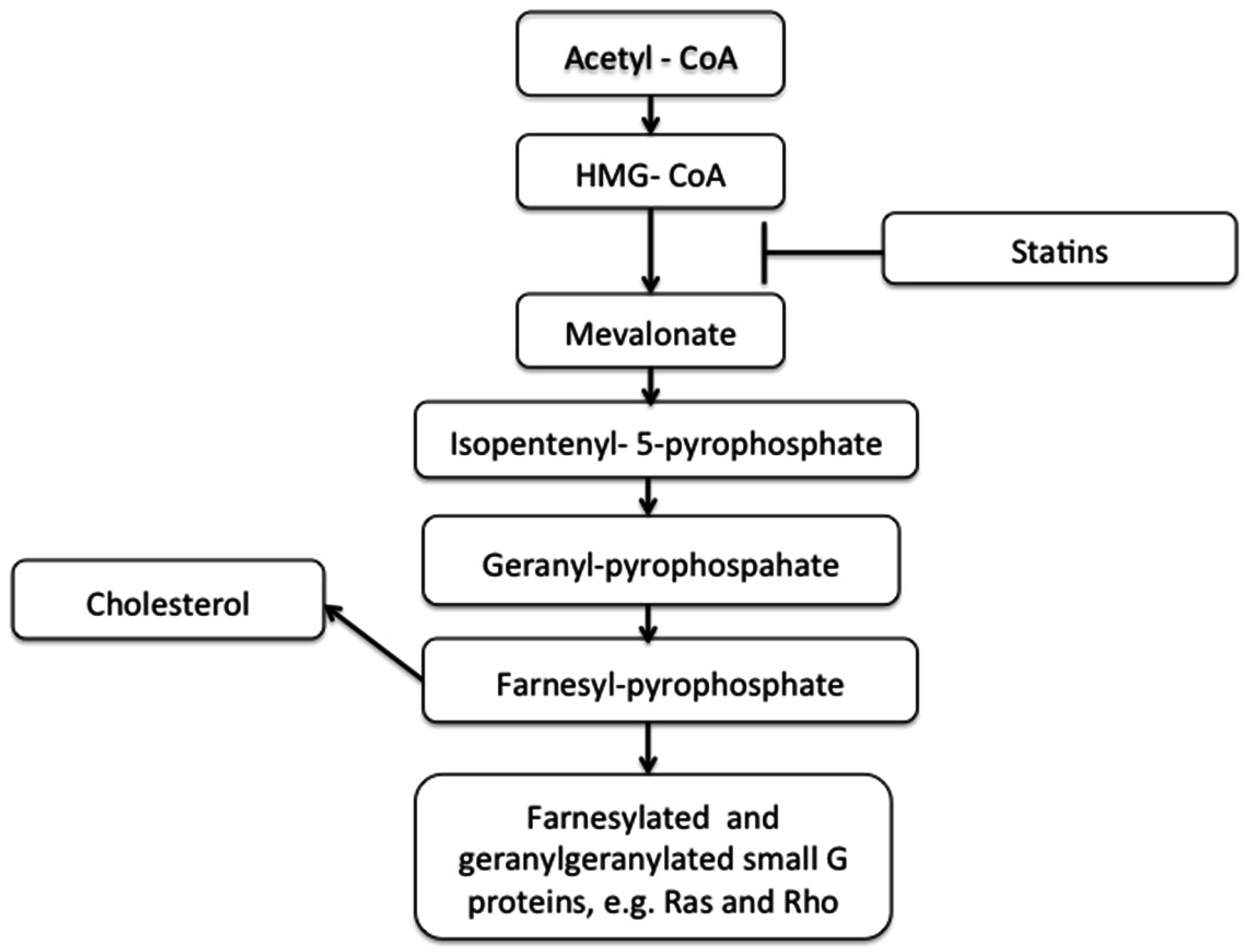
1.2. Cholesterol, Statins, and Cardiovascular Disease
1.3. Statins and Clinical Outcomes in COPD
2. Statins and Neutrophil Migration in COPD; In Vitro Studies
3. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Sevenoaks, M.J.; Stockley, R.A. Chronic obstructive pulmonary disease, inflammation and co-morbidity—A common inflammatory phenotype? Respir. Res. 2006, 7, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Parr, D.G.; White, A.J.; Bayley, D.L.; Guest, P.J.; Stockley, R.A. Inflammation in sputum relates to progression of disease in copd: A prospective study. Respir. Res. 2006, 7, 136. [Google Scholar] [CrossRef] [PubMed]
- Curkendall, S.M.; deLuise, C.; Jones, J.K.; Lanes, S.; Stang, M.R.; Goehring, E., Jr.; She, D. Cardiovascular disease in patients with chronic obstructive pulmonary disease, saskatchewan canada. Ann. Epidemiol. 2006, 16, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Feary, J.R.; Rodrigues, L.C.; Smith, C.J.; Hubbard, R.B.; Gibson, J.E. Prevalence of major comorbidities in subjects with copd and incidence of myocardial infarction and stroke: A comprehensive analysis using data from primary care. Thorax 2010, 65, 956–962. [Google Scholar] [CrossRef] [PubMed]
- Pitta, F.; Troosters, T.; Spruit, M.A.; Probst, V.S.; Decramer, M.; Gosselink, R. Characteristics of physical activities in daily life in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2005, 171, 972–977. [Google Scholar] [CrossRef] [PubMed]
- Mannino, D.M.; Thorn, D.; Swensen, A.; Holguin, F. Prevalence and outcomes of diabetes, hypertension and cardiovascular disease in copd. Eur. Respir. J. 2008, 32, 962–969. [Google Scholar] [CrossRef] [PubMed]
- Divo, M.; Cote, C.; de Torres, J.P.; Casanova, C.; Marin, J.M.; Pinto-Plata, V.; Zulueta, J.; Cabrera, C.; Zagaceta, J.; Hunninghake, G.; et al. Comorbidities and risk of mortality in patients with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2012, 186, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Lozano, R.; Naghavi, M.; Foreman, K.; Lim, S.; Shibuya, K.; Aboyans, V. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: A systematic analysis for the global burden of disease study 2010. Lancet 2012, 380, 2095–2128. [Google Scholar] [CrossRef]
- NICE. Chronic Obstructive Pulmonary Disease Costing Report. Implementing Nice Guidance. Available online: https://www.nice.org.uk/guidance/cg101/resources/costing-report-134511805 (accessed on 11 July 2016).
- Mannino, D.M.; Buist, A.S. Global burden of copd: Risk factors, prevalence, and future trends. Lancet 2007, 370, 765–773. [Google Scholar] [CrossRef]
- Salvi, S.; Barnes, P.J. Is exposure to biomass smoke the biggest risk factor for copd globally? Chest 2010, 138, 3–6. [Google Scholar] [CrossRef] [PubMed]
- Kurmi, O.P.; Semple, S.; Simkhada, P.; Smith, W.C.S.; Ayres, J.G. Copd and chronic bronchitis risk of indoor air pollution from solid fuel: A systematic review and meta-analysis. Thorax 2010, 65, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Ernst, P.; Saad, N.; Suissa, S. Inhaled corticosteroids in copd: The clinical evidence. Eur. Respir. J. 2015, 45, 525–537. [Google Scholar] [CrossRef] [PubMed]
- Pauwels, R.A.; Buist, A.S.; Calverley, P.M.; Jenkins, C.R.; Hurd, S.S.; Committee, G.S. Global strategy for the diagnosis, management and prevention of chronic obstructive pulmonary disease. Nhlbi/who global initiative for chronic obstructive lung disease (gold) workshop summary. Am. J. Respir. Crit. Care Med. 2001, 163, 1256–1276. [Google Scholar] [CrossRef] [PubMed]
- Stockley, R.A. Neutrophils and the pathogenesis of copd. Chest 2002, 121, 151S–155S. [Google Scholar] [CrossRef] [PubMed]
- Pilette, C.; Colinet, B.; Kiss, R.; André, S.; Kaltner, H.; Gabius, H.-J.; Delos, M.; Vaerman, J.-P.; Decramer, M.; Sibille, Y. Increased galectin-3 expression and intra-epithelial neutrophils in small airways in severe copd. Eur. Respir. J. 2007, 29, 914–922. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.B.; Daughton, D.; Robbins, R.A.; Ghafouri, M.A.; Oehlerking, M.; Rennard, S.I. Intraluminal airway inflammation in chronic bronchitis: Characterization and correlation with clinical parameters. Am. J. Respir. Crit. Care Med. 1989, 140, 1527–1537. [Google Scholar] [CrossRef] [PubMed]
- Damiano, V.; Tsang, A.; Kucich, U.; Abrams, W.; Rosenbloom, J.; Kimbel, P.; Fallahnejad, M.; Weinbaum, G. Immunolocalization of elastase in human emphysematous lungs. J. Clin. Invest. 1986, 78, 482. [Google Scholar] [CrossRef] [PubMed]
- Korkmaz, B.; Moreau, T.; Gauthier, F. Neutrophil elastase, proteinase 3 and cathepsin g: Physicochemical properties, activity and physiopathological functions. Biochimie 2008, 90, 227–242. [Google Scholar] [CrossRef] [PubMed]
- Nadel, J.A. Role of neutrophil elastase in hypersecretion during copd exacerbations, and proposed therapies. Chest 2000, 117, 386s–389s. [Google Scholar] [CrossRef] [PubMed]
- Borregaard, N. Neutrophils, from marrow to microbes. Immunity 2010, 33, 657–670. [Google Scholar] [CrossRef] [PubMed]
- Kansas, G.S. Selectins and their ligands: Current concepts and controversies. Blood 1996, 88, 3259–3287. [Google Scholar] [PubMed]
- Rickert, P.; Weiner, O.D.; Wang, F.; Bourne, H.R.; Servant, G. Leukocytes navigate by compass: Roles of pi3kgamma and its lipid products. Trends Cell Biol. 2000, 10, 466–473. [Google Scholar] [CrossRef]
- Weiner, O.D. Regulation of cell polarity during eukaryotic chemotaxis: The chemotactic compass. Curr. Opin. Cell Biol. 2002, 14, 196–202. [Google Scholar] [CrossRef]
- King, J.S.; Insall, R.H. Chemotaxis: Finding the way forward with dictyostelium. Trends Cell Biol. 2009, 19, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Stadtmann, A.; Block, H.; Volmering, S.; Abram, C.; Sohlbach, C.; Boras, M.; Lowell, C.A.; Zarbock, A. Cross-talk between shp1 and pipkiγ controls leukocyte recruitment. J. Immunol. 2015, 195, 1152–1161. [Google Scholar] [CrossRef] [PubMed]
- Kriebel, P.W.; Barr, V.A.; Parent, C.A. Adenylyl cyclase localization regulates streaming during chemotaxis. Cell 2003, 112, 549–560. [Google Scholar] [CrossRef]
- Wang, F. The signaling mechanisms underlying cell polarity and chemotaxis. Cold Spring Harb. Perspect. Biol. 2009, 1, a002980. [Google Scholar] [CrossRef] [PubMed]
- Bourdin, A.; Burgel, P.-R.; Chanez, P.; Garcia, G.; Perez, T.; Roche, N. Recent advances in copd: Pathophysiology, respiratory physiology and clinical aspects, including comorbidities. Eur. Respir. Rev. 2009, 18, 198–212. [Google Scholar] [CrossRef] [PubMed]
- Roth, M. Pathogenesis of copd. Part iii. Inflammation in copd. Int. J. Tuberc. Lung Dis. 2008, 12, 375–380. [Google Scholar] [PubMed]
- Kao, R.C.; Wehner, N.G.; Skubitz, K.M.; Gray, B.H.; Hoidal, J.R. Proteinase 3. A distinct human polymorphonuclear leukocyte proteinase that produces emphysema in hamsters. J. Clin. Invest. 1988, 82, 1963–1973. [Google Scholar] [CrossRef] [PubMed]
- Liou, T.G.; Campbell, E.J. Nonisotropic enzyme—Inhibitor interactions: A novel nonoxidative mechanism for quantum proteolysis by human neutrophils. Biochemistry 1995, 34, 16171–16177. [Google Scholar] [CrossRef] [PubMed]
- Stone, H.; McNab, G.; Wood, A.M.; Stockley, R.A.; Sapey, E. Variability of sputum inflammatory mediators in copd and alpha1-antitrypsin deficiency. Eur. Respir. J. 2012, 40, 561–569. [Google Scholar] [CrossRef] [PubMed]
- Underhill, D.M.; Ozinsky, A. Phagocytosis of microbes: Complexity in action. Annu. Rev. Immunol. 2002, 20, 825–852. [Google Scholar] [CrossRef] [PubMed]
- Amulic, B.; Cazalet, C.; Hayes, G.L.; Metzler, K.D.; Zychlinsky, A. Neutrophil function: From mechanisms to disease. Annu. Rev. Immunol. 2012, 30, 459–489. [Google Scholar] [CrossRef] [PubMed]
- Dahlgren, C.; Karlsson, A. Respiratory burst in human neutrophils. J. Immunol. Methods 1999, 232, 3–14. [Google Scholar] [CrossRef]
- Takeyama, K.; Dabbagh, K.; Jeong Shim, J.; Dao-Pick, T.; Ueki, I.F.; Nadel, J.A. Oxidative stress causes mucin synthesis via transactivation of epidermal growth factor receptor: Role of neutrophils. J. Immunol. 2000, 164, 1546–1552. [Google Scholar] [CrossRef] [PubMed]
- Voynow, J.A.; Young, L.R.; Wang, Y.; Horger, T.; Rose, M.C.; Fischer, B.M. Neutrophil elastase increases MUC5AC mRNA and protein expression in respiratory epithelial cells. Am. J. Physiol. 1999, 276, L835–L843. [Google Scholar] [PubMed]
- Takeyama, K.; Fahy, J.V.; Nadel, J.A. Relationship of epidermal growth factor receptors to goblet cell production in human bronchi. Am. J. Respir. Crit. Care Med. 2001, 163, 511–516. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.; Finnemann, S.C. Regulation of phagocytosis by rho gtpases. Small GTPases 2015, 6, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, M.J.; Radic, M. Neutrophil extracellular traps: Double-edged swords of innate immunity. J. Immunol. 2012, 189, 2689–2695. [Google Scholar] [CrossRef] [PubMed]
- Tashkin, D.P.; Clark, V.A.; Coulson, A.H.; Simmons, M.; Bourque, L.B.; Reems, C.; Detels, R.; Sayre, J.W.; Rokaw, S.N. The ucla population studies of chronic obstructive respiratory disease. Viii. Effects of smoking cessation on lung function; a prospective study of a free-living population. Am. Rev. Respir. Dis. 1984, 130, 707–715. [Google Scholar] [PubMed]
- Louhelainen, N.; Rytila, P.; Haahtela, T.; Kinnula, V.L.; Djukanovic, R. Persistance of oxidant and protease burden in the airways after smoking cessation. BMC Pulm. Med. 2009, 9. [Google Scholar] [CrossRef] [PubMed]
- Willemse, B.W.; ten Hacken, N.H.; Rutgers, B.; Lesman-Leegte, I.G.; Postma, D.S.; Timens, W. Effect of 1 year smoking cessation on airway inflammation in copd and asymtomatic smokers. Eur. Respir. J. 2005, 26, 835–845. [Google Scholar] [CrossRef] [PubMed]
- Lapperre, T.S.; Postma, D.S.; Gosman, M.M.; Snoeck-Strband, J.B.; ten Hacken, N.H.; Hiemstra, P.S.; Timens, W.; Sterk, P.J.; Mauad, T. Relation between duration of smoking cessation and bronchial inflammation in copd. Thorax 2006, 61, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Sapey, E.; Stockley, J.A.; Greenwood, H.; Ahmad, A.; Bayley, D.; Lord, J.M.; Insall, R.H.; Stockley, R.A. Behavioral and structural differences in migrating peripheral neutrophils from patients with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2011, 183, 1176–1186. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, S.; Rosowski, E.E.; Huttenlocjer, A. Neutrophil migration in infection and wound repair: Going forward in reverse. Nat. Rev. Immunol. 2016, 16, 378–391. [Google Scholar] [CrossRef] [PubMed]
- Liou, T.G.; Campbell, E.J. Quantum proteolysis resulting from release of single granules by human neutrophils: A novel, nonoxidative mechanism of extracellular proteolytic activity. J. Immunol. 1996, 157, 2624–2631. [Google Scholar] [PubMed]
- Clarenbach, C.F.; Senn, O.; Sievi, N.A.; Camen, G.; van Gestel, A.J.R.; Rossi, V.A.; Puhan, M.A.; Thurnheer, R.; Russi, E.W.; Kohler, M. Determinants of endothelial function in patients with COPD. Eur. Respir. J. 2013, 42, 1194–1204. [Google Scholar] [CrossRef] [PubMed]
- Carden, D.; Xiao, F.; Moak, C.; Willis, B.H.; Robinson-Jackson, S.; Alexander, S. Neutrophil elastase promotes lung microvascular injury and proteolysis of endothelial cadherins. Am. J. Phys. Heart Circ. Physiol. 1998, 275, H385–H392. [Google Scholar]
- Noguera, A.; Batle, S.; Miralles, C.; Iglesias, J.; Busquets, X.; MacNee, W.; Agustí, A.G.N. Enhanced neutrophil response in chronic obstructive pulmonary disease. Thorax 2001, 56, 432–437. [Google Scholar] [CrossRef] [PubMed]
- Vaitkus, M.; Lavinskiene, S.; Barkauskiene, D.; Bieksiene, K.; Jeroch, J.; Sakalauskas, R. Reactive oxygen species in peripheral blood and sputum neutrophils during bacterial and nonbacterial acute exacerbation of chronic obstructive pulmonary disease. Inflammation 2013, 36, 1485–1493. [Google Scholar] [CrossRef] [PubMed]
- Grabcanovic-Musija, F.; Obermayer, A.; Stoiber, W.; Krautgartner, W.-D.; Steinbacher, P.; Winterberg, N.; Bathke, A.C.; Klappacher, M.; Studnicka, M. Neutrophil extracellular trap (NET) formation characterises stable and exacerbated copd and correlates with airflow limitation. Respir. Res. 2015, 16, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Sangaletti, S.; Tripodo, C.; Chiodoni, C.; Guarnotta, C.; Cappetti, B.; Casalini, P.; Piconese, S.; Parenza, M.; Guiducci, C.; Vitali, C.; et al. Neutrophil extracellular traps mediate transfer of cytoplasmic neutrophil antigens to myeloid dendritic cells toward anca induction and associated autoimmunity. Blood 2012, 120, 3007–3018. [Google Scholar] [CrossRef] [PubMed]
- Low, T.B.; Greene, C.M.; O’Neill, S.J.; McElvaney, N.G. Quantification and evaluation of the role of antielastin autoantibodies in the emphysematous lung. Pulm. Med. 2001, 826160. [Google Scholar] [CrossRef] [PubMed]
- Carmona-Rivera, C.; Zhao, W.; Yalavarthi, S.; Kaplan, M.J. Neutrophil extracellular traps induce endothelial dysfunction in systemic lupus erythematosus through the activation of matrix metalloproteinase-2. Ann. Rheum. Dis. 2015, 74, 1417–1424. [Google Scholar] [CrossRef] [PubMed]
- Döring, Y.; Soehnlein, O.; Weber, C. Neutrophils cast nets in atherosclerosis: Employing peptidylarginine deiminase as a therapeutic target. Circ. Res. 2014, 114, 931–934. [Google Scholar] [CrossRef] [PubMed]
- Stockley, J.A.; Walton, G.M.; Sapey, E. Aberrant neutrophil functions in stable chronic obstructive pulmonary disease: The neutrophil as an immunotherapeutic target. Int. Immunopharmacol. 2013, 17, 1211–1217. [Google Scholar] [CrossRef] [PubMed]
- Mega, J.L.; Stitziel, N.O.; Smith, J.G.; Chasman, D.I.; Caulfield, M.J.; Devlin, J.J.; Nordio, F.; Hyde, C.L.; Cannon, C.P.; Sacks, F.M.; et al. Genetic risk, coronary heart disease events, and the clinical benefit of statin therapy: An analysis of primary and secondary prevention trials. Lancet 2015, 385, 2264–2271. [Google Scholar] [CrossRef]
- Baigent, C.; Keech, A.; Kearney, P.M.; Blackwell, L.; Buck, G.; Pollicino, C.; Kirby, A.; Sourjina, T.; Peto, R.; Collins, R.; et al. Efficacy and safety of cholesterol-lowering treatment: Prospective meta-analysis of data from 90056 participants in 14 randomised trials of statins. Lancet 2005, 366, 1267–1278. [Google Scholar]
- Mihaylova, B.; Emberson, J.; Blackwell, L.; Keech, A.; Simes, J.; Barnes, E.H.; Voysey, M.; Gray, A.; Collins, R.; Baigent, C. The effects of lowering LDL cholesterol with statin therapy in people at low risk of vascular disease: Meta-analysis of individual data from 27 randomised trials. Lancet 2012, 380, 581–590. [Google Scholar] [CrossRef]
- Sever, P.S.; Dahlöf, B.; Poulter, N.R.; Wedel, H.; Beevers, G.; Caulfield, M.; Collins, R.; Kjeldsen, S.E.; Kristinsson, A.; McInnes, G.T.; et al. Prevention of coronary and stroke events with atorvastatin in hypertensive patients who have average or lower-than-average cholesterol concentrations, in the Anglo-Scandinavian Cardiac Outcomes Trial--Lipid Lowering Arm (ASCOT-LLA): A multicentre randomised controlled trial. Lancet 2003, 361, 1149–1158. [Google Scholar] [PubMed]
- Ravnskov, U.; Diamond, D.M.; Hama, R.; Hamazaki, T.; Hammarskjöld, B.; Hynes, N.; Kendrick, M.; Langsjoen, P.H.; Malhotra, A.; Mascitelli, L.; et al. Lack of an association or an inverse association between low-density-lipoprotein cholesterol and mortality in the elderly: A systematic review. BMJ Open 2016, 6, e010401. [Google Scholar] [CrossRef] [PubMed]
- Higashio, H.; Satoh, Y.-I.; Saino, T. Mast cell degranulation is negatively regulated by the Munc13-4-binding small-guanosine triphosphatase Rab37. Sci. Rep. 2016, 6, 22539. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.X.-J.; Soto, I.; Guo, Y.-L.; Liu, Y. Control of secondary granule release in neutrophils by Ral GTPase. J. Biol. Chem. 2011, 286, 11724–11733. [Google Scholar] [CrossRef] [PubMed]
- Kleemann, R.; Verschuren, L.; de Rooij, B.-J.; Lindeman, J.; de Maat, M.M.; Szalai, A.J.; Princen, H.M.G.; Kooistra, T. Evidence for anti-inflammatory activity of statins and PPARα activators in human C-reactive protein transgenic mice in vivo and in cultured human hepatocytes in vitro. Blood 2004, 103, 4188–4194. [Google Scholar] [CrossRef] [PubMed]
- Dichtl, W.; Dulak, J.; Frick, M.; Alber, H.F.; Schwarzacher, S.P.; Ares, M.P.S.; Nilsson, J.; Pachinger, O.; Weidinger, F. HMG-CoA reductase inhibitors regulate inflammatory transcription factors in human endothelial and vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Kureishi, Y.; Zhengyu, L.; Ichiro, S.; Bialik, A.; Fulton, D.; Lefer, D.; Sessa, W.; Walsh, K. The HMG-CoA reductase inhibitor simvastatin activates the protein kinase Akt and promotes angiogenesis in normocholesterolemic animals. Nat. Med. 2000, 6, 1004–1010. [Google Scholar] [PubMed]
- Musial, J.; Undas, A.; Gajewski, P.; Jankowski, M.; Sydor, W.; Szczeklik, A. Anti-inflammatory effects of simvastatin in subjects with hypercholesterolemia. Int. J. Cardiol. 2001, 77, 247–253. [Google Scholar] [CrossRef]
- Diomede, L.; Albani, D.; Sottocorno, M.; Donati, M.B.; Bianchi, M.; Fruscella, P.; Salmona, M. In vivo anti-inflammatory effect of statins is mediated by nonsterol mevalonate products. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 1327–1332. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, M.; Einhaus, D.; Gold, E.S.; Aderem, A. Simvastatin augments lipopolysaccharide-induced proinflammatory responses in macrophages by differential regulation of the c-Fos and c-Jun transcription factors. J. Immunol. 2004, 172, 7377–7384. [Google Scholar] [CrossRef] [PubMed]
- Pruefer, D.; Scalia, R.; Lefer, A.M. Simvastatin inhibits leukocyte-endothelial cell interactions and protects against inflammatory processes in normocholesterolemic rats. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 2894–2900. [Google Scholar] [CrossRef] [PubMed]
- Durant, R.; Klouche, K.; Delbosc, S.; Morena, M.; Amigues, L.; Beraud, J.J.; Canaud, B.; Cristol, J.P. Superoxide anion overproduction in sepsis: Effects of vitamin e and simvastatin. Shock 2004, 22, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Kamio, K.; Liu, X.D.; Sugiura, H.; Togo, S.; Kawasaki, S.; Wang, X.; Ahn, Y.; Hogaboam, C.; Rennard, S.I. Statins inhibit matrix metalloproteinase release from human lung fibroblasts. Eur. Respir. J. 2010, 35, 637–646. [Google Scholar] [CrossRef] [PubMed]
- Hakamada-Taguchi, R.; Uehara, Y.; Kuribayashi, K.; Numabe, A.; Saito, K.; Negoro, H.; Fujita, T.; Toyo-oka, T.; Kato, T. Inhibition of hydroxymethylglutaryl-coenzyme a reductase reduces TH1 development and promotes TH2 development. Circ. Res. 2003, 93, 948–956. [Google Scholar] [CrossRef] [PubMed]
- Weitz-Schmidt, G.; Welzenbach, K.; Brinkmann, V.; Kamata, T.; Kallen, J.; Bruns, C.; Cottens, S.; Takada, Y.; Hommel, U. Statins selectively inhibit leukocyte function antigen-1 by binding to a novel regulatory integrin site. Nat. Med. 2001, 7, 687–692. [Google Scholar] [CrossRef] [PubMed]
- Kwak, B.; Mulhaupt, F.; Myit, S.; Mach, F. Statins as a newly recognized type of immunomodulator. Nat. Med. 2000, 6, 1399–1402. [Google Scholar] [PubMed]
- Romano, M.; Diomede, L.; Sironi, M.; Massimiliano, L.; Sottocorno, M.; Polentarutti, N.; Guglielmotti, A.; Albani, D.; Bruno, A.; Fruscella, P.; et al. Inhibition of monocyte chemotactic protein-1 synthesis by statins. Lab. Invest. 2000, 80, 1095–1100. [Google Scholar] [CrossRef] [PubMed]
- Sever, P.S.; Poulter, N.R.; Chang, C.L.; Thom, S.A.; Hughes, A.D.; Welsh, P.; Sattar, N. Evaluation of C-reactive protein before and on-treatment as a predictor of benefit of atorvastatin: A cohort analysis from the Anglo-Scandinavian Cardiac Outcomes Trial lipid-lowering arm. J. Am. Coll. Cardiol. 2013, 62, 717–729. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, R. The role of tnf in cardiovascular disease. Pharmacol. Res. 1999, 40, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Niwa, S.; Totsuka, T.; Hayashi, S. Inhibitory effect of fluvastatin, an HMG-CoA reductase inhibitor, on the expression of adhesion molecules on human monocyte cell line. Int. J. Immunopharmacol. 1996, 18, 669–675. [Google Scholar] [CrossRef]
- Takeuchi, S.; Kawashima, S.; Rikitake, Y.; Ueyama, T.; Inoue, N.; Hirata, K.-I.; Yokoyama, M. Cerivastatin suppresses lipopolysaccharide-induced ICAM-1 expression through inhibition of Rho GTPase in BAEC. Biochem. Biophys. Res. Commun. 2000, 269, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Antoniades, C.; Bakogiannis, C.; Tousoulis, D.; Reilly, S.; Zhang, M.-H.; Paschalis, A.; Antonopoulos, A.S.; Demosthenous, M.; Miliou, A.; Psarros, C.; et al. Preoperative atorvastatin treatment in CABG patients rapidly improves vein graft redox state by inhibition of Rac1 and NADPH-oxidase activity. Circulation 2010, 122, S66–S73. [Google Scholar] [CrossRef] [PubMed]
- Crisby, M.; Nordin-Fredriksson, G.; Shah, P.K.; Yano, J.; Zhu, J.; Nilsson, J. Pravastatin treatment increases collagen content and decreases lipid content, inflammation, metalloproteinases, and cell death in human carotid plaques. Circulation 2001, 103, 926–933. [Google Scholar] [CrossRef] [PubMed]
- Soehnlein, O. Multiple roles for neutrophils in atherosclerosis. Circ. Res. 2012, 110, 875–888. [Google Scholar] [CrossRef] [PubMed]
- Hartwig, H.; Silvestre Roig, C.; Daemen, M.; Lutgens, E.; Soehnlein, O. Neutrophils in atherosclerosis. A brief overview. Hämostaseologie 2015, 35, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Hiasa, Y.; Ohara, Y.; Miyazaki, S.; Ogura, R.; Suzuki, N.; Hosokawa, S.; Kishi, K.; Ohtani, R. Relationship of admission neutrophil count to microvascular injury, left ventricular dilation, and long-term outcome in patients treated with primary angioplasty for acute myocardial infarction. Circ. J. 2008, 72, 867–872. [Google Scholar] [CrossRef] [PubMed]
- Kaya, M.G.; Yalcin, R.; Okyay, K.; Poyraz, F.; Bayraktar, N.; Pasaoglu, H.; Boyaci, B.; Cengel, A. Potential role of plasma myeloperoxidase level in predicting long-term outcome of acute myocardial infarction. Texas Heart Inst. J. 2012, 39, 500–506. [Google Scholar]
- Awasthi, D.; Nagarkoti, S.; Kumar, A.; Dubey, M.; Singh, A.K.; Pathak, P.; Chandra, T.; Barthwal, M.K.; Dikshit, M. Oxidized LDL induced extracellular trap formation in human neutrophils via TLR-PKC-IRAK-MAPK and NADPH-Oxidase activation. Free Radic. Biol. Med. 2016, 93, 190–203. [Google Scholar] [CrossRef] [PubMed]
- Sedgwick, J.B.; Hwang, Y.S.; Gerbyshak, H.A.; Kita, H.; Busse, W.W. Oxidized low-density lipoprotein activates migration and degranulation of human granulocytes. Am. J. Respir. Cell Mol. Biol. 2003, 29, 702–709. [Google Scholar] [CrossRef] [PubMed]
- Silveira, A.; Dominical, V.; Lazarini, M.; Costa, F.; Conran, N. Simvastatin abrogates inflamed neutrophil adhesive properties, in association with the inhibition of Mac-1 integrin expression and modulation of Rho kinase activity. Inflamm. Res. 2013, 62, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Al-Ani, B. Simvastatin inhibits neutrophil degranulation induced by anti-neutrophil cytoplasm auto-antibodies and N-formyl-methionine-leucine-phenylalanine (fMLP) peptide. Saudi Med. J. 2013, 34, 477–483. [Google Scholar] [PubMed]
- Kinsella, A.; Raza, A.; Kennedy, S.; Fan, Y.; Wood, A.E.; Watson, R.W. The impact of high-dose statin therapy on transendothelial neutrophil migration and serum cholesterol levels in healthy male volunteers. Eur. J. Clin. Pharmacol. 2011, 67, 1103–1108. [Google Scholar] [CrossRef] [PubMed]
- Al-Ghoul, W.M.; Kim, M.S.; Fazal, N.; Azim, A.C.; Ali, A. Evidence for simvastatin anti-inflammatory actions based on quantitative analyses of netosis and other inflammation/oxidation markers. Results Immunol. 2014, 4, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Black-Shinn, J.L.; Kinney, G.L.; Wise, A.L.; Regan, E.A.; Make, B.; Krantz, M.J.; Barr, R.G.; Murphy, J.R.; Lynch, D.; Silverman, E.K.; et al. Cardiovascular disease is associated with copd severity and reduced functional status and quality of life. COPD 2014, 11, 546–551. [Google Scholar] [CrossRef] [PubMed]
- Keddissi, J.I.; Younis, W.G.; Chbeir, E.A.; Daher, N.N.; Dernaika, T.A.; Kinasewitz, G.T. The use of statins and lung function in current and former smokers. Chest 2007, 132, 1764–1771. [Google Scholar] [CrossRef] [PubMed]
- Alexeeff, S.E.; Litonjua, A.A.; Sparrow, D.; Vokonas, P.S.; Schwartz, J. Statin use reduces decline in lung function. Am. J. Respir. Crit. Care Med. 2007, 176, 742–747. [Google Scholar] [CrossRef] [PubMed]
- Frost, F.J.; Petersen, H.; Tollestrup, K.; Skipper, B. Influenza and COPD mortality protection as pleiotropic, dose-dependent effects of statins. Chest 2007, 131, 1006–1012. [Google Scholar] [CrossRef] [PubMed]
- Van Gestel, Y.R.; Hoeks, S.E.; Sin, D.D.; Simsek, C.; Welten, G.M.; Schouten, O.; Stam, H.; Mertens, F.W.; van Domburg, R.T.; Poldermans, D. Effect of statin therapy on mortality in patients with peripheral arterial disease and comparison of those with versus without associated chronic obstructive pulmonary disease. Am. J. Cardiol. 2008, 102, 192–196. [Google Scholar] [CrossRef] [PubMed]
- Cao, C.; Wu, Y.; Xu, Z.; Lv, D.; Zhang, C.; Lai, T.; Li, W.; Shen, H. The effect of statins on chronic obstructive pulmonary disease exacerbation and mortality: A systematic review and meta-analysis of observational research. Sci. Rep. 2015, 5, 16461. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-H.; Lee, D.-S.; Kim, E.-K.; Choe, K.-H.; Oh, Y.-M.; Shim, T.-S.; Kim, S.-E.; Lee, Y.-S.; Lee, S.-D. Simvastatin inhibits cigarette smoking—Induced emphysema and pulmonary hypertension in rat lungs. Am. J. Respir. Crit. Care Med. 2005, 172, 987–993. [Google Scholar] [CrossRef] [PubMed]
- Neukamm, A.; Høiseth, A.D.; Einvik, G.; Lehmann, S.; Hagve, T.A.; Søyseth, V.; Omland, T. Rosuvastatin treatment in stable chronic obstructive pulmonary disease (RODEO): A randomized controlled trial. J. Intern. Med. 2015, 278, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Citgez, E.; van der Palen, J.; Koehorst-ter Huurne, K.; Movig, K.; van der Valk, P.; Brusse-Keizer, M. Statins and morbidity and mortality in copd in the comic study: A prospective copd cohort study. BMJ Open Respir. Res. 2016, 3, e000142. [Google Scholar] [CrossRef] [PubMed]
- Howard, M.L.; Vincent, A.H. Statin effects on exacerbation rates, mortality, and inflammatory markers in patients with chronic obstructive pulmonary disease: A review of prospective studies. Pharmacother. J. Hum. Pharmacol. Drug Ther. 2016, 36, 536–547. [Google Scholar] [CrossRef] [PubMed]
- Patel, J.M.; Thickett, D.R.; Gao, F.; Sapey, E. Statins for sepsis: Distinguishing signal from the noise when designing clinical trials. Am. J. Respir. Crit. Care Med. 2013, 188, 874. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, J.R.; Barnard, J.W.; Grigoryev, D.N.; Ma, S.-F.; Tuder, R.M.; Garcia, J.G.N. Simvastatin attenuates vascular leak and inflammation in murine inflammatory lung injury. Am. J. Phys. Lung Cell. Mol. Physiol. 2005, 288, L1026–L1032. [Google Scholar] [CrossRef] [PubMed]
- Steiner, S.; Speidl, W.S.; Pleiner, J.; Seidinger, D.; Zorn, G.; Kaun, C.; Wojta, J.; Huber, K.; Minar, E.; Wolzt, M.; et al. Simvastatin blunts endotoxin-induced tissue factor in vivo. Circulation 2005, 111, 1841–1846. [Google Scholar] [CrossRef] [PubMed]
- Criner, G.J.; Connett, J.E.; Aaron, S.D.; Albert, R.K.; Bailey, W.C.; Casaburi, R.; Cooper, J.A.D.; Curtis, J.L.; Dransfield, M.T.; Han, M.K.; et al. Simvastatin for the prevention of exacerbations in moderate-to-severe COPD. N. Engl. J. Med. 2014, 370, 2201–2210. [Google Scholar] [CrossRef] [PubMed]
- Rezk, N.A.; Elewa, A. Anti inflammatory effects of statin in copd. Egypt. J. Chest Dis. Tuberc. 2013, 62, 65–69. [Google Scholar] [CrossRef]
- Sapey, E.; Greenwood, H.; Walton, G.; Mann, E.; Love, A.; Aaronson, N.; Insall, R.H.; Stockley, R.A.; Lord, J.M. Phosphoinositide 3-kinase inhibition restores neutrophil accuracy in the elderly: Toward targeted treatments for immunosenescence. Blood 2014, 123, 239–248. [Google Scholar] [CrossRef] [PubMed]
- Corsini, A.; Bellosta, S.; Baetta, R.; Fumagalli, R.; Paoletti, R.; Bernini, F. New insights into the pharmacodynamic and pharmakinetic properties of statins. Pharmacol. Ther. 1999, 84, 413–428. [Google Scholar] [CrossRef]
- Chow, O.A.; von Kockritz-Blickwede, M.; Bright, A.T.; Hensler, M.E.; Zinkernagel, A.S.; Cogen, A.L.; Gallo, R.L.; Monestier, M.; Wang, Y.; Glass, C.K.; et al. Statins enhance formation of phagocyte extracellular traps. Cell Host Microbe 2010, 8, 445–454. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Effect | Model/Cell-Type | Reference |
|---|---|---|
| Increased NF-IκB expression | Human hepatocytes | [67] |
| Human endothelial cells | [68] | |
| Phosphorylation of Akt | Human endothelial cells | [69] |
| Decreased TNFα production | Human volunteers | [70] |
| Decreased IL-6 production | Human volunteers | [70] |
| Mice | [71] | |
| Increased IL-12 production | Mouse peritoneal macrophages | [72] |
| Reduced plasma C-reactive protein | Human volunteers | [70] |
| Human hepatocytes | [67] | |
| Reduced expression of P-Selectin | Rat mesenteric endothelium | [73] |
| Reduced superoxide production | Human monocytes | [74] |
| Inhibition of matrix metalloproteinase production | Human lung fibroblasts | [75] |
| Regulation of Th1/Th2 polarisation | Mice | [76] |
| Block of LFA-1 ICAM interaction | Various: isolated proteins, cell lines, murine model | [77] |
| Inhibition of MHC expression | Human macrophages & endothelial cells | [78] |
| Inhibition of monocyte chemotactic protein-1 synthesis | Human peripheral blood mononuclear cells | [79] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Walton, G.M.; Stockley, J.A.; Griffiths, D.; Sadhra, C.S.; Purvis, T.; Sapey, E. Repurposing Treatments to Enhance Innate Immunity. Can Statins Improve Neutrophil Functions and Clinical Outcomes in COPD? J. Clin. Med. 2016, 5, 89. https://doi.org/10.3390/jcm5100089
Walton GM, Stockley JA, Griffiths D, Sadhra CS, Purvis T, Sapey E. Repurposing Treatments to Enhance Innate Immunity. Can Statins Improve Neutrophil Functions and Clinical Outcomes in COPD? Journal of Clinical Medicine. 2016; 5(10):89. https://doi.org/10.3390/jcm5100089
Chicago/Turabian StyleWalton, Georgia M., James A. Stockley, Diane Griffiths, Charandeep S. Sadhra, Thomas Purvis, and Elizabeth Sapey. 2016. "Repurposing Treatments to Enhance Innate Immunity. Can Statins Improve Neutrophil Functions and Clinical Outcomes in COPD?" Journal of Clinical Medicine 5, no. 10: 89. https://doi.org/10.3390/jcm5100089