
A Systematic Review of the Role of Dysfunctional Wound Healing in the Pathogenesis and Treatment of Idiopathic Pulmonary Fibrosis


Abstract
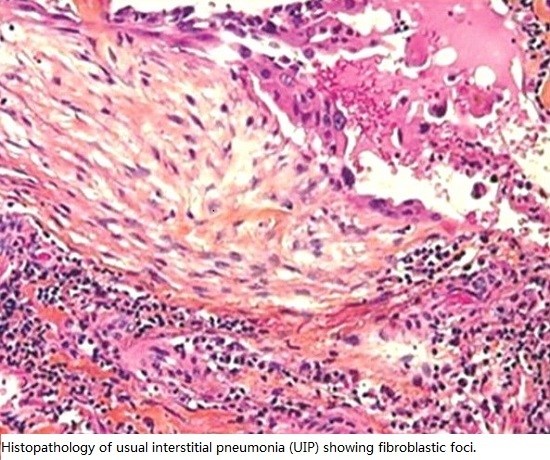
:
1. Overview of the Wound-Healing Process
1.1. Homeostasis Phase
1.2. Inflammatory and Cell Migration Phase
1.3. Proliferation and Extracellular Matrix (ECM) Remodeling Phase
1.4. Chronic Wound Healing
1.5. Etiology
2. Overview of Idiopathic Pulmonary Fibrosis (IPF)
2.1. Prevalence/Incidence
2.2. Treatment Guidelines
2.3. Diagnosis
3. Potential Treatment, Targets of Intervention
3.1. Non-Pharmacologic Management
3.1.1. Home Pulse Oximetry
3.1.2. Pulmonary Rehabilitation
3.1.3. Lung Transplantation
3.2. Pharmacologic Management
3.2.1. Corticosteroids
3.2.2. Anticoagulation Trials in IPF
3.2.3. N-Aetylcysteine
3.2.4. Nintedanib
Mode of Action
Clinical Efficacy
Adverse Effects
3.2.5. Pirfenidone
Mode of Action and In Vivo Activity
Clinical Efficacy
Approval
Adverse Effects
3.2.6. Supplemental Oxygen Therapy
3.3. Lung Injury/Epithelial Cell Death
3.3.1. The Adult Lung Epithelium
3.3.2. The Injured Epithelium and IPF
3.4. Clotting/Coagulation
Coagulation in IPF
3.5. Immune Activation
3.5.1. Fibroblast Accumulation/Myofibroblast Differentiation
3.5.2. Role of Epigenetics in IPF Fibroblast/Myofibroblast Activity
3.6. Remodeling/Matrix
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Velnar, T.; Bailey, T.; Smrkolj, V. The wound healing process: An overview of the cellular and molecular mechanisms. J. Int. Med. Res. 2009, 37, 1528–1542. [Google Scholar] [CrossRef] [PubMed]
- Loots, M.A.; Lamme, E.N.; Zeegelaar, J.; Mekkes, J.R.; Bos, J.D.; Middelkoop, E. Differences in cellular infiltrate and extracellular matrix of chronic diabetic and venous ulcers versus acute wounds. J. Investig. Dermatol. 1998, 111, 850–857. [Google Scholar] [CrossRef] [PubMed]
- Rodero, M.P.; Khosrotehrani, K. Skin wound healing modulation by macrophages. Int. J. Clin. Exp. Pathol. 2010, 3, 643–653. [Google Scholar] [PubMed]
- Gosain, A.; DiPietro, L.A. Aging and wound healing. World J. Surg. 2004, 28, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Tamama, K.; Kerpedjieva, S.S. Acceleration of wound healing by multiple growth factors and cytokines secreted from multipotential stromal cells/mesenchymal stem cells. Adv. Wound Care 2012, 1, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Barrientos, S.; Brem, H.; Stojadinovic, O.; Tomic-Canic, M. Clinical application of growth factors and cytokines in wound healing. Wound Repair Regen. 2014, 22, 569–578. [Google Scholar] [CrossRef] [PubMed]
- Gurtner, G.C.; Werner, S.; Barrandon, Y.; Longaker, M.T. Wound repair and regeneration. Nature 2008, 453, 314–321. [Google Scholar] [CrossRef] [PubMed]
- Wilgus, T.A.; Roy, S.; McDaniel, J.C. Neutrophils and wound repair: Positive actions and negative reactions. Adv. Wound Care 2013, 2, 379–388. [Google Scholar] [CrossRef] [PubMed]
- Mittal, M.; Siddiqui, M.R.; Tran, K.; Reddy, S.P.; Malik, A.B. Reactive oxygen species in inflammation and tissue injury. Antioxid. Redox Signal. 2014, 20, 1126–1167. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; DiPietro, L.A. Factors affecting wound healing. J. Dent. Res. 2010, 89, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Borthwick, L.A.; Corris, P.A.; Mahida, R.; Walker, A.; Gardner, A.; Suwara, M.; Johnson, G.E.; Moisey, E.J.; Brodlie, M.; Ward, C.; et al. Tnfalpha from classically activated macrophages accentuates epithelial to mesenchymal transition in obliterative bronchiolitis. Am. J. Transplant. 2013, 13, 621–633. [Google Scholar] [CrossRef] [PubMed]
- Herrera, I.; Cisneros, J.; Maldonado, M.; Ramirez, R.; Ortiz-Quintero, B.; Anso, E.; Chandel, N.S.; Selman, M.; Pardo, A. Matrix metalloproteinase (mmp)-1 induces lung alveolar epithelial cell migration and proliferation, protects from apoptosis, and represses mitochondrial oxygen consumption. J. Biol. Chem. 2013, 288, 25964–25975. [Google Scholar] [CrossRef] [PubMed]
- Dancer, R.C.A.; Wood, A.M.; Thickett, D.R. Metalloproteinases in idiopathic pulmonary fibrosis. Eur. Respir. J. 2011, 38, 1461–1467. [Google Scholar] [CrossRef] [PubMed]
- Clarke, D.L.; Carruthers, A.M.; Mustelin, T.; Murray, L.A. Matrix regulation of idiopathic pulmonary fibrosis: The role of enzymes. Fibrogenesis Tissue Repair 2013, 6, 20. [Google Scholar] [CrossRef] [PubMed]
- González-López, A.; Albaiceta, G.M. Repair after acute lung injury: Molecular mechanisms and therapeutic opportunities. Crit. Care 2012, 16, 209. [Google Scholar] [CrossRef] [PubMed]
- Crosby, L.M.; Waters, C.M. Epithelial repair mechanisms in the lung. Am. J. Physiol. Lung Cell. Mol. Physiol. 2010, 298, L715–L731. [Google Scholar] [CrossRef] [PubMed]
- Meltzer, E.B.; Noble, P.W. Idiopathic pulmonary fibrosis. Orphanet J. Rare Dis. 2008, 3, 8. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, I.E.; Heinzelmann, K.; Verleden, S.; Eickelberg, O. Characteristic patterns in the fibrotic lung. Comparing idiopathic pulmonary fibrosis with chronic lung allograft dysfunction. Ann. Am. Thorac. Soc. 2015, 12, S34–S41. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Collard, H.R.; Egan, J.J.; Martinez, F.J.; Behr, J.; Brown, K.K.; Colby, T.V.; Cordier, J.-F.; Flaherty, K.R.; Lasky, J.A.; et al. An official ats/ers/jrs/alat statement: Idiopathic pulmonary fibrosis: Evidence-based guidelines for diagnosis and management. Am. J. Respir. Crit. Care Med. 2011, 183, 788–824. [Google Scholar] [CrossRef] [PubMed]
- Scott, J.; Johnston, I.; Britton, J. What causes cryptogenic fibrosing alveolitis? A case-control study of environmental exposure to dust. BMJ 1990, 301, 1015–1017. [Google Scholar] [CrossRef] [PubMed]
- Iwai, K.; Mori, T.; Yamada, N.; Yamaguchi, M.; Hosoda, Y. Idiopathic pulmonary fibrosis. Epidemiologic approaches to occupational exposure. Am. J. Respir. Crit. Care Med. 1994, 150, 670–675. [Google Scholar] [CrossRef] [PubMed]
- Ley, B.; Collard, H.R. Epidemiology of idiopathic pulmonary fibrosis. Clin. Epidemiol. 2013, 5, 483–492. [Google Scholar] [CrossRef] [PubMed]
- Nalysnyk, L.; Cid-Ruzafa, J.; Rotella, P.; Esser, D. Incidence and prevalence of idiopathic pulmonary fibrosis: Review of the literature. Eur. Respir. Rev. 2012, 21, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, R.; Lewis, S.; Richards, K.; Johnston, I.; Britton, J. Occupational exposure to metal or wood dust and aetiology of cryptogenic fibrosing alveolitis. Lancet (London, England) 1996, 347, 284–289. [Google Scholar] [CrossRef]
- Behr, J. The diagnosis and treatment of idiopathic pulmonary fibrosis. Deutsches Ärzteblatt Int. 2013, 110, 875–881. [Google Scholar]
- Coultas, D.B.; Zumwalt, R.E.; Black, W.C.; Sobonya, R.E. The epidemiology of interstitial lung diseases. Am. J. Respir. Crit. Care Med. 1994, 150, 967–972. [Google Scholar] [CrossRef] [PubMed]
- Fernandez Perez, E.R.; Daniels, C.E.; Schroeder, D.R.; St Sauver, J.; Hartman, T.E.; Bartholmai, B.J.; Yi, E.S.; Ryu, J.H. Incidence, prevalence, and clinical course of idiopathic pulmonary fibrosis: A population-based study. Chest 2010, 137, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Kolek, V. Epidemiology of cryptogenic fibrosing alveolitis in moravia and silesia. Acta Univ. Palacki. Olomuc. Fac. Med. 1994, 137, 49–50. [Google Scholar] [CrossRef]
- Hodgson, U.; Laitinen, T.; Tukiainen, P. Nationwide prevalence of sporadic and familial idiopathic pulmonary fibrosis: Evidence of founder effect among multiplex families in finland. Thorax 2002, 57, 338–342. [Google Scholar] [CrossRef] [PubMed]
- Karakatsani, A.; Papakosta, D.; Rapti, A.; Antoniou, K.M.; Dimadi, M.; Markopoulou, A.; Latsi, P.; Polychronopoulos, V.; Birba, G.; Ch, L.; et al. Epidemiology of interstitial lung diseases in greece. Respir. Med. 2009, 103, 1122–1129. [Google Scholar] [CrossRef] [PubMed]
- Gribbin, J.; Hubbard, R.B.; Le Jeune, I.; Smith, C.J.; West, J.; Tata, L.J. Incidence and mortality of idiopathic pulmonary fibrosis and sarcoidosis in the UK. Thorax 2006, 61, 980–985. [Google Scholar] [CrossRef] [PubMed]
- Navaratnam, V.; Fleming, K.M.; West, J.; Smith, C.J.; Jenkins, R.G.; Fogarty, A.; Hubbard, R.B. The rising incidence of idiopathic pulmonary fibrosis in the U.K. Thorax 2011, 66, 462–467. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.C.; Wang, C.Y.; Lu, H.M.; Chen, L.; Teng, N.C.; Yan, Y.H.; Wang, J.Y.; Chang, Y.T.; Chao, T.T.; Lin, H.I.; et al. Idiopathic pulmonary fibrosis in taiwan—A population-based study. Respir. Med. 2012, 106, 1566–1574. [Google Scholar] [CrossRef] [PubMed]
- Ohno, S.; Nakaya, T.; Bando, M.; Sugiyama, Y. Idiopathic pulmonary fibrosis—Results from a Japanese nationwide epidemiological survey using individual clinical records. Respirology (Carlton, Victoria) 2008, 13, 926–928. [Google Scholar] [CrossRef] [PubMed]
- Meuret, G.; Fueter, R.; Gloor, F. Early stage of fulminant idiopathic pulmonary fibrosis cured by intense combination therapy using cyclophosphamide, vincristine, and prednisone. Respir. Int Rev Thorac. Dis. 1978, 36, 228–233. [Google Scholar] [CrossRef]
- Raghu, G.; Depaso, W.J.; Cain, K.; Hammar, S.P.; Wetzel, C.E.; Dreis, D.F.; Hutchinson, J.; Pardee, N.E.; Winterbauer, R.H. Azathioprine combined with prednisone in the treatment of idiopathic pulmonary fibrosis: A prospective double-blind, randomized, placebo-controlled clinical trial. Am. Rev. Respir. Dis. 1991, 144, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Rochwerg, B.; Schünemann, H.J.; Raghu, G. Idiopathic pulmonary fibrosis—Clinical management guided by the evidence-based grade approach: What arguments can be made against transparency in guideline development? BMC Med. 2016, 14, 22. [Google Scholar] [CrossRef] [PubMed]
- Meyer, K.C. Diagnosis and management of interstitial lung disease. Transl. Respir. Med. 2014, 2, 4. [Google Scholar] [CrossRef] [PubMed]
- King, T.E. Nonspecific interstitial pneumonia and systemic sclerosis. Am. J. Respir. Crit. Care Med. 2002, 165, 1578–1579. [Google Scholar] [CrossRef] [PubMed]
- Tansey, D.; Wells, A.U.; Colby, T.V.; Ip, S.; Nikolakoupolou, A.; du Bois, R.M.; Hansell, D.M.; Nicholson, A.G. Variations in histological patterns of interstitial pneumonia between connective tissue disorders and their relationship to prognosis. Histopathology 2004, 44, 585–596. [Google Scholar] [CrossRef] [PubMed]
- American Thoracic Society. Idiopathic pulmonary fibrosis: Diagnosis and treatment. Am. J. Respir. Crit. Care Med. 2000, 161, 646–664. [Google Scholar]
- Verleden, S.E.; Todd, J.L.; Sato, M.; Palmer, S.M.; Martinu, T.; Pavlisko, E.N.; Vos, R.; Neyrinck, A.; Van Raemdonck, D.; Saito, T.; et al. Impact of clad phenotype on survival after lung retransplantation: A multicenter study. Am. J. Transplant. 2015, 15, 2223–2230. [Google Scholar] [CrossRef] [PubMed]
- Kaarteenaho, R. The current position of surgical lung biopsy in the diagnosis of idiopathic pulmonary fibrosis. Respir. Res. 2013, 14, 43. [Google Scholar] [CrossRef] [PubMed]
- Maher, T.M. Pirfenidone in idiopathic pulmonary fibrosis. Drugs Today (Barcelona, Spain) 2010, 46, 473–482. [Google Scholar] [CrossRef] [PubMed]
- Cottin, V.; Maher, T. Long-term clinical and real-world experience with pirfenidone in the treatment of idiopathic pulmonary fibrosis. Eur. Respir. Rev. 2015, 24, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Fala, L. Ofev (nintedanib): First tyrosine kinase inhibitor approved for the treatment of patients with idiopathic pulmonary fibrosis. Am. Health Drug Benefits 2015, 8, 101–104. [Google Scholar] [PubMed]
- Gross, T.J.; Hunninghake, G.W. Idiopathic pulmonary fibrosis. N. Engl. J. Med. 2001, 345, 517–525. [Google Scholar] [CrossRef] [PubMed]
- Egan, J.J. Follow-up and nonpharmacological management of the idiopathic pulmonary fibrosis patient. Eur. Respir. Rev. 2011, 20, 114–117. [Google Scholar] [CrossRef] [PubMed]
- Adamali, H.I.; Anwar, M.S.; Russell, A.-M.; Egan, J.J. Non-pharmacological treatment of idiopathic pulmonary fibrosis. Curr. Respir. Care Rep. 2012, 1, 208–215. [Google Scholar] [CrossRef]
- American Thoracic Society; European Respiratory Society. American Thoracic Society/European Respiratory Society International Multidisciplinary Consensus Classification of the Idiopathic Interstitial Pneumonias. This joint statement of the American Thoracic Society (ATS), and the European Respiratory Society (ERS) was adopted by the ATS board of directors, June 2001 and by the ERS Executive Committee, June 2001. Am. J. Respir. Crit. Care Med. 2002, 165, 277–304. [Google Scholar]
- Martinez, F.J.; Flaherty, K. Pulmonary function testing in idiopathic interstitial pneumonias. Proc. Am. Thorac. Soc. 2006, 3, 315–321. [Google Scholar] [CrossRef] [PubMed]
- Christie, J.D.; Edwards, L.B.; Kucheryavaya, A.Y.; Benden, C.; Dipchand, A.I.; Dobbels, F.; Kirk, R.; Rahmel, A.O.; Stehlik, J.; Hertz, M.I. The registry of the international society for heart and lung transplantation: 29th adult lung and heart-lung transplant report-2012. J. Heart Lung Transplant. 2012, 31, 1073–1086. [Google Scholar] [CrossRef] [PubMed]
- Hartert, M.; Senbaklavaci, Ö.; Gohrbandt, B.; Fischer, B.M.; Buhl, R.; Vahl, C.-F. Lung transplantation: A treatment option in end-stage lung disease. Deutsches Ärzteblatt Int. 2014, 111, 107–116. [Google Scholar]
- Spagnolo, P.; Tonelli, R.; Cocconcelli, E.; Stefani, A.; Richeldi, L. Idiopathic pulmonary fibrosis: Diagnostic pitfalls and therapeutic challenges. Multidiscip. Respir. Med. 2012, 7, 42. [Google Scholar] [CrossRef] [PubMed]
- Tzouvelekis, A.; Bonella, F.; Spagnolo, P. Update on therapeutic management of idiopathic pulmonary fibrosis. Ther. Clin. Risk Manag. 2015, 11, 359–370. [Google Scholar] [PubMed]
- Tulek, B.; Kiyan, E.; Toy, H.; Kiyici, A.; Narin, C.; Suerdem, M. Anti-inflammatory and anti-fibrotic effects of sirolimus on bleomycin-induced pulmonary fibrosis in rats. Clin. Investig. Med. 2011, 34, E341. [Google Scholar] [PubMed]
- Myllärniemi, M.; Kaarteenaho, R. Pharmacological treatment of idiopathic pulmonary fibrosis—Preclinical and clinical studies of pirfenidone, nintedanib, and N-acetylcysteine. Eur. Clin. Respir. J. 2015. [Google Scholar] [CrossRef]
- Ahluwalia, N.; Shea, B.S.; Tager, A.M. New therapeutic targets in idiopathic pulmonary fibrosis. Aiming to rein in runaway wound-healing responses. Am. J. Respir. Crit. Care Med. 2014, 190, 867–878. [Google Scholar] [CrossRef] [PubMed]
- Misumi, S.; Lynch, D.A. Idiopathic pulmonary fibrosis/usual interstitial pneumonia. Proc. Am. Thorac. Soc. 2006, 3, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Luppi, F.; Cerri, S.; Beghè, B.; Fabbri, L.M.; Richeldi, L. Corticosteroid and immunomodulatory agents in idiopathic pulmonary fibrosis. Respir. Med. 2004, 98, 1035–1044. [Google Scholar] [CrossRef] [PubMed]
- Crooks, M.G.; Hart, S.P. Coagulation and anticoagulation in idiopathic pulmonary fibrosis. Eur. Respir. Rev. 2015, 24, 392–399. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.S.; Wynn, T.A. Pulmonary fibrosis: Pathogenesis, etiology and regulation. Mucosal Immunol. 2009, 2, 103–121. [Google Scholar] [CrossRef] [PubMed]
- Chambers, R.C. Abnormal wound healing responses in pulmonary fibrosis: Focus on coagulation signalling. Eur. Respir. Rev. 2008, 17, 130–137. [Google Scholar] [CrossRef]
- Juarez, M.M.; Chan, A.L.; Norris, A.G.; Morrissey, B.M.; Albertson, T.E. Acute exacerbation of idiopathic pulmonary fibrosis—A review of current and novel pharmacotherapies. J. Thorac. Dis. 2015, 7, 499–519. [Google Scholar]
- Huan, C.; Yang, T.; Liang, J.; Xie, T.; Cheng, L.; Liu, N.; Kurkciyan, A.; Monterrosa Mena, J.; Wang, C.; Dai, H.; et al. Methylation-mediated bmper expression in fibroblast activation in vitro and lung fibrosis in mice in vivo. Sci. Rep. 2015, 5, 14910. [Google Scholar] [CrossRef] [PubMed]
- Wollin, L.; Wex, E.; Pautsch, A.; Schnapp, G.; Hostettler, K.E.; Stowasser, S.; Kolb, M. Mode of action of nintedanib in the treatment of idiopathic pulmonary fibrosis. Eur. Respir. J. 2015, 45, 1434–1445. [Google Scholar] [CrossRef] [PubMed]
- Richeldi, L.; du Bois, R.M.; Raghu, G.; Azuma, A.; Brown, K.K.; Costabel, U.; Cottin, V.; Flaherty, K.R.; Hansell, D.M.; Inoue, Y.; et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N. Engl. J. Med. 2014, 370, 2071–2082. [Google Scholar] [CrossRef] [PubMed]
- Lehtonen, S.T.; Veijola, A.; Karvonen, H.; Lappi-Blanco, E.; Sormunen, R.; Korpela, S.; Zagai, U.; Sköld, M.C.; Kaarteenaho, R. Pirfenidone and nintedanib modulate properties of fibroblasts and myofibroblasts in idiopathic pulmonary fibrosis. Respir. Res. 2016, 17, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Kreuter, M.; Bonella, F.; Wijsenbeek, M.; Maher, T.M.; Spagnolo, P. Pharmacological treatment of idiopathic pulmonary fibrosis: Current approaches, unsolved issues, and future perspectives. BioMed Res. Int. 2015, 2015, 10. [Google Scholar] [CrossRef] [PubMed]
- Cho, M.E.; Kopp, J.B. Pirfenidone: An anti-fibrotic and cytoprotective agent as therapy for progressive kidney disease. Expert Opin. Investig. Drugs 2010, 19, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Iyer, S.N.; Wild, J.S.; Schiedt, M.J.; Hyde, D.M.; Margolin, S.B.; Giri, S.N. Dietary intake of pirfenidone ameliorates bleomycin-induced lung fibrosis in hamsters. J. Lab. Clin. Med. 1995, 125, 779–785. [Google Scholar] [PubMed]
- Gan, Y.; Herzog, E.L.; Gomer, R.H. Pirfenidone treatment of idiopathic pulmonary fibrosis. Ther. Clin. Risk Manag. 2011, 7, 39–47. [Google Scholar] [PubMed]
- Kolilekas, L.; Manali, E.; Vlami, K.A.; Lyberopoulos, P.; Triantafillidou, C.; Kagouridis, K.; Baou, K.; Gyftopoulos, S.; Vougas, K.N.; Karakatsani, A.; et al. Sleep oxygen desaturation predicts survival in idiopathic pulmonary fibrosis. J. Clin. Sleep Med. 2013, 9, 593–601. [Google Scholar] [CrossRef] [PubMed]
- Ozalevli, S.; Karaali, H.K.; Ilgin, D.; Ucan, E.S. Effect of home-based pulmonary rehabilitation in patients with idiopathic pulmonary fibrosis. Multidiscip. Respir. Med. 2010, 5, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Miki, K.; Maekura, R.; Hiraga, T.; Okuda, Y.; Okamoto, T.; Hirotani, A.; Ogura, T. Impairments and prognostic factors for survival in patients with idiopathic pulmonary fibrosis. Respir. Med. 2003, 97, 482–490. [Google Scholar] [CrossRef] [PubMed]
- Sakai, N.; Tager, A.M. Fibrosis of two: Epithelial cell-fibroblast interactions in pulmonary fibrosis. Biochim. Biophys. Acta 2013, 1832, 911–921. [Google Scholar] [CrossRef] [PubMed]
- Noble, P.W. Epithelial fibroblast triggering and interactions in pulmonary fibrosis. Eur. Respir. Rev. 2008, 17, 123–129. [Google Scholar] [CrossRef]
- Chen, H.; Matsumoto, K.; Brockway, B.L.; Rackley, C.R.; Liang, J.; Lee, J.-H.; Jiang, D.; Noble, P.W.; Randell, S.H.; Kim, C.F.; et al. Airway epithelial progenitors are region specific and show differential responses to bleomycin-induced lung injury. Stem Cell. (Dayton, OH) 2012, 30, 1948–1960. [Google Scholar] [CrossRef] [PubMed]
- Selman, M.; Pardo, A. Role of epithelial cells in idiopathic pulmonary fibrosis. Proc. Am. Thorac. Soc. 2006, 3, 364–372. [Google Scholar] [CrossRef] [PubMed]
- Pelaseyed, T.; Bergström, J.H.; Gustafsson, J.K.; Ermund, A.; Birchenough, G.M.H.; Schütte, A.; van der Post, S.; Svensson, F.; Rodríguez-Piñeiro, A.M.; Nyström, E.E.L.; et al. The mucus and mucins of the goblet cells and enterocytes provide the first defense line of the gastrointestinal tract and interact with the immune system. Immunol. Rev. 2014, 260, 8–20. [Google Scholar] [CrossRef] [PubMed]
- Wolters, P.J.; Collard, H.R.; Jones, K.D. Pathogenesis of idiopathic pulmonary fibrosis. Annu. Rev. Pathol. 2014, 9, 157–179. [Google Scholar] [CrossRef] [PubMed]
- Camelo, A.; Dunmore, R.; Sleeman, M.A.; Clarke, D.L. The epithelium in idiopathic pulmonary fibrosis: Breaking the barrier. Front. Pharm. 2013, 4, 173. [Google Scholar] [CrossRef] [PubMed]
- Armanios, M. Telomerase and idiopathic pulmonary fibrosis. Mutat. Res. 2012, 730, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Tanjore, H.; Blackwell, T.S.; Lawson, W.E. Emerging evidence for endoplasmic reticulum stress in the pathogenesis of idiopathic pulmonary fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 302, L721–L729. [Google Scholar] [CrossRef] [PubMed]
- Schultz, G.S.; Wysocki, A. Interactions between extracellular matrix and growth factors in wound healing. Wound Repair Regen. 2009, 17, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Mercer, P.F.; Chambers, R.C. Coagulation and coagulation signalling in fibrosis. Biochim. Biophys. Acta 2013, 1832, 1018–1027. [Google Scholar] [CrossRef] [PubMed]
- Sebag, S.C.; Bastarache, J.A.; Ware, L.B. Therapeutic modulation of coagulation and fibrinolysis in acute lung injury and the acute respiratory distress syndrome. Curr. Pharm. Biotechnol. 2011, 12, 1481–1496. [Google Scholar] [CrossRef]
- Wynn, T.A.; Ramalingam, T.R. Mechanisms of fibrosis: Therapeutic translation for fibrotic disease. Nat. Med. 2012, 18, 1028–1040. [Google Scholar] [CrossRef] [PubMed]
- Cheresh, P.; Kim, S.J.; Tulasiram, S.; Kamp, D.W. Oxidative stress and pulmonary fibrosis. Biochim. Biophys. Acta 2013, 1832, 1028–1040. [Google Scholar] [CrossRef] [PubMed]
- Bocchino, M.; Agnese, S.; Fagone, E.; Svegliati, S.; Grieco, D.; Vancheri, C.; Gabrielli, A.; Sanduzzi, A.; Avvedimento, E.V. Reactive oxygen species are required for maintenance and differentiation of primary lung fibroblasts in idiopathic pulmonary fibrosis. PLoS ONE 2010, 5, e14003. [Google Scholar] [CrossRef] [PubMed]
- Ding, N.-H.; Li, J.J.; Sun, L.-Q. Molecular mechanisms and treatment of radiation-induced lung fibrosis. Curr. Drug Targets 2013, 14, 1247–1356. [Google Scholar] [CrossRef]
- Huebener, P.; Schwabe, R.F. Regulation of wound healing and organ fibrosis by toll-like receptors. Biochim. Biophys. Acta 2013, 1832, 1005–1017. [Google Scholar] [CrossRef] [PubMed]
- Millien, V.O.; Lu, W.; Shaw, J.; Yuan, X.; Mak, G.; Roberts, L.; Song, L.-Z.; Knight, J.M.; Creighton, C.J.; Luong, A.; et al. Cleavage of fibrinogen by proteinases elicits allergic responses through toll-like receptor 4. Science (New York) 2013, 341, 792–796. [Google Scholar] [CrossRef] [PubMed]
- Todd, N.W.; Luzina, I.G.; Atamas, S.P. Molecular and cellular mechanisms of pulmonary fibrosis. Fibrogenesis Tissue Repair 2012, 5, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Zoz, D.F.; Lawson, W.E.; Blackwell, T.S. Idiopathic pulmonary fibrosis: A disorder of epithelial cell dysfunction. Am. J. Med. Sci. 2011, 341, 435–438. [Google Scholar] [CrossRef] [PubMed]
- Kendall, R.T.; Feghali-Bostwick, C.A. Fibroblasts in fibrosis: Novel roles and mediators. Front. Pharm. 2014, 5, 123. [Google Scholar] [CrossRef] [PubMed]
- Neary, R.; Watson, C.J.; Baugh, J.A. Epigenetics and the overhealing wound: The role of DNA methylation in fibrosis. Fibrogenesis Tissue Repair 2015, 8, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Vancheri, C. Idiopathic pulmonary fibrosis and cancer: Do they really look similar? BMC Med. 2015, 13, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Robinson, C.M.; Watson, C.J.; Baugh, J.A. Epigenetics within the matrix: A neo-regulator of fibrotic disease. Epigenetics 2012, 7, 987–993. [Google Scholar] [CrossRef] [PubMed]
- Prasad, S.; Hogaboam, C.M.; Jarai, G. Deficient repair response of ipf fibroblasts in a co-culture model of epithelial injury and repair. Fibrogenesis Tissue Repair 2014, 7, 7. [Google Scholar] [CrossRef] [PubMed]
- Selman, M.; Pardo, A. Idiopathic pulmonary fibrosis: An epithelial/fibroblastic cross-talk disorder. Respir. Res. 2002, 3, 3. [Google Scholar] [CrossRef] [PubMed]
- Pardo, A.; Selman, M. Role of matrix metaloproteases in idiopathic pulmonary fibrosis. Fibrogenesis Tissue Repair 2012, 5, S9. [Google Scholar]
- Henderson, N.C.; Sheppard, D. Integrin-mediated regulation of tgfβ in fibrosis. Biochim. Biophys. Acta 2013, 1832, 891–896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, D.; Liang, J.; Noble, P.W. Regulation of non-infectious lung injury, inflammation, and repair by the extracellular matrix glycosaminoglycan hyaluronan. Anat Rec. (Hoboken) 2010, 293, 982–985. [Google Scholar] [CrossRef] [PubMed]
- White, E.S. Lung extracellular matrix and fibroblast function. Ann. Am. Thorac. Soc. 2015, 12, S30–S33. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Country and Study Period First Author (Ref.) | IPF Prevalence/100,000 Population | IPF Incidence/100,000 Population | Case Detection and Study Method | ||||
|---|---|---|---|---|---|---|---|
| All | Male | Female | All | Male | Female | ||
| USA | |||||||
| 1988–1990 Coultas et al. [26] | 20.2 | 13.2 | 10.7 | 7.4 | Population-based | ||
| 1997–2005 Fernandez Perez et al. [27] | 27.9–63.0 | ILD registry Population-based medical record linkage system (2000 ATS/ERS criteria) | |||||
| Europe | |||||||
| Czech Republic 1981–1990 Kolek et al. [28] | 12.1 | 0.94 | Clinical registry | ||||
| Finland 1997–1998 Hodgson et al. [29] | 16–18 | Pulmonary clinic database | |||||
| Greece 2004 Karakatsani et al. [30] | 3.38 | 0.93 | National survey of pulmonologists (2000 ATS/ERS criteria) | ||||
| UK 1991–2003 Gribbin et al. [31] | 4.6 | 5.69 | 3.44 | National-wide primary care database | |||
| UK 2000–2009 Navaratnam et al. [32] | 7.44 | 9.46 | National-wide primary care database | ||||
| Asia | |||||||
| Taiwan 1997–2007 Lai et al. [33] | 0.7–6.4 | 0.6–1.4 | Taiwanese national health insurance system | ||||
| Japan 2005 Ohno et al.[34] | 2.9 | Medical benefits records | |||||
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Betensley, A.; Sharif, R.; Karamichos, D. A Systematic Review of the Role of Dysfunctional Wound Healing in the Pathogenesis and Treatment of Idiopathic Pulmonary Fibrosis. J. Clin. Med. 2017, 6, 2. https://doi.org/10.3390/jcm6010002
Betensley A, Sharif R, Karamichos D. A Systematic Review of the Role of Dysfunctional Wound Healing in the Pathogenesis and Treatment of Idiopathic Pulmonary Fibrosis. Journal of Clinical Medicine. 2017; 6(1):2. https://doi.org/10.3390/jcm6010002
Chicago/Turabian StyleBetensley, Alan, Rabab Sharif, and Dimitrios Karamichos. 2017. "A Systematic Review of the Role of Dysfunctional Wound Healing in the Pathogenesis and Treatment of Idiopathic Pulmonary Fibrosis" Journal of Clinical Medicine 6, no. 1: 2. https://doi.org/10.3390/jcm6010002