Viral Oncology: Molecular Biology and Pathogenesis

Abstract
:1. Introduction
2. Epstein-Barr Virus
2.1. EBV Establishes Latency in Resting Memory B Cells
2.2. EBV Oncogenic Proteins
2.3. EBV and T Cell Lymphoproliferative Disorders
2.4. EBV and Epithelial Malignancies
2.5. EBV and B-Cell Lymphoproliferative Disorders
2.6. Therapeutic Options
3. Human Herpesvirus-8
3.1. HHV-8 Life Cycle
3.2. HHV-8 Oncogenic Proteins
3.3. HHV-8-Associated Tumors
3.4. Therapeutic Options
4. Merkel Cell Polyomavirus
4.1. MCPyV Genome
4.2. MCPyV Oncogenic Factors
4.3. Therapeutic Options
5. Human Papillomavirus
5.1. Structure and Genome
5.2. Pathogenesis and Carcinogenesis
5.3. Cervical Cancer
5.4. Oropharyngeal Cancer
5.5. Penile Cancer
5.6. Anal Cancer
5.7. Vulvar & Vaginal Cancer
5.8. Preventative Vaccines
5.9. Therapeutic Immunotherapy
6. Hepatitis B Virus
6.1. HBV Genome
6.2. Direct Mechanism of HBV Oncogenesis
6.3. Indirect Mechanism of HBV Oncogenesis
6.4. Therapeutic Options
7. Hepatitis C Virus
7.1. HCV Genome
7.2. Direct Mechanism of HCV Oncogenesis
7.3. Indirect Mechanism of HCV Oncogenesis
7.4. Therapeutic Options
8. Human T-Cell Lymphotropic Virus-1
8.1. HTLV-1 Molecular Biology
8.2. HTLV-1 Oncogenic Proteins
8.3. ATLL Transformation
8.4. Therapeutic Options
9. Conclusions
Author Contributions
Conflicts of Interest
References
- Epstein, M.A.; Achong, B.G.; Barr, Y.M. Virus particles in cultured lymphoblasts from Burkitt’s lymphoma. Lancet 1964, 1, 702–703. [Google Scholar] [CrossRef]
- Rous, P. A sarcoma of the fowl transmissible by an agent separable from the tumor cells. J. Exp. Med. 1911, 13, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Rous, P. A transmissible avian neoplasm (sarcoma of the common fowl). J. Exp. Med. 1910, 12, 696–705. [Google Scholar] [CrossRef] [PubMed]
- Zur Hausen, H. The search for infectious causes of human cancers: Where and why. Virology 2009, 392, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Bouvard, V.; Baan, R.; Straif, K.; Grosse, Y.; Secretan, B.; El Ghissassi, F.; Benbrahim-Tallaa, L.; Guha, N.; Freeman, C.; Galichet, L.; et al. A review of human carcinogens—Part B: Biological agents. Lancet Oncol. 2009, 10, 321–322. [Google Scholar] [CrossRef]
- Boyle, P.; Levin, B. World Cancer Report 2008; International Agency for Research on Cancer: Lyon, France, 2008. [Google Scholar]
- De Martel, C.; Ferlay, J.; Franceschi, S.; Vignat, J.; Bray, F.; Forman, D.; Plummer, M. Global burden of cancers attributable to infections in 2008: A review and synthetic analysis. Lancet Oncol. 2012, 13, 607–615. [Google Scholar] [CrossRef]
- Moore, P.S.; Chang, Y. Why do viruses cause cancer? Highlights of the first century of human tumour virology. Nat. Rev. Cancer 2010, 10, 878–889. [Google Scholar] [CrossRef] [PubMed]
- Parsonnet, J. Microbes and Malignancy: Infection as a Cause of Human Cancers; Oxford University Press: New York, NY, USA, 1999; pp. 3–18. ISBN 978-0195104011. [Google Scholar]
- Zur Hausen, H. Oncogenic DNA viruses. Oncogene 2001, 20, 7820–7823. [Google Scholar] [CrossRef] [PubMed]
- Akram, N.; Imran, M.; Noreen, M.; Ahmed, F.; Atif, M.; Fatima, Z.; Bilal Waqar, A. Oncogenic Role of Tumor Viruses in Humans. Viral Immunol. 2017, 30, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.J. The common mechanisms of transformation by the small DNA tumor viruses: The inactivation of tumor suppressor gene products: P53. Virology 2009, 384, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Shackelford, J.; Pagano, J.S. Tumor viruses and cell signaling pathways: Deubiquitination versus ubiquitination. Mol. Cell. Biol. 2004, 24, 5089–5093. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.T.; Kyo, S.; Laimins, L.A. Telomerase activation by human papillomavirus type 16 E6 protein: Induction of human telomerase reverse transcriptase expression through Myc and GC-rich Sp1 binding sites. J. Virol. 2001, 75, 5559–5566. [Google Scholar] [CrossRef] [PubMed]
- Klingelhutz, A.J.; Foster, S.A.; McDougall, J.K. Telomerase activation by the E6 gene product of human papillomavirus type 16. Nature 1996, 380, 79–82. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.C.; Borah, S.; Robertson, E.S. Latency-associated nuclear antigen of Kaposi’s sarcoma-associated herpesvirus up-regulates transcription of human telomerase reverse transcriptase promoter through interaction with transcription factor Sp1. J. Virol. 2004, 78, 10348–10359. [Google Scholar] [CrossRef] [PubMed]
- Kataoka, H.; Tahara, H.; Watanabe, T.; Sugawara, M.; Ide, T.; Goto, M.; Furuichi, Y.; Sugimoto, M. Immortalization of immunologically committed Epstein-Barr virus-transformed human B-lymphoblastoid cell lines accompanied by a strong telomerase activity. Differentiation 1997, 62, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Terrin, L.; Dal Col, J.; Rampazzo, E.; Zancai, P.; Pedrotti, M.; Ammirabile, G.; Bergamin, S.; Rizzo, S.; Dolcetti, R.; De Rossi, A. Latent membrane protein 1 of Epstein-Barr virus activates the hTERT promoter and enhances telomerase activity in B lymphocytes. J. Virol. 2008, 82, 10175–10187. [Google Scholar] [CrossRef] [PubMed]
- Buchkovich, N.J.; Yu, Y.; Zampieri, C.A.; Alwine, J.C. The TORrid affairs of viruses: Effects of mammalian DNA viruses on the PI3K-Akt-mTOR signalling pathway. Nat. Rev. Microbiol. 2008, 6, 266–275. [Google Scholar] [CrossRef] [PubMed]
- Godfrey, A.; Anderson, J.; Papanastasiou, A.; Takeuchi, Y.; Boshoff, C. Inhibiting primary effusion lymphoma by lentiviral vectors encoding short hairpin RNA. Blood 2005, 105, 2510–2518. [Google Scholar] [CrossRef] [PubMed]
- Yasunaga, J.; Matsuoka, M. Leukaemogenic mechanism of human T-cell leukaemia virus type I. Rev. Med. Virol. 2007, 17, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Mosialos, G.; Birkenbach, M.; Yalamanchili, R.; VanArsdale, T.; Ware, C.; Kieff, E. The Epstein-Barr virus transforming protein LMP1 engages signaling proteins for the tumor necrosis factor receptor family. Cell 1995, 80, 389–399. [Google Scholar] [CrossRef]
- Liu, L.; Eby, M.T.; Rathore, N.; Sinha, S.K.; Kumar, A.; Chaudhary, P.M. The human herpes virus 8-encoded viral FLICE inhibitory protein physically associates with and persistently activates the Ikappa B kinase complex. J. Biol. Chem. 2002, 277, 13745–13751. [Google Scholar] [CrossRef] [PubMed]
- Fujimuro, M.; Wu, F.Y.; ApRhys, C.; Kajumbula, H.; Young, D.B.; Hayward, G.S.; Hayward, S.D. A novel viral mechanism for dysregulation of beta-catenin in Kaposi’s sarcoma-associated herpesvirus latency. Nat. Med. 2003, 9, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Bergonzini, V.; Salata, C.; Calistri, A.; Parolin, C.; Palù, G. View and review on viral oncology research. Infect. Agent Cancer 2010, 5, 11. [Google Scholar] [CrossRef] [PubMed]
- McFadden, K.; Luftig, M.A. Interplay between DNA tumor viruses and the host DNA damage response. Curr. Top. Microbiol. Immunol. 2013, 371, 229–257. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.; Tho, L.M.; Xu, N.; Gillespie, D.A. The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Adv. Cancer Res. 2010, 108, 73–112. [Google Scholar] [CrossRef] [PubMed]
- Bartek, J.; Bartkova, J.; Lukas, J. DNA damage signalling guards against activated oncogenes and tumour progression. Oncogene 2007, 26, 7773–7779. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.H.; Chen, C.J.; Lai, M.S.; Hsu, H.M.; Wu, T.C.; Kong, M.S.; Liang, D.C.; Shau, W.Y.; Chen, D.S. Universal hepatitis B vaccination in Taiwan and the incidence of hepatocellular carcinoma in children. Taiwan Childhood Hepatoma Study Group. N. Engl. J. Med. 1997, 336, 1855–1859. [Google Scholar] [CrossRef] [PubMed]
- Lavanchy, D. Hepatitis B virus epidemiology, disease burden, treatment, and current and emerging prevention and control measures. J. Viral. Hepat. 2004, 11, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Goldie, S.J.; Kohli, M.; Grima, D.; Weinstein, M.C.; Wright, T.C.; Bosch, F.X.; Franco, E. Projected clinical benefits and cost-effectiveness of a human papillomavirus 16/18 vaccine. J. Natl. Cancer Inst. 2004, 96, 604–615. [Google Scholar] [CrossRef] [PubMed]
- Schiller, J.T.; Lowy, D.R. Virus infection and human cancer: An overview. Recent Results Cancer Res. 2014, 193, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Shinozaki-Ushiku, A.; Kunita, A.; Fukayama, M. Update on Epstein-Barr virus and gastric cancer (review). Int. J. Oncol. 2015, 46, 1421–1434. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.; Chen, K.; Young, K.H. Epstein-Barr virus-positive T/NK-cell lymphoproliferative disorders. Exp. Mol. Med. 2015, 47, e133. [Google Scholar] [CrossRef] [PubMed]
- Jácome, A.A.; Lima, E.M.; Kazzi, A.I.; Chaves, G.F.; Mendonça, D.C.; Maciel, M.M.; Santos, J.S. Epstein-Barr virus-positive gastric cancer: A distinct molecular subtype of the disease? Rev. Soc. Bras. Med. Trop. 2016, 49, 150–157. [Google Scholar] [CrossRef] [PubMed]
- Castillo, J.J.; Beltran, B.E.; Miranda, R.N.; Young, K.H.; Chavez, J.C.; Sotomayor, E.M. EBV-positive diffuse large B-cell lymphoma of the elderly: 2016 update on diagnosis, risk-stratification, and management. Am. J. Hematol. 2016, 91, 529–537. [Google Scholar] [CrossRef] [PubMed]
- Pannone, G.; Zamparese, R.; Pace, M.; Pedicillo, M.C.; Cagiano, S.; Somma, P.; Errico, M.E.; Donofrio, V.; Franco, R.; De Chiara, A.; et al. The role of EBV in the pathogenesis of Burkitt’s Lymphoma: An Italian hospital based survey. Infect. Agent Cancer 2014, 9, 34. [Google Scholar] [CrossRef] [PubMed]
- Pavlovic, A.; Glavina Durdov, M.; Capkun, V.; Jakelic Pitesa, J.; Bozic Sakic, M. Classical Hodgkin Lymphoma with Positive Epstein-Barr Virus Status is Associated with More FOXP3 Regulatory T Cells. Med. Sci. Monit. 2016, 22, 2340–2346. [Google Scholar] [CrossRef] [PubMed]
- Carbone, A.; Gloghini, A.; Dotti, G. EBV-associated lymphoproliferative disorders: Classification and treatment. Oncologist 2008, 13, 577–585. [Google Scholar] [CrossRef] [PubMed]
- Hudnall, S.D.; Ge, Y.; Wei, L.; Yang, N.P.; Wang, H.Q.; Chen, T. Distribution and phenotype of Epstein-Barr virus-infected cells in human pharyngeal tonsils. Mod. Pathol. 2005, 18, 519–527. [Google Scholar] [CrossRef] [PubMed]
- Tabiasco, J.; Vercellone, A.; Meggetto, F.; Hudrisier, D.; Brousset, P.; Fournié, J.J. Acquisition of viral receptor by NK cells through immunological synapse. J. Immunol. 2003, 170, 5993–5998. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, S.H.; Campo, E.; Pileri, S.A.; Harris, N.L.; Stein, H.; Siebert, R.; Advani, R.; Ghielmini, M.; Salles, G.A.; Zelenetz, A.D.; et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 2016, 127, 2375–2390. [Google Scholar] [CrossRef] [PubMed]
- Petrara, M.R.; Giunco, S.; Serraino, D.; Dolcetti, R.; De Rossi, A. Post-transplant lymphoproliferative disorders: From epidemiology to pathogenesis-driven treatment. Cancer Lett. 2015, 369, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Tse, E.; Kwong, Y.L. Epstein Barr virus-associated lymphoproliferative diseases: The virus as a therapeutic target. Exp. Mol. Med. 2015, 47, e136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neparidze, N.; Lacy, J. Malignancies associated with epstein-barr virus: Pathobiology, clinical features, and evolving treatments. Clin. Adv. Hematol. Oncol. 2014, 12, 358–371. [Google Scholar] [PubMed]
- Jaffe, E.S.; Nicolae, A.; Pittaluga, S. Peripheral T-cell and NK-cell lymphomas in the WHO classification: Pearls and pitfalls. Mod. Pathol. 2013, 26, S71–S87. [Google Scholar] [CrossRef] [PubMed]
- Tse, E.; Kwong, Y.L. The diagnosis and management of NK/T-cell lymphomas. J. Hematol. Oncol. 2017, 10, 85. [Google Scholar] [CrossRef] [PubMed]
- Sivachandran, N.; Dawson, C.W.; Young, L.S.; Liu, F.F.; Middeldorp, J.; Frappier, L. Contributions of the Epstein-Barr virus EBNA1 protein to gastric carcinoma. J. Virol. 2012, 86, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.S.; Kim, D.H.; Roh, J.K.; Middeldorp, J.M.; Kim, Y.S.; Kim, S.; Han, S.; Kim, C.W.; Lee, B.L.; Kim, W.H.; et al. Epstein-Barr virus-encoded BARF1 promotes proliferation of gastric carcinoma cells through regulation of NF-κB. J. Virol. 2013, 87, 10515–10523. [Google Scholar] [CrossRef] [PubMed]
- Breda, E.; Catarino, R.J.; Azevedo, I.; Lobão, M.; Monteiro, E.; Medeiros, R. Epstein-Barr virus detection in nasopharyngeal carcinoma: Implications in a low-risk area. Braz. J. Otorhinolaryngol. 2010, 76, 310–315. [Google Scholar] [CrossRef] [PubMed]
- Chu, E.A.; Wu, J.M.; Tunkel, D.E.; Ishman, S.L. Nasopharyngeal carcinoma: The role of the Epstein-Barr virus. Medscape J. Med. 2008, 10, 165. [Google Scholar] [PubMed]
- Liu, Y.; Yang, W.; Pan, Y.; Ji, J.; Lu, Z.; Ke, Y. Genome-wide analysis of Epstein-Barr virus (EBV) isolated from EBV-associated gastric carcinoma (EBVaGC). Oncotarget 2016, 7, 4903–4914. [Google Scholar] [CrossRef] [PubMed]
- Roughan, J.E.; Thorley-Lawson, D.A. The intersection of Epstein-Barr virus with the germinal center. J. Virol. 2009, 83, 3968–3976. [Google Scholar] [CrossRef] [PubMed]
- Coleman, C.B.; Wohlford, E.M.; Smith, N.A.; King, C.A.; Ritchie, J.A.; Baresel, P.C.; Kimura, H.; Rochford, R. Epstein-Barr virus type 2 latently infects T cells, inducing an atypical activation characterized by expression of lymphotactic cytokines. J. Virol. 2015, 89, 2301–2312. [Google Scholar] [CrossRef] [PubMed]
- Frappier, L. Contributions of Epstein-Barr nuclear antigen 1 (EBNA1) to cell immortalization and survival. Viruses 2012, 4, 1537–1547. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.; De Matteo, E.; Narbaitz, M.; Carreño, F.A.; Preciado, M.V.; Chabay, P.A. Epstein-Barr virus presence in pediatric diffuse large B-cell lymphoma reveals a particular association and latency patterns: Analysis of viral role in tumor microenvironment. Int. J. Cancer 2013, 132, 1572–1580. [Google Scholar] [CrossRef] [PubMed]
- Rowe, M.; Fitzsimmons, L.; Bell, A.I. Epstein-Barr virus and Burkitt lymphoma. Chin. J. Cancer 2014, 33, 609–619. [Google Scholar] [CrossRef] [PubMed]
- Grömminger, S.; Mautner, J.; Bornkamm, G.W. Burkitt lymphoma: The role of Epstein-Barr virus revisited. Br. J. Haematol. 2012, 156, 719–729. [Google Scholar] [CrossRef] [PubMed]
- Young, L.S.; Dawson, C.W. Epstein-Barr virus and nasopharyngeal carcinoma. Chin. J. Cancer 2014, 33, 581–590. [Google Scholar] [CrossRef] [PubMed]
- Chen, J. Roles of the PI3K/Akt pathway in Epstein-Barr virus-induced cancers and therapeutic implications. World J. Virol. 2012, 1, 154–161. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, M.; Ikuta, K.; Yanagihara, K.; Tajima, M.; Kuratsune, H.; Kurata, T.; Sairenji, T. Effect of transforming growth factor-beta1 on the cell growth and Epstein-Barr virus reactivation in EBV-infected epithelial cell lines. Virology 2001, 288, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Hino, R.; Uozaki, H.; Inoue, Y.; Shintani, Y.; Ushiku, T.; Sakatani, T.; Takada, K.; Fukayama, M. Survival advantage of EBV-associated gastric carcinoma: Survivin up-regulation by viral latent membrane protein 2A. Cancer Res. 2008, 68, 1427–1435. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Gao, Y.; Luo, B.; Zhao, Y. Construction and Antiapoptosis Activities of Recombinant Adenoviral Expression Vector Carrying EBV Latent Membrane Protein 2A. Gastroenterol. Res. Pract. 2011, 2011, 182832. [Google Scholar] [CrossRef] [PubMed]
- Pal, A.D.; Basak, N.P.; Banerjee, A.S.; Banerjee, S. Epstein-Barr virus latent membrane protein-2A alters mitochondrial dynamics promoting cellular migration mediated by Notch signaling pathway. Carcinogenesis 2014, 35, 1592–1601. [Google Scholar] [CrossRef] [PubMed]
- Hino, R.; Uozaki, H.; Murakami, N.; Ushiku, T.; Shinozaki, A.; Ishikawa, S.; Morikawa, T.; Nakaya, T.; Sakatani, T.; Takada, K.; et al. Activation of DNA methyltransferase 1 by EBV latent membrane protein 2A leads to promoter hypermethylation of PTEN gene in gastric carcinoma. Cancer Res. 2009, 69, 2766–2774. [Google Scholar] [CrossRef] [PubMed]
- Gruhne, B.; Sompallae, R.; Marescotti, D.; Kamranvar, S.A.; Gastaldello, S.; Masucci, M.G. The Epstein-Barr virus nuclear antigen-1 promotes genomic instability via induction of reactive oxygen species. Proc. Natl. Acad. Sci. USA 2009, 106, 2313–2318. [Google Scholar] [CrossRef] [PubMed]
- Kamranvar, S.A.; Masucci, M.G. The Epstein-Barr virus nuclear antigen-1 promotes telomere dysfunction via induction of oxidative stress. Leukemia 2011, 25, 1017–1025. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, M.; Holthaus, A.M.; Calderwood, M.A.; Lai, C.Y.; Krastins, B.; Sarracino, D.; Johannsen, E. The EBNA3 family of Epstein-Barr virus nuclear proteins associates with the USP46/USP12 deubiquitination complexes to regulate lymphoblastoid cell line growth. PLoS Pathog. 2015, 11, e1004822. [Google Scholar] [CrossRef] [PubMed]
- Shinozaki, A.; Sakatani, T.; Ushiku, T.; Hino, R.; Isogai, M.; Ishikawa, S.; Uozaki, H.; Takada, K.; Fukayama, M. Downregulation of microRNA-200 in EBV-associated gastric carcinoma. Cancer Res. 2010, 70, 4719–4727. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.S.; Pal, A.D.; Banerjee, S. Epstein-Barr virus-encoded small non-coding RNAs induce cancer cell chemoresistance and migration. Virology 2013, 443, 294–305. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.K.; Perrine, S.P.; Faller, D.V. Advances in Virus-Directed Therapeutics against Epstein-Barr Virus-Associated Malignancies. Adv. Virol. 2012, 2012, 509296. [Google Scholar] [CrossRef] [PubMed]
- Lima, R.T.; Nascimento, M.S.J.; Vasconcelos, M.H. EBV-associated cancers: Strategies for targeting the virus. In Microbial Pathogens and Strategies for Combating Them: Science, Technology and Education; Méndez-Vilas, A., Ed.; Formatex Research Center: Badajoz, Spain, 2013; Volume 3, pp. 1608–1618. ISBN 978-84-942134-1-0. [Google Scholar]
- Liu, D.; Mamorska-Dyga, A. Syk inhibitors in clinical development for hematological malignancies. J. Hematol. Oncol. 2017, 10, 145. [Google Scholar] [CrossRef] [PubMed]
- Hutajulu, S.H.; Kurnianda, J.; Tan, I.B.; Middeldorp, J.M. Therapeutic implications of Epstein-Barr virus infection for the treatment of nasopharyngeal carcinoma. Ther. Clin. Risk Manag. 2014, 10, 721–736. [Google Scholar] [CrossRef] [PubMed]
- Kanakry, J.A.; Ambinder, R.F. EBV-related lymphomas: New approaches to treatment. Curr. Treat. Options Oncol. 2013, 14, 224–236. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Yang, L.; Jiang, W.; Wang, X.; Liao, W.; Tan, G.; Liao, Y.; Qiu, Y.; Feng, D.; Tang, F.; et al. Therapeutic evaluation of Epstein-Barr virus-encoded latent membrane protein-1 targeted DNAzyme for treating of nasopharyngeal carcinomas. Mol. Ther. 2014, 22, 371–377. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.Y.; Kim, J.O.; Lee, S.K.; Chae, H.S.; Kang, J.H. LY294002 may overcome 5-FU resistance via down-regulation of activated p-AKT in Epstein-Barr virus-positive gastric cancer cells. BMC Cancer 2010, 10, 425. [Google Scholar] [CrossRef] [PubMed]
- Cen, O.; Longnecker, R. Rapamycin reverses splenomegaly and inhibits tumor development in a transgenic model of Epstein-Barr virus-related Burkitt’s lymphoma. Mol. Cancer Ther. 2011, 10, 679–686. [Google Scholar] [CrossRef] [PubMed]
- Manara, M.C.; Nicoletti, G.; Zambelli, D.; Ventura, S.; Guerzoni, C.; Landuzzi, L.; Lollini, P.L.; Maira, S.M.; García-Echeverría, C.; Mercuri, M.; et al. NVP-BEZ235 as a new therapeutic option for sarcomas. Clin. Cancer Res. 2010, 16, 530–540. [Google Scholar] [CrossRef] [PubMed]
- Bhende, P.M.; Park, S.I.; Lim, M.S.; Dittmer, D.P.; Damania, B. The dual PI3K/mTOR inhibitor, NVP-BEZ235, is efficacious against follicular lymphoma. Leukemia 2010, 24, 1781–1784. [Google Scholar] [CrossRef] [PubMed]
- Mahadevan, D.; Chiorean, E.G.; Harris, W.B.; Von Hoff, D.D.; Stejskal-Barnett, A.; Qi, W.; Anthony, S.P.; Younger, A.E.; Rensvold, D.M.; Cordova, F.; et al. Phase I pharmacokinetic and pharmacodynamic study of the pan-PI3K/mTORC vascular targeted pro-drug SF1126 in patients with advanced solid tumours and B-cell malignancies. Eur. J. Cancer 2012, 48, 3319–3327. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Qiu, Y.; Kong, D. Class I phosphatidylinositol 3-kinase inhibitors for cancer therapy. Acta Pharm. Sin. B 2017, 7, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Sychev, Z.E.; Hu, A.; DiMaio, T.A.; Gitter, A.; Camp, N.D.; Noble, W.S.; Wolf-Yadlin, A.; Lagunoff, M. Integrated systems biology analysis of KSHV latent infection reveals viral induction and reliance on peroxisome mediated lipid metabolism. PLoS Pathog. 2017, 13, e1006256. [Google Scholar] [CrossRef] [PubMed]
- De Paoli, P.; Carbone, A. Kaposi’s Sarcoma Herpesvirus: Twenty years after its discovery. Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 1288–1294. [Google Scholar] [PubMed]
- Wakeman, B.S.; Izumiya, Y.; Speck, S.H. Identification of Novel Kaposi’s Sarcoma-Associated Herpesvirus Orf50 Transcripts: Discovery of New RTA Isoforms with Variable Transactivation Potential. J. Virol. 2017, 91, e01434-16. [Google Scholar] [CrossRef] [PubMed]
- Dow, D.E.; Cunningham, C.K.; Buchanan, A.M. A Review of Human Herpesvirus 8, the Kaposi’s Sarcoma-Associated Herpesvirus, in the Pediatric Population. J. Pediatr. Infect. Dis. Soc. 2014, 3, 66–76. [Google Scholar] [CrossRef] [PubMed]
- Kaposi, M. Idiopathisches multiples Pigmentsarkomen der Haut. Arch. Dermatol. Syph. 1872, 4, 265–273. [Google Scholar] [CrossRef]
- Beral, V.; Peterman, T.A.; Berkelman, R.L.; Jaffe, H.W. Kaposi’s sarcoma among persons with AIDS: A sexually transmitted infection? Lancet 1990, 335, 123–128. [Google Scholar] [CrossRef]
- Beral, V.; Bull, D.; Darby, S.; Weller, I.; Carne, C.; Beecham, M.; Jaffe, H. Risk of Kaposi’s sarcoma and sexual practices associated with faecal contact in homosexual or bisexual men with AIDS. Lancet 1992, 339, 632–635. [Google Scholar] [CrossRef]
- Peterman, T.A.; Jaffe, H.W.; Beral, V. Epidemiologic clues to the etiology of Kaposi’s sarcoma. AIDS 1993, 7, 605–611. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.; Cesarman, E.; Pessin, M.S.; Lee, F.; Culpepper, J.; Knowles, D.M.; Moore, P.S. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi’s sarcoma. Science 1994, 266, 1865–1869. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.; Low, G. Human Herpes Virus-8-Associated Multicentric Castleman’s Disease in a Human Immunodeficiency Virus-Positive Patient with a Previous History of Kaposi’s Sarcoma. J. Clin. Imaging Sci. 2015, 5, 59. [Google Scholar] [CrossRef] [PubMed]
- Okada, S.; Goto, H.; Yotsumoto, M. Current status of treatment for primary effusion lymphoma. Intractable Rare Dis. Res. 2014, 3, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Thakker, S.; Verma, S.C. Co-infections and Pathogenesis of KSHV-Associated Malignancies. Front. Microbiol. 2016, 7, 151. [Google Scholar] [CrossRef] [PubMed]
- Gantt, S.; Casper, C. Human herpesvirus 8-associated neoplasms: The roles of viral replication and antiviral treatment. Curr. Opin. Infect. Dis. 2011, 24, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Hu, H.; He, Z.; Liang, D.; Sun, R.; Lan, K. Fine-Tuning of the Kaposi’s Sarcoma-Associated Herpesvirus Life Cycle in Neighboring Cells through the RTA-JAG1-Notch Pathway. PLoS Pathog. 2016, 12, e1005900. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.S.; De Leo, A.; Wang, Z.; Kerekovic, A.; Hills, R.; Lieberman, P.M. BET-Inhibitors Disrupt Rad21-Dependent Conformational Control of KSHV Latency. PLoS Pathog. 2017, 13, e1006100. [Google Scholar] [CrossRef] [PubMed]
- Balistreri, G.; Viiliäinen, J.; Turunen, M.; Diaz, R.; Lyly, L.; Pekkonen, P.; Rantala, J.; Ojala, K.; Sarek, G.; Teesalu, M.; et al. Oncogenic Herpesvirus Utilizes Stress-Induced Cell Cycle Checkpoints for Efficient Lytic Replication. PLoS Pathog. 2016, 12, e1005424. [Google Scholar] [CrossRef] [PubMed]
- Purushothaman, P.; Dabral, P.; Gupta, N.; Sarkar, R.; Verma, S.C. KSHV Genome Replication and Maintenance. Front. Microbiol. 2016, 7, 54. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, L.D. Human herpesvirus-8: Kaposi sarcoma, multicentric Castleman disease, and primary effusion lymphoma. Hematol. Am. Soc. Hematol. Educ. Program 2013, 2013, 103–108. [Google Scholar] [CrossRef] [PubMed]
- Cavallin, L.E.; Goldschmidt-Clermont, P.; Mesri, E.A. Molecular and cellular mechanisms of KSHV oncogenesis of Kaposi’s sarcoma associated with HIV/AIDS. PLoS Pathog. 2014, 10, e1004154. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.C.; Cesarman, E. NF-κB as a target for oncogenic viruses. Curr. Top. Microbiol. Immunol. 2011, 349, 197–244. [Google Scholar] [CrossRef] [PubMed]
- Knowlton, E.R.; Rappocciolo, G.; Piazza, P.; Lepone, L.M.; Nadgir, S.V.; Bullotta, A.; Berendam, S.J.; Li, J.; Reinhart, T.A.; Jenkins, F.J.; et al. Human herpesvirus 8 induces polyfunctional B lymphocytes that drive Kaposi’s sarcoma. MBio 2014, 5, e01277-14. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Guidelines on the Treatment of Skin and Oral HIV-Associated Conditions in Children and Adults; World Health Organization: Geneva, Switzerland, 2014. [Google Scholar]
- Takahashi-Makise, N.; Suzu, S.; Hiyoshi, M.; Ohsugi, T.; Katano, H.; Umezawa, K.; Okada, S. Biscoclaurine alkaloid cepharanthine inhibits the growth of primary effusion lymphoma in vitro and in vivo and induces apoptosis via suppression of the NF-kappaB pathway. Int. J. Cancer 2009, 125, 1464–1472. [Google Scholar] [CrossRef] [PubMed]
- Matsuno, T.; Kariya, R.; Yano, S.; Morino-Koga, S.; Taura, M.; Suico, M.A.; Shimauchi, Y.; Matsuyama, S.; Okamoto, Y.; Shuto, T.; et al. Diethyldithiocarbamate induces apoptosis in HHV-8-infected primary effusion lymphoma cells via inhibition of the NF-κB pathway. Int. J. Oncol. 2012, 40, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Goto, H.; Kariya, R.; Shimamoto, M.; Kudo, E.; Taura, M.; Katano, H.; Okada, S. Antitumor effect of berberine against primary effusion lymphoma via inhibition of NF-κB pathway. Cancer Sci. 2012, 103, 775–781. [Google Scholar] [CrossRef] [PubMed]
- Gopalakrishnan, R.; Matta, H.; Chaudhary, P.M. A purine scaffold HSP90 inhibitor BIIB021 has selective activity against KSHV-associated primary effusion lymphoma and blocks vFLIP K13-induced NF-κB. Clin. Cancer Res. 2013, 19, 5016–5026. [Google Scholar] [CrossRef] [PubMed]
- Shigemi, Z.; Furukawa, Y.; Hosokawa, K.; Minami, S.; Matsuhiro, J.; Nakata, S.; Watanabe, T.; Kagawa, H.; Nakagawa, K.; Takeda, H.; et al. Diallyl trisulfide induces apoptosis by suppressing NF-κB signaling through destabilization of TRAF6 in primary effusion lymphoma. Int. J. Oncol. 2016, 48, 293–304. [Google Scholar] [CrossRef] [PubMed]
- Uldrick, T.S.; Gonçalves, P.H.; Wyvill, K.M.; Peer, C.J.; Bernstein, W.; Aleman, K.; Polizzotto, M.N.; Venzon, D.; Steinberg, S.M.; Marshall, V.; et al. A Phase Ib Study of Sorafenib (BAY 43-9006) in Patients with Kaposi Sarcoma. Oncologist 2017, 22, 505-e49. [Google Scholar] [CrossRef] [PubMed]
- Dittmer, D.P.; Damania, B. Kaposi sarcoma-associated herpesvirus: Immunobiology, oncogenesis, and therapy. J. Clin. Investig. 2016, 126, 3165–3175. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, S.; Ashlock, B.M.; Natkunam, Y.; Sujoy, V.; Chapman, J.R.; Ramos, J.C.; Mesri, E.A.; Lossos, I.S. CD30 targeting with brentuximab vedotin: A novel therapeutic approach to primary effusion lymphoma. Blood 2013, 122, 1233–1242. [Google Scholar] [CrossRef] [PubMed]
- Koon, H.B.; Krown, S.E.; Lee, J.Y.; Honda, K.; Rapisuwon, S.; Wang, Z.; Aboulafia, D.; Reid, E.G.; Rudek, M.A.; Dezube, B.J.; et al. Phase II trial of imatinib in AIDS-associated Kaposi’s sarcoma: AIDS Malignancy Consortium Protocol 042. J. Clin. Oncol. 2014, 32, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Cao, W.; Vyboh, K.; Routy, B.; Chababi-Atallah, M.; Lemire, B.; Routy, J.P. Imatinib for highly chemoresistant Kaposi sarcoma in a patient with long-term HIV control: A case report and literature review. Curr. Oncol. 2015, 22, e395. [Google Scholar] [CrossRef] [PubMed]
- Casper, C.; Krantz, E.M.; Corey, L.; Kuntz, S.R.; Wang, J.; Selke, S.; Hamilton, S.; Huang, M.L.; Wald, A. Valganciclovir for suppression of human herpesvirus-8 replication: A randomized, double-blind, placebo-controlled, crossover trial. J. Infect. Dis. 2008, 198, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Glesby, M.J.; Hoover, D.R.; Weng, S.; Graham, N.M.; Phair, J.P.; Detels, R.; Ho, M.; Saah, A.J. Use of antiherpes drugs and the risk of Kaposi’s sarcoma: Data from the Multicenter AIDS Cohort Study. J. Infect. Dis. 1996, 173, 1477–1480. [Google Scholar] [CrossRef] [PubMed]
- Mocroft, A.; Youle, M.; Gazzard, B.; Morcinek, J.; Halai, R.; Phillips, A.N. Anti-herpesvirus treatment and risk of Kaposi’s sarcoma in HIV infection. Royal Free/Chelsea and Westminster Hospitals Collaborative Group. AIDS 1996, 10, 1101–1105. [Google Scholar] [PubMed]
- Krown, S.E.; Dittmer, D.P.; Cesarman, E. Pilot study of oral valganciclovir therapy in patients with classic Kaposi sarcoma. J. Infect. Dis. 2011, 203, 1082–1086. [Google Scholar] [CrossRef] [PubMed]
- Morfeldt, L.; Torssander, J. Long-term remission of Kaposi’s sarcoma following foscarnet treatment in HIV-infected patients. Scand. J. Infect. Dis. 1994, 26, 749–752. [Google Scholar] [CrossRef] [PubMed]
- Mazzi, R.; Parisi, S.G.; Sarmati, L.; Uccella, I.; Nicastri, E.; Carolo, G.; Gatti, F.; Concia, E.; Andreoni, M. Efficacy of cidofovir on human herpesvirus 8 viraemia and Kaposi’s sarcoma progression in two patients with AIDS. AIDS 2001, 15, 2061–2062. [Google Scholar] [CrossRef] [PubMed]
- Bossini, N.; Sandrini, S.; Setti, G.; Luppi, M.; Maiorca, P.; Maffei, C.; Cancarini, G. Successful treatment with liposomal doxorubicin and foscarnet in a patient with widespread Kaposi’s sarcoma and human herpes virus 8-related, serious hemophagocytic syndrome, after renal transplantation. G. Ital. Nefrol. 2005, 22, 281–286. [Google Scholar] [PubMed]
- Moyo, T.K.; Richards, K.L.; Damania, B. Use of cidofovir for the treatment of HIV-negative human herpes virus-8-associated primary effusion lymphoma. Clin. Adv. Hematol. Oncol. 2010, 8, 372–374. [Google Scholar] [PubMed]
- Little, R.F.; Merced-Galindez, F.; Staskus, K.; Whitby, D.; Aoki, Y.; Humphrey, R.; Pluda, J.M.; Marshall, V.; Walters, M.; Welles, L.; et al. A pilot study of cidofovir in patients with kaposi sarcoma. J. Infect. Dis. 2003, 187, 149–153. [Google Scholar] [CrossRef] [PubMed]
- Cattamanchi, A.; Saracino, M.; Selke, S.; Huang, M.L.; Magaret, A.; Celum, C.; Corey, L.; Wald, A.; Casper, C. Treatment with valacyclovir, famciclovir, or antiretrovirals reduces human herpesvirus-8 replication in HIV-1 seropositive men. J. Med. Virol. 2011, 83, 1696–1703. [Google Scholar] [CrossRef] [PubMed]
- Sergerie, Y.; Boivin, G. Evaluation of susceptibility of human herpesvirus 8 to antiviral drugs by quantitative real-time PCR. J. Clin. Microbiol. 2003, 41, 3897–3900. [Google Scholar] [CrossRef] [PubMed]
- Friedrichs, C.; Neyts, J.; Gaspar, G.; De Clercq, E.; Wutzler, P. Evaluation of antiviral activity against human herpesvirus 8 (HHV-8) and Epstein-Barr virus (EBV) by a quantitative real-time PCR assay. Antivir. Res. 2004, 62, 121–123. [Google Scholar] [CrossRef] [PubMed]
- Johne, R.; Buck, C.B.; Allander, T.; Atwood, W.J.; Garcea, R.L.; Imperiale, M.J.; Major, E.O.; Ramqvist, T.; Norkin, L.C. Taxonomical developments in the family Polyomaviridae. Arch. Virol. 2011, 156, 1627–1634. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.; Shuda, M.; Chang, Y.; Moore, P.S. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science 2008, 319, 1096–1100. [Google Scholar] [CrossRef] [PubMed]
- DeCaprio, J.A.; Garcea, R.L. A cornucopia of human polyomaviruses. Nat. Rev. Microbiol. 2013, 11, 264–276. [Google Scholar] [CrossRef] [PubMed]
- Moens, U.; Rasheed, K.; Abdulsalam, I.; Sveinbjørnsson, B. The role of Merkel cell polyomavirus and other human polyomaviruses in emerging hallmarks of cancer. Viruses 2015, 7, 1871–1901. [Google Scholar] [CrossRef] [PubMed]
- Spurgeon, M.E.; Lambert, P.F. Merkel cell polyomavirus: A newly discovered human virus with oncogenic potential. Virology 2013, 435, 118–130. [Google Scholar] [CrossRef] [PubMed]
- Shuda, M.; Guastafierro, A.; Geng, X.; Shuda, Y.; Ostrowski, S.M.; Lukianov, S.; Jenkins, F.J.; Honda, K.; Maricich, S.M.; Moore, P.S.; et al. Merkel Cell Polyomavirus Small T Antigen Induces Cancer and Embryonic Merkel Cell Proliferation in a Transgenic Mouse Model. PLoS ONE 2015, 10, e0142329. [Google Scholar] [CrossRef] [PubMed]
- Samimi, M.; Gardair, C.; Nicol, J.T.; Arnold, F.; Touzé, A.; Coursaget, P. Merkel cell polyomavirus in merkel cell carcinoma: Clinical and therapeutic perspectives. Semin. Oncol. 2015, 42, 347–358. [Google Scholar] [CrossRef] [PubMed]
- Álvarez-Argüelles, M.E.; Melón, S.; Rojo, S.; Fernandez-Blázquez, A.; Boga, J.A.; Palacio, A.; Vivanco, B.; de Oña, M. Detection and quantification of Merkel cell polyomavirus. Analysis of Merkel cell carcinoma cases from 1977 to 2015. J. Med. Virol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Rodig, S.J.; Cheng, J.; Wardzala, J.; DoRosario, A.; Scanlon, J.J.; Laga, A.C.; Martinez-Fernandez, A.; Barletta, J.A.; Bellizzi, A.M.; Sadasivam, S.; et al. Improved detection suggests all Merkel cell carcinomas harbor Merkel polyomavirus. J. Clin. Investig. 2012, 122, 4645–4653. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, T.L.; Dennis, S.; Kachare, S.D.; Vohra, N.A.; Wong, J.H.; Zervos, E.E. Dramatic Increase in the Incidence and Mortality from Merkel Cell Carcinoma in the United States. Am. Surg. 2015, 81, 802–806. [Google Scholar] [PubMed]
- Paik, J.Y.; Hall, G.; Clarkson, A.; Lee, L.; Toon, C.; Colebatch, A.; Chou, A.; Gill, A.J. Immunohistochemistry for Merkel cell polyomavirus is highly specific but not sensitive for the diagnosis of Merkel cell carcinoma in the Australian population. Hum. Pathol. 2011, 42, 1385–1390. [Google Scholar] [CrossRef] [PubMed]
- Schowalter, R.M.; Pastrana, D.V.; Buck, C.B. Glycosaminoglycans and sialylated glycans sequentially facilitate Merkel cell polyomavirus infectious entry. PLoS Pathog. 2011, 7, e1002161. [Google Scholar] [CrossRef] [PubMed]
- Foulongne, V.; Courgnaud, V.; Champeau, W.; Segondy, M. Detection of Merkel cell polyomavirus on environmental surfaces. J. Med. Virol. 2011, 83, 1435–1439. [Google Scholar] [CrossRef] [PubMed]
- Babakir-Mina, M.; Ciccozzi, M.; Lo Presti, A.; Greco, F.; Perno, C.F.; Ciotti, M. Identification of Merkel cell polyomavirus in the lower respiratory tract of Italian patients. J. Med. Virol. 2010, 82, 505–509. [Google Scholar] [CrossRef] [PubMed]
- Bialasiewicz, S.; Lambert, S.B.; Whiley, D.M.; Nissen, M.D.; Sloots, T.P. Merkel cell polyomavirus DNA in respiratory specimens from children and adults. Emerg. Infect. Dis. 2009, 15, 492–494. [Google Scholar] [CrossRef] [PubMed]
- Goh, S.; Lindau, C.; Tiveljung-Lindell, A.; Allander, T. Merkel cell polyomavirus in respiratory tract secretions. Emerg. Infect. Dis. 2009, 15, 489–491. [Google Scholar] [CrossRef] [PubMed]
- Kantola, K.; Sadeghi, M.; Lahtinen, A.; Koskenvuo, M.; Aaltonen, L.M.; Möttönen, M.; Rahiala, J.; Saarinen-Pihkala, U.; Riikonen, P.; Jartti, T.; et al. Merkel cell polyomavirus DNA in tumor-free tonsillar tissues and upper respiratory tract samples: Implications for respiratory transmission and latency. J. Clin. Virol. 2009, 45, 292–295. [Google Scholar] [CrossRef] [PubMed]
- Loyo, M.; Guerrero-Preston, R.; Brait, M.; Hoque, M.O.; Chuang, A.; Kim, M.S.; Sharma, R.; Liégeois, N.J.; Koch, W.M.; Califano, J.A.; et al. Quantitative detection of Merkel cell virus in human tissues and possible mode of transmission. Int. J. Cancer 2010, 126, 2991–2996. [Google Scholar] [CrossRef] [PubMed]
- Sharp, C.P.; Norja, P.; Anthony, I.; Bell, J.E.; Simmonds, P. Reactivation and mutation of newly discovered WU, KI, and Merkel cell carcinoma polyomaviruses in immunosuppressed individuals. J. Infect. Dis. 2009, 199, 398–404. [Google Scholar] [CrossRef] [PubMed]
- Shuda, M.; Arora, R.; Kwun, H.J.; Feng, H.; Sarid, R.; Fernández-Figueras, M.T.; Tolstov, Y.; Gjoerup, O.; Mansukhani, M.M.; Swerdlow, S.H.; et al. Human Merkel cell polyomavirus infection I. MCV T antigen expression in Merkel cell carcinoma, lymphoid tissues and lymphoid tumors. Int. J. Cancer 2009, 125, 1243–1249. [Google Scholar] [CrossRef] [PubMed]
- Toracchio, S.; Foyle, A.; Sroller, V.; Reed, J.A.; Wu, J.; Kozinetz, C.A.; Butel, J.S. Lymphotropism of Merkel cell polyomavirus infection, Nova Scotia, Canada. Emerg. Infect. Dis. 2010, 16, 1702–1709. [Google Scholar] [CrossRef] [PubMed]
- Bofill-Mas, S.; Rodriguez-Manzano, J.; Calgua, B.; Carratala, A.; Girones, R. Newly described human polyomaviruses Merkel cell, KI and WU are present in urban sewage and may represent potential environmental contaminants. Virol. J. 2010, 7, 141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Husseiny, M.I.; Anastasi, B.; Singer, J.; Lacey, S.F. A comparative study of Merkel cell, BK and JC polyomavirus infections in renal transplant recipients and healthy subjects. J. Clin. Virol. 2010, 49, 137–140. [Google Scholar] [CrossRef] [PubMed]
- Laude, H.C.; Jonchère, B.; Maubec, E.; Carlotti, A.; Marinho, E.; Couturaud, B.; Peter, M.; Sastre-Garau, X.; Avril, M.F.; Dupin, N.; et al. Distinct merkel cell polyomavirus molecular features in tumour and non tumour specimens from patients with merkel cell carcinoma. PLoS Pathog. 2010, 6, e1001076. [Google Scholar] [CrossRef] [PubMed]
- Mertz, K.D.; Junt, T.; Schmid, M.; Pfaltz, M.; Kempf, W. Inflammatory monocytes are a reservoir for Merkel cell polyomavirus. J. Investig. Dermatol. 2010, 130, 1146–1151. [Google Scholar] [CrossRef] [PubMed]
- Campello, C.; Comar, M.; D’Agaro, P.; Minicozzi, A.; Rodella, L.; Poli, A. A molecular case-control study of the Merkel cell polyomavirus in colon cancer. J. Med. Virol. 2011, 83, 721–724. [Google Scholar] [CrossRef] [PubMed]
- Falchook, G.S.; Rady, P.; Konopinski, J.C.; Busaidy, N.; Hess, K.; Hymes, S.; Nguyen, H.P.; Prieto, V.G.; Bustinza-Linares, E.; Lin, Q.; et al. Merkel cell polyomavirus and human papilloma virus in proliferative skin lesions arising in patients treated with BRAF inhibitors. Arch. Dermatol. Res. 2016, 308, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.H.; Cohen, D.N.; Rady, P.L.; Tyring, S.K. BRAF inhibitor-associated cutaneous squamous cell carcinoma: New mechanistic insight, emerging evidence for viral involvement and perspectives on clinical management. Br. J. Dermatol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Wieland, U.; Mauch, C.; Kreuter, A.; Krieg, T.; Pfister, H. Merkel cell polyomavirus DNA in persons without merkel cell carcinoma. Emerg. Infect. Dis. 2009, 15, 1496–1498. [Google Scholar] [CrossRef] [PubMed]
- Schowalter, R.M.; Pastrana, D.V.; Pumphrey, K.A.; Moyer, A.L.; Buck, C.B. Merkel cell polyomavirus and two previously unknown polyomaviruses are chronically shed from human skin. Cell Host Microbe 2010, 7, 509–515. [Google Scholar] [CrossRef] [PubMed]
- Tolstov, Y.L.; Pastrana, D.V.; Feng, H.; Becker, J.C.; Jenkins, F.J.; Moschos, S.; Chang, Y.; Buck, C.B.; Moore, P.S. Human Merkel cell polyomavirus infection II. MCV is a common human infection that can be detected by conformational capsid epitope immunoassays. Int. J. Cancer 2009, 125, 1250–1256. [Google Scholar] [CrossRef] [PubMed]
- Tolstov, Y.L.; Knauer, A.; Chen, J.G.; Kensler, T.W.; Kingsley, L.A.; Moore, P.S.; Chang, Y. Asymptomatic primary Merkel cell polyomavirus infection among adults. Emerg. Infect. Dis. 2011, 17, 1371–1380. [Google Scholar] [CrossRef] [PubMed]
- Gjoerup, O.; Chang, Y. Update on human polyomaviruses and cancer. Adv. Cancer Res. 2010, 106, 1–51. [Google Scholar] [CrossRef] [PubMed]
- Shuda, M.; Feng, H.; Kwun, H.J.; Rosen, S.T.; Gjoerup, O.; Moore, P.S.; Chang, Y. T antigen mutations are a human tumor-specific signature for Merkel cell polyomavirus. Proc. Natl. Acad. Sci. USA 2008, 105, 16272–16277. [Google Scholar] [CrossRef] [PubMed]
- Carter, J.J.; Daugherty, M.D.; Qi, X.; Bheda-Malge, A.; Wipf, G.C.; Robinson, K.; Roman, A.; Malik, H.S.; Galloway, D.A. Identification of an overprinting gene in Merkel cell polyomavirus provides evolutionary insight into the birth of viral genes. Proc. Natl. Acad. Sci. USA 2013, 110, 12744–12749. [Google Scholar] [CrossRef] [PubMed]
- Houben, R.; Shuda, M.; Weinkam, R.; Schrama, D.; Feng, H.; Chang, Y.; Moore, P.S.; Becker, J.C. Merkel cell polyomavirus-infected Merkel cell carcinoma cells require expression of viral T antigens. J. Virol. 2010, 84, 7064–7072. [Google Scholar] [CrossRef] [PubMed]
- Zerrahn, J.; Knippschild, U.; Winkler, T.; Deppert, W. Independent expression of the transforming amino-terminal domain of SV40 large I antigen from an alternatively spliced third SV40 early mRNA. EMBO J. 1993, 12, 4739–4746. [Google Scholar] [PubMed]
- Comerford, S.A.; Schultz, N.; Hinnant, E.A.; Klapproth, S.; Hammer, R.E. Comparative analysis of SV40 17kT and LT function in vivo demonstrates that LT’s C-terminus re-programs hepatic gene expression and is necessary for tumorigenesis in the liver. Oncogenesis 2012, 1, e28. [Google Scholar] [CrossRef] [PubMed]
- Stakaitytė, G.; Wood, J.J.; Knight, L.M.; Abdul-Sada, H.; Adzahar, N.S.; Nwogu, N.; Macdonald, A.; Whitehouse, A. Merkel cell polyomavirus: Molecular insights into the most recently discovered human tumour virus. Cancers 2014, 6, 1267–1297. [Google Scholar] [CrossRef] [PubMed]
- Pastrana, D.V.; Tolstov, Y.L.; Becker, J.C.; Moore, P.S.; Chang, Y.; Buck, C.B. Quantitation of human seroresponsiveness to Merkel cell polyomavirus. PLoS Pathog. 2009, 5, e1000578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Touzé, A.; Le Bidre, E.; Laude, H.; Fleury, M.J.; Cazal, R.; Arnold, F.; Carlotti, A.; Maubec, E.; Aubin, F.; Avril, M.F.; et al. High levels of antibodies against merkel cell polyomavirus identify a subset of patients with merkel cell carcinoma with better clinical outcome. J. Clin. Oncol. 2011, 29, 1612–1619. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Sato, Y.; Watanabe, D.; Ito, H.; Shimonohara, N.; Tsuji, T.; Nakajima, N.; Suzuki, Y.; Matsuo, K.; Nakagawa, H.; et al. Nuclear localization of Merkel cell polyomavirus large T antigen in Merkel cell carcinoma. Virology 2010, 398, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Schowalter, R.M.; Buck, C.B. The Merkel cell polyomavirus minor capsid protein. PLoS Pathog. 2013, 9, e1003558. [Google Scholar] [CrossRef] [PubMed]
- Kassem, A.; Schöpflin, A.; Diaz, C.; Weyers, W.; Stickeler, E.; Werner, M.; Zur Hausen, A. Frequent detection of Merkel cell polyomavirus in human Merkel cell carcinomas and identification of a unique deletion in the VP1 gene. Cancer Res. 2008, 68, 5009–5013. [Google Scholar] [CrossRef] [PubMed]
- Becker, J.C.; Houben, R.; Ugurel, S.; Trefzer, U.; Pföhler, C.; Schrama, D. MC polyomavirus is frequently present in Merkel cell carcinoma of European patients. J. Investig. Dermatol. 2009, 129, 248–250. [Google Scholar] [CrossRef] [PubMed]
- Sastre-Garau, X.; Peter, M.; Avril, M.F.; Laude, H.; Couturier, J.; Rozenberg, F.; Almeida, A.; Boitier, F.; Carlotti, A.; Couturaud, B.; et al. Merkel cell carcinoma of the skin: Pathological and molecular evidence for a causative role of MCV in oncogenesis. J. Pathol. 2009, 218, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; MacDonald, M.; You, J. Merkel cell polyomavirus infection and Merkel cell carcinoma. Curr. Opin. Virol. 2016, 20, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Calder, K.B.; Smoller, B.R. New insights into merkel cell carcinoma. Adv. Anat. Pathol. 2010, 17, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Demetriou, S.K.; Ona-Vu, K.; Sullivan, E.M.; Dong, T.K.; Hsu, S.W.; Oh, D.H. Defective DNA repair and cell cycle arrest in cells expressing Merkel cell polyomavirus T antigen. Int. J. Cancer 2012, 131, 1818–1827. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, M.; Wieland, U.; Kreuter, A.; Pawlita, M. C-terminal deletions of Merkel cell polyomavirus large T-antigen, a highly specific surrogate marker for virally induced malignancy. Int. J. Cancer 2012, 131, 2863–2868. [Google Scholar] [CrossRef] [PubMed]
- Fischer, N.; Brandner, J.; Fuchs, F.; Moll, I.; Grundhoff, A. Detection of Merkel cell polyomavirus (MCPyV) in Merkel cell carcinoma cell lines: Cell morphology and growth phenotype do not reflect presence of the virus. Int. J. Cancer 2010, 126, 2133–2142. [Google Scholar] [CrossRef] [PubMed]
- Katano, H.; Ito, H.; Suzuki, Y.; Nakamura, T.; Sato, Y.; Tsuji, T.; Matsuo, K.; Nakagawa, H.; Sata, T. Detection of Merkel cell polyomavirus in Merkel cell carcinoma and Kaposi’s sarcoma. J. Med. Virol. 2009, 81, 1951–1958. [Google Scholar] [CrossRef] [PubMed]
- Wendzicki, J.A.; Moore, P.S.; Chang, Y. Large T and small T antigens of Merkel cell polyomavirus. Curr. Opin. Virol. 2015, 11, 38–43. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, C.S.; Cantalupo, P.; Pipas, J.M. The molecular chaperone activity of simian virus 40 large T antigen is required to disrupt Rb-E2F family complexes by an ATP-dependent mechanism. Mol. Cell. Biol. 2000, 20, 6233–6243. [Google Scholar] [CrossRef] [PubMed]
- Shuda, M.; Kwun, H.J.; Feng, H.; Chang, Y.; Moore, P.S. Human Merkel cell polyomavirus small T antigen is an oncoprotein targeting the 4E-BP1 translation regulator. J. Clin. Investig. 2011, 121, 3623–3634. [Google Scholar] [CrossRef] [PubMed]
- Burnett, P.E.; Barrow, R.K.; Cohen, N.A.; Snyder, S.H.; Sabatini, D.M. RAFT1 phosphorylation of the translational regulators p70 S6 kinase and 4E-BP1. Proc. Natl. Acad. Sci. USA 1998, 95, 1432–1437. [Google Scholar] [CrossRef] [PubMed]
- Kwun, H.J.; Shuda, M.; Feng, H.; Camacho, C.J.; Moore, P.S.; Chang, Y. Merkel cell polyomavirus small T antigen controls viral replication and oncoprotein expression by targeting the cellular ubiquitin ligase SCFFbw7. Cell Host Microbe 2013, 14, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.H.; Simonette, R.A.; Nguyen, H.P.; Rady, P.L.; Tyring, S.K. Merkel cell polyomavirus in Merkel cell carcinogenesis: Small T antigen-mediates c-Jun phosphorylation. Virus Genes 2016, 52, 397–399. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.Y.; Ke, H.; Hall, R.P.; Zhang, J.Y. c-Jun promotes whereas JunB inhibits epidermal neoplasia. J. Investig. Dermatol. 2011, 131, 1149–1158. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.H.; Simonette, R.A.; Nguyen, H.P.; Rady, P.L.; Tyring, S.K. Small T-antigen of the TS-associated polyomavirus activates factors implicated in the MAPK pathway. J. Eur. Acad. Dermatol. Venereol. 2016, 30, 1061–1062. [Google Scholar] [CrossRef] [PubMed]
- Kwun, H.J.; Shuda, M.; Camacho, C.J.; Gamper, A.M.; Thant, M.; Chang, Y.; Moore, P.S. Restricted protein phosphatase 2A targeting by Merkel cell polyomavirus small T antigen. J. Virol. 2015, 89, 4191–4200. [Google Scholar] [CrossRef] [PubMed]
- Sihto, H.; Kukko, H.; Koljonen, V.; Sankila, R.; Böhling, T.; Joensuu, H. Merkel cell polyomavirus infection, large T antigen, retinoblastoma protein and outcome in Merkel cell carcinoma. Clin. Cancer Res. 2011, 17, 4806–4813. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, X.; Diaz, J.; Tsang, S.H.; Buck, C.B.; You, J. Merkel cell polyomavirus large T antigen disrupts host genomic integrity and inhibits cellular proliferation. J. Virol. 2013, 87, 9173–9188. [Google Scholar] [CrossRef] [PubMed]
- Borchert, S.; Czech-Sioli, M.; Neumann, F.; Schmidt, C.; Wimmer, P.; Dobner, T.; Grundhoff, A.; Fischer, N. High-affinity Rb binding, p53 inhibition, subcellular localization, and transformation by wild-type or tumor-derived shortened Merkel cell polyomavirus large T antigens. J. Virol. 2014, 88, 3144–3160. [Google Scholar] [CrossRef] [PubMed]
- Sihto, H.; Kukko, H.; Koljonen, V.; Sankila, R.; Böhling, T.; Joensuu, H. Clinical factors associated with Merkel cell polyomavirus infection in Merkel cell carcinoma. J. Natl. Cancer Inst. 2009, 101, 938–945. [Google Scholar] [CrossRef] [PubMed]
- Arora, R.; Shuda, M.; Guastafierro, A.; Feng, H.; Toptan, T.; Tolstov, Y.; Normolle, D.; Vollmer, L.L.; Vogt, A.; Dömling, A.; et al. Survivin is a therapeutic target in Merkel cell carcinoma. Sci. Transl. Med. 2012, 4, 133ra56. [Google Scholar] [CrossRef] [PubMed]
- Dresang, L.R.; Guastafierro, A.; Arora, R.; Normolle, D.; Chang, Y.; Moore, P.S. Response of Merkel cell polyomavirus-positive merkel cell carcinoma xenografts to a survivin inhibitor. PLoS ONE 2013, 8, e80543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, Q.; Gomez, B.P.; Viscidi, R.P.; Peng, S.; He, L.; Ma, B.; Wu, T.C.; Hung, C.F. Development of a DNA vaccine targeting Merkel cell polyomavirus. Vaccine 2012, 30, 1322–1329. [Google Scholar] [CrossRef] [PubMed]
- Gomez, B.; He, L.; Tsai, Y.C.; Wu, T.C.; Viscidi, R.P.; Hung, C.F. Creation of a Merkel cell polyomavirus small T antigen-expressing murine tumor model and a DNA vaccine targeting small T antigen. Cell Biosci. 2013, 3, 29. [Google Scholar] [CrossRef] [PubMed]
- Iyer, J.G.; Afanasiev, O.K.; McClurkan, C.; Paulson, K.; Nagase, K.; Jing, L.; Marshak, J.O.; Dong, L.; Carter, J.; Lai, I.; et al. Merkel cell polyomavirus-specific CD8⁺ and CD4⁺ T-cell responses identified in Merkel cell carcinomas and blood. Clin. Cancer Res. 2011, 17, 6671–6680. [Google Scholar] [CrossRef] [PubMed]
- Paulson, K.G.; Tegeder, A.; Willmes, C.; Iyer, J.G.; Afanasiev, O.K.; Schrama, D.; Koba, S.; Thibodeau, R.; Nagase, K.; Simonson, W.T.; et al. Downregulation of MHC-I expression is prevalent but reversible in Merkel cell carcinoma. Cancer Immunol. Res. 2014, 2, 1071–1079. [Google Scholar] [CrossRef] [PubMed]
- Shahzad, N.; Shuda, M.; Gheit, T.; Kwun, H.J.; Cornet, I.; Saidj, D.; Zannetti, C.; Hasan, U.; Chang, Y.; Moore, P.S.; et al. The T antigen locus of Merkel cell polyomavirus downregulates human Toll-like receptor 9 expression. J. Virol. 2013, 87, 13009–13019. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, D.A.; Abdul-Sada, H.; Knight, L.M.; Jackson, B.R.; Richards, K.; Prescott, E.L.; Peach, A.H.; Blair, G.E.; Macdonald, A.; Whitehouse, A. Merkel cell polyomavirus small T antigen targets the NEMO adaptor protein to disrupt inflammatory signaling. J. Virol. 2013, 87, 13853–13867. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, H.L.; Russell, J.; Hamid, O.; Bhatia, S.; Terheyden, P.; D’Angelo, S.P.; Shih, K.C.; Lebbé, C.; Linette, G.P.; Milella, M.; et al. Avelumab in patients with chemotherapy-refractory metastatic Merkel cell carcinoma: A multicentre, single-group, open-label, phase 2 trial. Lancet Oncol. 2016, 17, 1374–1385. [Google Scholar] [CrossRef]
- Aveluma (BAVENCIO). Available online: https://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm547965.htm (accessed on 20 September 2017).
- Nghiem, P.T.; Bhatia, S.; Lipson, E.J.; Kudchadkar, R.R.; Miller, N.J.; Annamalai, L.; Berry, S.; Chartash, E.K.; Daud, A.; Fling, S.P.; et al. PD-1 Blockade with Pembrolizumab in Advanced Merkel-Cell Carcinoma. N. Engl. J. Med. 2016, 374, 2542–2552. [Google Scholar] [CrossRef] [PubMed]
- Mantripragada, K.; Birnbaum, A. Response to Anti-PD-1 Therapy in Metastatic Merkel Cell Carcinoma Metastatic to the Heart and Pancreas. Cureus 2015, 7, e403. [Google Scholar] [CrossRef] [PubMed]
- Walocko, F.M.; Scheier, B.Y.; Harms, P.W.; Fecher, L.A.; Lao, C.D. Metastatic Merkel cell carcinoma response to nivolumab. J. Immunother. Cancer 2016, 4, 79. [Google Scholar] [CrossRef] [PubMed]
- Gao, G.; Smith, D.I. Human Papillomavirus and the Development of Different Cancers. Cytogenet. Genome Res. 2016, 150, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Brianti, P.; De Flammineis, E.; Mercuri, S.R. Review of HPV-related diseases and cancers. New Microbiol. 2017, 40, 80–85. [Google Scholar] [PubMed]
- Handler, M.Z.; Handler, N.S.; Majewski, S.; Schwartz, R.A. Human papillomavirus vaccine trials and tribulations: Clinical perspectives. J. Am. Acad. Dermatol. 2015, 73, 743–756. [Google Scholar] [CrossRef] [PubMed]
- Doorbar, J.; Quint, W.; Banks, L.; Bravo, I.G.; Stoler, M.; Broker, T.R.; Stanley, M.A. The biology and life-cycle of human papillomaviruses. Vaccine 2012, 30, F55–F70. [Google Scholar] [CrossRef] [PubMed]
- Forman, D.; de Martel, C.; Lacey, C.J.; Soerjomataram, I.; Lortet-Tieulent, J.; Bruni, L.; Vignat, J.; Ferlay, J.; Bray, F.; Plummer, M.; et al. Global burden of human papillomavirus and related diseases. Vaccine 2012, 30, F12–F23. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, J.C.; Calonje, E. Cutaneous manifestations of human papillomaviruses: A review. Acta Dermatovenerol. Alp. Pannonica Adriat. 2011, 20, 145–154. [Google Scholar] [PubMed]
- Kash, N.; Lee, M.A.; Kollipara, R.; Downing, C.; Guidry, J.; Tyring, S.K. Safety and Efficacy Data on Vaccines and Immunization to Human Papillomavirus. J. Clin. Med. 2015, 4, 614–633. [Google Scholar] [CrossRef] [PubMed]
- Zur Hausen, H. Papillomaviruses in the causation of human cancers-a brief historical account. Virology 2009, 384, 260–265. [Google Scholar] [CrossRef] [PubMed]
- Grce, M.; Mravak-Stipetić, M. Human papillomavirus-associated diseases. Clin. Dermatol. 2014, 32, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Blake, D.R.; Middleman, A.B. Human Papillomavirus Vaccine Update. Pediatr. Clin. N. Am. 2017, 64, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Lowy, D.R. HPV vaccination to prevent cervical cancer and other HPV-associated disease: From basic science to effective interventions. J. Clin. Investig. 2016, 126, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Handisurya, A.; Schellenbacher, C.; Kirnbauer, R. Diseases caused by human papillomaviruses (HPV). J. Dtsch. Dermatol. Ges. 2009, 7, 453–466. [Google Scholar] [CrossRef] [PubMed]
- Bernard, H.U.; Burk, R.D.; Chen, Z.; van Doorslaer, K.; zur Hausen, H.; de Villiers, E.M. Classification of papillomaviruses (PVs) based on 189 PV types and proposal of taxonomic amendments. Virology 2010, 401, 70–79. [Google Scholar] [CrossRef] [PubMed]
- De Villiers, E.M.; Fauquet, C.; Broker, T.R.; Bernard, H.U.; zur Hausen, H. Classification of papillomaviruses. Virology 2004, 324, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Aksoy, P.; Gottschalk, E.Y.; Meneses, P.I. HPV entry into cells. Mutat. Res. Rev. Mutat. Res. 2017, 772, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Panatto, D.; Amicizia, D.; Bragazzi, N.L.; Rizzitelli, E.; Tramalloni, D.; Valle, I.; Gasparini, R. Human Papillomavirus Vaccine: State of the Art and Future Perspectives. Adv. Protein Chem. Struct. Biol. 2015, 101, 231–322. [Google Scholar] [CrossRef] [PubMed]
- Doorbar, J.; Egawa, N.; Griffin, H.; Kranjec, C.; Murakami, I. Human papillomavirus molecular biology and disease association. Rev. Med. Virol. 2015, 25, 2–23. [Google Scholar] [CrossRef] [PubMed]
- Scheffner, M.; Werness, B.A.; Huibregtse, J.M.; Levine, A.J.; Howley, P.M. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell 1990, 63, 1129–1136. [Google Scholar] [CrossRef]
- Scheffner, M.; Huibregtse, J.M.; Vierstra, R.D.; Howley, P.M. The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell 1993, 75, 495–505. [Google Scholar] [CrossRef]
- Chen, J. Signaling pathways in HPV-associated cancers and therapeutic implications. Rev. Med. Virol. 2015, 25, 24–53. [Google Scholar] [CrossRef] [PubMed]
- Burd, E.M. Human papillomavirus and cervical cancer. Clin. Microbiol. Rev. 2003, 16, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Crosbie, E.J.; Einstein, M.H.; Franceschi, S.; Kitchener, H.C. Human papillomavirus and cervical cancer. Lancet 2013, 382, 889–899. [Google Scholar] [CrossRef]
- Stanley, M. Pathology and epidemiology of HPV infection in females. Gynecol. Oncol. 2010, 117, S5–S10. [Google Scholar] [CrossRef] [PubMed]
- Moody, C.A.; Laimins, L.A. Human papillomavirus oncoproteins: Pathways to transformation. Nat. Rev. Cancer 2010, 10, 550–560. [Google Scholar] [CrossRef] [PubMed]
- Pullos, A.N.; Castilho, R.M.; Squarize, C.H. HPV Infection of the Head and Neck Region and Its Stem Cells. J. Dent. Res. 2015, 94, 1532–1543. [Google Scholar] [CrossRef] [PubMed]
- Walboomers, J.M.; Jacobs, M.V.; Manos, M.M.; Bosch, F.X.; Kummer, J.A.; Shah, K.V.; Snijders, P.J.; Peto, J.; Meijer, C.J.; Muñoz, N. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J. Pathol. 1999, 189, 12–19. [Google Scholar] [CrossRef]
- Zur Hausen, H. Papillomaviruses and cancer: From basic studies to clinical application. Nat. Rev. Cancer 2002, 2, 342–350. [Google Scholar] [CrossRef] [PubMed]
- Olthof, N.C.; Huebbers, C.U.; Kolligs, J.; Henfling, M.; Ramaekers, F.C.; Cornet, I.; van Lent-Albrechts, J.A.; Stegmann, A.P.; Silling, S.; Wieland, U.; et al. Viral load, gene expression and mapping of viral integration sites in HPV16-associated HNSCC cell lines. Int. J. Cancer 2015, 136, E207–E218. [Google Scholar] [CrossRef] [PubMed]
- Cancer Stat Facts: Cervix Uteri Cancer. Available online: https://seer.cancer.gov/statfacts/html/cervix.html (accessed on 14 October 2017).
- International Collaboration of Epidemiological Studies of Cervical Cancer. Comparison of risk factors for invasive squamous cell carcinoma and adenocarcinoma of the cervix: Collaborative reanalysis of individual data on 8097 women with squamous cell carcinoma and 1374 women with adenocarcinoma from 12 epidemiological studies. Int. J. Cancer 2007, 120, 885–891. [Google Scholar] [CrossRef]
- Ries, L.A.G.; Melbert, D.; Krapcho, M.; Mariotto, A.; Miller, B.A.; Feuer, E.J.; Clegg, L.; Horner, M.J.; Howlader, N.; Eisner, M.P.; et al. SEER Cancer Statistics Review, 1975–2004; National Cancer Institute: Bethesda, MD, USA, 2007; Based on November 2006 SEER Data Submission, Posted to the SEER Web Site, 2007. Available online: http://seer.cancer.gov/csr/1975_2004/ (accessed on 20 October 2017).
- Li, N.; Franceschi, S.; Howell-Jones, R.; Snijders, P.J.; Clifford, G.M. Human papillomavirus type distribution in 30,848 invasive cervical cancers worldwide: Variation by geographical region, histological type and year of publication. Int. J. Cancer 2011, 128, 927–935. [Google Scholar] [CrossRef] [PubMed]
- Guan, P.; Clifford, G.M.; Franceschi, S. Human papillomavirus types in glandular lesions of the cervix: A meta-analysis of published studies. Int. J. Cancer 2013, 132, 248–250. [Google Scholar] [CrossRef] [PubMed]
- Schiffman, M.; Castle, P.E.; Jeronimo, J.; Rodriguez, A.C.; Wacholder, S. Human papillomavirus and cervical cancer. Lancet 2007, 370, 890–907. [Google Scholar] [CrossRef]
- Wright, T.C., Jr.; Massad, L.S.; Dunton, C.J.; Spitzer, M.; Wilkinson, E.J.; Solomon, D.; 2006 American Society for Colposcopy and Cervical Pathology-sponsored Consensus Conference. 2006 consensus guidelines for the management of women with cervical intraepithelial neoplasia or adenocarcinoma in situ. Am. J. Obstet. Gynecol. 2003, 197, 340–345. [Google Scholar] [CrossRef] [PubMed]
- Vega-Peña, A.; Illades-Aguiar, B.; Flores-Alfaro, E.; López-Bayghen, E.; Leyva-Vázquez, M.A.; Castañeda-Saucedo, E.; Alarcón-Romero, L.D.C. Risk of progression of early cervical lesions is associated with integration and persistence of HPV-16 and expression of E6, Ki-67, and telomerase. J. Cytol. 2013, 30, 226–232. [Google Scholar] [CrossRef] [PubMed]
- Prigge, E.S.; von Knebel Doeberitz, M.; Reuschenbach, M. Clinical relevance and implications of HPV-induced neoplasia in different anatomical locations. Mutat. Res. Rev. Mutat. Res. 2017, 772, 51–66. [Google Scholar] [CrossRef] [PubMed]
- Moyer, V.A.; U.S. Preventive Services Task Force. Screening for cervical cancer: U.S. Preventive Services Task Force recommendation statement. Ann. Intern. Med. 2012, 156, 880–891. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.B.; Liu, I.Y.; Gornbein, J.A.; Nguyen, C.T. HPV-Positive Oropharyngeal Carcinoma: A Systematic Review of Treatment and Prognosis. Otolaryngol. Head Neck Surg. 2015, 153, 758–769. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, A.K.; Engels, E.A.; Pfeiffer, R.M.; Hernandez, B.Y.; Xiao, W.; Kim, E.; Jiang, B.; Goodman, M.T.; Sibug-Saber, M.; Cozen, W.; et al. Human papillomavirus and rising oropharyngeal cancer incidence in the United States. J. Clin. Oncol. 2011, 29, 4294–4301. [Google Scholar] [CrossRef] [PubMed]
- Kreimer, A.R.; Clifford, G.M.; Boyle, P.; Franceschi, S. Human papillomavirus types in head and neck squamous cell carcinomas worldwide: A systematic review. Cancer Epidemiol. Biomark. Prev. 2005, 14, 467–475. [Google Scholar] [CrossRef] [PubMed]
- Leemans, C.R.; Braakhuis, B.J.; Brakenhoff, R.H. The molecular biology of head and neck cancer. Nat. Rev. Cancer 2011, 11, 9–22. [Google Scholar] [CrossRef] [PubMed]
- St Guily, J.L.; Jacquard, A.C.; Prétet, J.L.; Haesebaert, J.; Beby-Defaux, A.; Clavel, C.; Agius, G.; Birembaut, P.; Okaïs, C.; Léocmach, Y.; et al. Human papillomavirus genotype distribution in oropharynx and oral cavity cancer in France—The EDiTH VI study. J. Clin. Virol. 2011, 51, 100–104. [Google Scholar] [CrossRef] [PubMed]
- Mellin, H.; Friesland, S.; Auer, G.; Dalianis, T.; Munck-Wikland, E. Human papillomavirus and DNA ploidy in tonsillar cancer—Correlation to prognosis. Anticancer Res. 2003, 23, 2821–2828. [Google Scholar] [PubMed]
- Deng, Z.; Hasegawa, M.; Aoki, K.; Matayoshi, S.; Kiyuna, A.; Yamashita, Y.; Uehara, T.; Agena, S.; Maeda, H.; Xie, M.; et al. A comprehensive evaluation of human papillomavirus positive status and p16INK4a overexpression as a prognostic biomarker in head and neck squamous cell carcinoma. Int. J. Oncol. 2014, 45, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Owadally, W.; Hurt, C.; Timmins, H.; Parsons, E.; Townsend, S.; Patterson, J.; Hutcheson, K.; Powell, N.; Beasley, M.; Palaniappan, N.; et al. PATHOS: A phase II/III trial of risk-stratified, reduced intensity adjuvant treatment in patients undergoing transoral surgery for Human papillomavirus (HPV) positive oropharyngeal cancer. BMC Cancer 2015, 15, 602. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.M.; Felix, C.; Wang, P.C.; Hsu, S.; Basehart, V.; Garst, J.; Beron, P.; Wong, D.; Rosove, M.H.; Rao, S.; et al. Reduced-dose radiotherapy for human papillomavirus-associated squamous-cell carcinoma of the oropharynx: A single-arm, phase 2 study. Lancet Oncol. 2017, 18, 803–811. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2016. CA Cancer J. Clin. 2016, 66, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Backes, D.M.; Kurman, R.J.; Pimenta, J.M.; Smith, J.S. Systematic review of human papillomavirus prevalence in invasive penile cancer. Cancer Causes Control 2009, 20, 449–457. [Google Scholar] [CrossRef] [PubMed]
- Spiess, P.E.; Dhillon, J.; Baumgarten, A.S.; Johnstone, P.A.; Giuliano, A.R. Pathophysiological basis of human papillomavirus in penile cancer: Key to prevention and delivery of more effective therapies. CA Cancer J. Clin. 2016, 66, 481–495. [Google Scholar] [CrossRef] [PubMed]
- Stratton, K.L.; Culkin, D.J. A Contemporary Review of HPV and Penile Cancer. Oncology 2016, 30, 245–249. [Google Scholar] [PubMed]
- Clark, P.E.; Spiess, P.E.; Agarwal, N.; Biagioli, M.C.; Eisenberger, M.A.; Greenberg, R.E.; Herr, H.W.; Inman, B.A.; Kuban, D.A.; Kuzel, T.M.; et al. Penile cancer: Clinical Practice Guidelines in Oncology. J. Natl. Compr. Canc. Netw. 2013, 11, 594–615. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Laversanne, M.; Brewster, D.H.; Gombe Mbalawa, C.; Kohler, B.; Piñeros, M.; Steliarova-Foucher, E.; Swaminathan, R.; Antoni, S.; et al. Cancer Incidence in Five Continents: Inclusion criteria, highlights from Volume X and the global status of cancer registration. Int. J. Cancer 2015, 137, 2060–2071. [Google Scholar] [CrossRef] [PubMed]
- Stanley, M.A.; Winder, D.M.; Sterling, J.C.; Goon, P.K. HPV infection, anal intra-epithelial neoplasia (AIN) and anal cancer: Current issues. BMC Cancer 2012, 12, 398. [Google Scholar] [CrossRef] [PubMed]
- Abramowitz, L.; Jacquard, A.C.; Jaroud, F.; Haesebaert, J.; Siproudhis, L.; Pradat, P.; Aynaud, O.; Leocmach, Y.; Soubeyrand, B.; Dachez, R.; et al. Human papillomavirus genotype distribution in anal cancer in France: The EDiTH V study. Int. J. Cancer 2011, 129, 433–439. [Google Scholar] [CrossRef] [PubMed]
- Goodman, M.T.; Shvetsov, Y.B.; McDuffie, K.; Wilkens, L.R.; Zhu, X.; Thompson, P.J.; Ning, L.; Killeen, J.; Kamemoto, L.; Hernandez, B.Y. Sequential acquisition of human papillomavirus (HPV) infection of the anus and cervix: The Hawaii HPV Cohort Study. J. Infect. Dis. 2010, 201, 1331–1339. [Google Scholar] [CrossRef] [PubMed]
- Moscicki, A.B.; Schiffman, M.; Burchell, A.; Albero, G.; Giuliano, A.R.; Goodman, M.T.; Kjaer, S.K.; Palefsky, J. Updating the natural history of human papillomavirus and anogenital cancers. Vaccine 2012, 30, F24–F33. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2017. CA Cancer J. Clin. 2017, 67, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Buchanan, T.R.; Graybill, W.S.; Pierce, J.Y. Morbidity and mortality of vulvar and vaginal cancers: Impact of 2-, 4-, and 9-valent HPV vaccines. Hum. Vaccines Immunother. 2016, 12, 1352–1356. [Google Scholar] [CrossRef] [PubMed]
- Massad, L.S. Outcomes after diagnosis of vaginal intraepithelial neoplasia. J. Low. Genit. Tract Dis. 2008, 12, 16–19. [Google Scholar] [CrossRef] [PubMed]
- Skapa, P.; Robová, H.; Rob, L.; Zámečník, J. Review of precancerous vulvar lesions. Cesk. Patol. 2012, 48, 15–21. [Google Scholar] [PubMed]
- Cao, H.; Wang, S.; Zhang, Z.; Lou, J. Prognostic Value of Overexpressed p16INK4a in Vulvar Cancer: A Meta-Analysis. PLoS ONE 2016, 11, e0152459. [Google Scholar] [CrossRef] [PubMed]
- Signorelli, C.; Odone, A.; Ciorba, V.; Cella, P.; Audisio, R.A.; Lombardi, A.; Mariani, L.; Mennini, F.S.; Pecorelli, S.; Rezza, G.; et al. Human papillomavirus 9-valent vaccine for cancer prevention: A systematic review of the available evidence. Epidemiol. Infect. 2017, 145, 1962–1982. [Google Scholar] [CrossRef] [PubMed]
- Cuzick, J. Gardasil 9 joins the fight against cervix cancer. Expert Rev. Vaccines 2015, 14, 1047–1049. [Google Scholar] [CrossRef] [PubMed]
- Meites, E.; Kempe, A.; Markowitz, L.E. Use of a 2-Dose Schedule for Human Papillomavirus Vaccination-Updated Recommendations of the Advisory Committee on Immunization Practices. Am. J. Transpl. 2017, 17, 834–837. [Google Scholar] [CrossRef] [PubMed]
- Harper, D.M.; DeMars, L.R. HPV vaccines-A review of the first decade. Gynecol. Oncol. 2017, 146, 196–204. [Google Scholar] [CrossRef] [PubMed]
- Campo, M.S.; Roden, R.B. Papillomavirus prophylactic vaccines: Established successes, new approaches. J. Virol. 2010, 84, 1214–1220. [Google Scholar] [CrossRef] [PubMed]
- Schiller, J.T.; Castellsagué, X.; Garland, S.M. A review of clinical trials of human papillomavirus prophylactic vaccines. Vaccine 2012, 30, F123–F138. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.; Farmer, E.; Wu, T.C.; Hung, C.F. Perspectives for therapeutic HPV vaccine development. J. Biomed. Sci. 2016, 23, 75. [Google Scholar] [CrossRef] [PubMed]
- Miles, B.A.; Monk, B.J.; Safran, H.P. Mechanistic insights into ADXS11-001 human papillomavirus-associated cancer immunotherapy. Gynecol. Oncol. Res. Pract. 2017, 4, 9. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, M.; Stites, D.P.; Farhat, S.; Sisler, J.R.; Moss, B.; Kong, F.; Moscicki, A.B.; Palefsky, J.M. Cytotoxic T lymphocyte responses to E6 and E7 proteins of human papillomavirus type 16: Relationship to cervical intraepithelial neoplasia. J. Infect. Dis. 1997, 175, 927–931. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Kim, H.J. Current status and future prospects for human papillomavirus vaccines. Arch. Pharm. Res. 2017, 40, 1050–1063. [Google Scholar] [CrossRef] [PubMed]
- Franco, E.; Bagnato, B.; Marino, M.G.; Meleleo, C.; Serino, L.; Zaratti, L. Hepatitis B: Epidemiology and prevention in developing countries. World J. Hepatol. 2012, 4, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Ott, J.J.; Stevens, G.A.; Groeger, J.; Wiersma, S.T. Global epidemiology of hepatitis B virus infection: New estimates of age-specific HBsAg seroprevalence and endemicity. Vaccine 2012, 30, 2212–2219. [Google Scholar] [CrossRef] [PubMed]
- Croagh, C.M.; Lubel, J.S. Natural history of chronic hepatitis B: Phases in a complex relationship. World J. Gastroenterol. 2014, 20, 10395–10404. [Google Scholar] [CrossRef] [PubMed]
- Papastergiou, V.; Lombardi, R.; MacDonald, D.; Tsochatzis, E.A. Global Epidemiology of Hepatitis B Virus (HBV) Infection. Curr. Hepatol. Rep. 2015, 14, 171–178. [Google Scholar] [CrossRef]
- Wu, S.Y.; Lan, S.H.; Liu, H.S. Autophagy and microRNA in hepatitis B virus-related hepatocellular carcinoma. World J. Gastroenterol. 2016, 22, 176–187. [Google Scholar] [CrossRef] [PubMed]
- Niederhauser, C. Reducing the risk of hepatitis B virus transfusion-transmitted infection. J. Blood Med. 2011, 2, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Bozza, C.; Cinausero, M.; Iacono, D.; Puglisi, F. Hepatitis B and cancer: A practical guide for the oncologist. Crit. Rev. Oncol. Hematol. 2016, 98, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Schinzari, V.; Barnaba, V.; Piconese, S. Chronic hepatitis B virus and hepatitis C virus infections and cancer: Synergy between viral and host factors. Clin. Microbiol. Infect. 2015, 21, 969–974. [Google Scholar] [CrossRef] [PubMed]
- Perz, J.F.; Armstrong, G.L.; Farrington, L.A.; Hutin, Y.J.; Bell, B.P. The contributions of hepatitis B virus and hepatitis C virus infections to cirrhosis and primary liver cancer worldwide. J. Hepatol. 2006, 45, 529–538. [Google Scholar] [CrossRef] [PubMed]
- Saitta, C.; Tripodi, G.; Barbera, A.; Bertuccio, A.; Smedile, A.; Ciancio, A.; Raffa, G.; Sangiovanni, A.; Navarra, G.; Raimondo, G.; et al. Hepatitis B virus (HBV) DNA integration in patients with occult HBV infection and hepatocellular carcinoma. Liver Int. 2015, 35, 2311–2317. [Google Scholar] [CrossRef] [PubMed]
- Geng, M.; Xin, X.; Bi, L.Q.; Zhou, L.T.; Liu, X.H. Molecular mechanism of hepatitis B virus X protein function in hepatocarcinogenesis. World J. Gastroenterol. 2015, 21, 10732–10738. [Google Scholar] [CrossRef] [PubMed]
- Marcucci, F.; Spada, E.; Mele, A.; Caserta, C.A.; Pulsoni, A. The association of hepatitis B virus infection with B-cell non-Hodgkin lymphoma—A review. Am. J. Blood Res. 2012, 2, 18–28. [Google Scholar] [PubMed]
- Ye, Y.F.; Xiang, Y.Q.; Fang, F.; Gao, R.; Zhang, L.F.; Xie, S.H.; Liu, Z.; Du, J.L.; Chen, S.H.; Hong, M.H.; et al. Hepatitis B virus infection and risk of nasopharyngeal carcinoma in southern China. Cancer Epidemiol. Biomark. Prev. 2015, 24, 1766–1773. [Google Scholar] [CrossRef] [PubMed]
- Datta, S. An overview of molecular epidemiology of hepatitis B virus (HBV) in India. Virol. J. 2008, 19, 156. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, S. Hepatitis B virus: Significance of genotypes. J. Viral Hepat. 2005, 12, 111–124. [Google Scholar] [CrossRef] [PubMed]
- El-Serag, H.B. Epidemiology of viral hepatitis and hepatocellular carcinoma. Gastroenterology 2012, 142, 1264–1273. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.J.; Yang, H.I.; Iloeje, U.H. Hepatitis B virus DNA levels and outcomes in chronic hepatitis B. Hepatology 2009, 49, S72–S84. [Google Scholar] [CrossRef] [PubMed]
- Ni, Y.H.; Chang, M.H.; Wang, K.J.; Hsu, H.Y.; Chen, H.L.; Kao, J.H.; Yeh, S.H.; Jeng, Y.M.; Tsai, K.S.; Chen, D.S. Clinical relevance of hepatitis B virus genotype in children with chronic infection and hepatocellular carcinoma. Gastroenterology 2004, 127, 1733–1738. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.I.; Yeh, S.H.; Chen, P.J.; Iloeje, U.H.; Jen, C.L.; Su, J.; Wang, L.Y.; Lu, S.N.; You, S.L.; Chen, D.S.; et al. Associations between hepatitis B virus genotype and mutants and the risk of hepatocellular carcinoma. J. Natl. Cancer Inst. 2008, 100, 1134–1143. [Google Scholar] [CrossRef] [PubMed]
- Fang, Z.L.; Sabin, C.A.; Dong, B.Q.; Ge, L.Y.; Wei, S.C.; Chen, Q.Y.; Fang, K.X.; Yang, J.Y.; Wang, X.Y.; Harrison, T.J. HBV A1762T, G1764A mutations are a valuable biomarker for identifying a subset of male HBsAg carriers at extremely high risk of hepatocellular carcinoma: A prospective study. Am. J. Gastroenterol. 2008, 103, 2254–2262. [Google Scholar] [CrossRef] [PubMed]
- Lau, C.C.; Sun, T.; Ching, A.K.; He, M.; Li, J.W.; Wong, A.M.; Co, N.N.; Chan, A.W.; Li, P.S.; Lung, R.W.; et al. Viral–human chimeric transcript predisposes risk to liver cancer development and progression. Cancer Cell 2014, 25, 335–349. [Google Scholar] [CrossRef] [PubMed]
- Sung, W.K.; Zheng, H.; Li, S.; Chen, R.; Liu, X.; Li, Y.; Lee, N.P.; Lee, W.H.; Ariyaratne, P.N.; Tennakoon, C.; et al. Genome-wide survey of recurrent HBV integration in hepatocellular carcinoma. Nat. Genet. 2012, 44, 765–769. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Ghosh, S.; Dasgupta, D.; Ghosh, A.; Datta, S.; Sikdar, N.; Datta, S.; Chowdhury, A.; Banerjee, S. Hepatitis B Virus X Protein Upregulates hELG1/ATAD5 Expression through E2F1 in Hepatocellular Carcinoma. Int. J. Biol. Sci. 2016, 12, 30–41. [Google Scholar] [CrossRef] [PubMed]
- Lamontagne, J.; Steel, L.F.; Bouchard, M.J. Hepatitis B virus and microRNAs: Complex interactions affecting hepatitis B virus replication and hepatitis B virus-associated diseases. World J. Gastroenterol. 2015, 21, 7375–7399. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; You, X.; Chi, X.; Wang, T.; Ye, L.; Niu, J.; Zhang, X. Hepatitis B virus X protein mutant HBxΔ127 promotes proliferation of hepatoma cells through up-regulating miR-215 targeting PTPRT. Biochem. Biophys. Res. Commun. 2014, 444, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Tsai, W.L.; Chung, R.T. Viral hepatocarcinogenesis. Oncogene 2010, 29, 2309–2324. [Google Scholar] [CrossRef] [PubMed]
- Cheng, B.; Zheng, Y.; Guo, X.; Wang, Y.; Liu, C. Hepatitis B viral X protein alters the biological features and expressions of DNA repair enzymes in LO2 cells. Liver Int. 2010, 30, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Kew, M.C. Hepatitis B virus x protein in the pathogenesis of hepatitis B virus-induced hepatocellular carcinoma. J. Gastroenterol. Hepatol. 2011, 26, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Singh, R.; Massey, A.C.; Kane, S.S.; Kaushik, S.; Grant, T.; Xiang, Y.; Cuervo, A.M.; Czaja, M.J. Loss of Macroautophagy Promotes or Prevents Fibroblast Apoptosis Depending on the Death Stimulus. J. Biol. Chem. 2008, 283, 4766–4777. [Google Scholar] [CrossRef] [PubMed]
- Sir, D.; Tian, Y.; Chen, W.; Ann, D.K.; Yen, T.-S.B.; Ou, J.J. The early autophagic pathway is activated by hepatitis B virus and required for viral DNA replication. Proc. Natl. Acad. Sci. USA 2010, 107, 4383–4388. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Sir, D.; Kuo, C.; Ann, D.K.; Ou, J.J. Autophagy Required for Hepatitis B Virus Replication in Transgenic Mice. J. Virol. 2011, 85, 13453–13456. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.T.; Chen, G.G.; Hu, B.G.; Zhang, Z.Y.; Yun, J.P.; He, M.L.; Lai, P.B.S. Hepatitis B virus x protein induces autophagy via activating death-associated protein kinase. J. Viral Hepat. 2014, 21, 642–649. [Google Scholar] [CrossRef] [PubMed]
- Wirth, M.; Joachim, J.; Tooze, S.A. Autophagosome formation—The role of ULK1 and Beclin1-PI3KC3 complexes in setting the stage. Semin. Cancer Biol. 2013, 23, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Levin-Salomon, V.; Bialik, S.; Kimchi, A. DAP-kinase and autophagy. Apoptosis 2014, 19, 346–356. [Google Scholar] [CrossRef] [PubMed]
- Takamura, A.; Komatsu, M.; Hara, T.; Sakamoto, A.; Kishi, C.; Waguri, S.; Eishi, Y.; Hino, O.; Tanaka, K.; Mizushima, N. Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 2011, 25, 795–800. [Google Scholar] [CrossRef] [PubMed]
- Kotsafti, A.; Farinati, F.; Cardin, R.; Cillo, U.; Nitti, D.; Bortolami, M. Autophagy and apoptosis-related genes in chronic liver disease and hepatocellular carcinoma. BMC Gastroenterol. 2012, 12, 118. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Zhang, H.; Gu, C.; Yin, J.; He, Y.; Xie, J.; Cao, G. Associations Between Hepatitis B Virus Mutations and the Risk of Hepatocellular Carcinoma: A Meta-Analysis. J. Natl. Cancer Inst. 2009, 101, 1066–1082. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.W.; Yang, F.C.; Lu, H.Q.; Zhang, J.S. Hepatocellular carcinoma and hepatitis B surface protein. World J. Gastroenterol. 2016, 22, 1943–1952. [Google Scholar] [CrossRef] [PubMed]
- Pollicino, T.; Cacciola, I.; Saffioti, F.; Raimondo, G. Hepatitis B virus PreS/S gene variants: Pathobiology and clinical implications. J. Hepatol. 2014, 61, 408–417. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, Inflammation, and Cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [PubMed]
- Hanada, T.; Kobayashi, T.; Chinen, T.; Saeki, K.; Takaki, H.; Koga, K.; Minoda, Y.; Sanada, T.; Yoshioka, T.; Mimata, H.; et al. IFNgamma-dependent, spontaneous development of colorectal carcinomas in SOCS1-deficient mice. J. Exp. Med. 2006, 203, 1391–1397. [Google Scholar] [CrossRef] [PubMed]
- DeNardo, D.G.; Barreto, J.B.; Andreu, P.; Vasquez, L.; Tawfik, D.; Kolhatkar, N.; Coussens, L.M. CD4(+) T cells regulate pulmonary metastasis of mammary carcinomas by enhancing protumor properties of macrophages. Cancer Cell 2009, 16, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Yi, T.; Kortylewski, M.; Pardoll, D.M.; Zeng, D.; Yu, H. IL-17 can promote tumor growth through an IL-6-Stat3 signaling pathway. J. Exp. Med. 2009, 206, 1457–1464. [Google Scholar] [CrossRef] [PubMed]
- Luedde, T.; Schwabe, R.F. NF-κB in the liver—Linking injury, fibrosis and hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zheng, B.; Han, Q.; Zhang, C.; Tian, Z.; Zhang, J. Targeting blockage of STAT3 inhibits hepatitis B virus-related hepatocellular carcinoma. Cancer Biol. Ther. 2016, 17, 449–456. [Google Scholar] [CrossRef] [PubMed]
- He, G.; Yu, G.Y.; Temkin, V.; Ogata, H.; Kuntzen, C.; Sakurai, T.; Sieghart, W.; Peck-Radosavljevic, M.; Leffert, H.L.; Karin, M. Hepatocyte IKKβ/NF-κB inhibits tumor promotion and progression by preventing oxidative stress-driven STAT3 activation. Cancer Cell 2010, 17, 286–297. [Google Scholar] [CrossRef] [PubMed]
- Mohandas, K.M. Hepatitis B associated hepatocellular carcinoma: Epidemiology, diagnosis and treatment. Hepat. B Annu. 2004, 1, 140–152. [Google Scholar]
- Zhang, Y.Q.; Guo, J.S. Antiviral therapies for hepatitis B virus-related hepatocellular carcinoma. World J. Gastroenterol. 2015, 21, 3860–3866. [Google Scholar] [CrossRef] [PubMed]
- Raza, A.; Sood, G.K. Hepatocellular carcinoma review: Current treatment, and evidence-based medicine. World J. Gastroenterol. 2014, 20, 4115–4127. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.L.; Forner, A.; et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.L.; Kang, Y.K.; Chen, Z.; Tsao, C.J.; Qin, S.; Kim, J.S.; Luo, R.; Feng, J.; Ye, S.; Yang, T.S.; et al. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: A phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2009, 10, 25–34. [Google Scholar] [CrossRef]
- Bruix, J.; Qin, S.; Merle, P.; Granito, A.; Huang, Y.H.; Bodoky, G.; Pracht, M.; Yokosuka, O.; Rosmorduc, O.; Breder, V.; et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 389, 56–66. [Google Scholar] [CrossRef]
- Niu, B.; Hann, H.W. Hepatitis B Virus–Related Hepatocellular Carcinoma: Carcinogenesis, Prevention, and Treatment. In Updates in Liver Cancer; Abdeldayem, H., Ed.; InTech: Rijeka, Croatia, 2017. [Google Scholar]
- Sarin, S.K.; Kumar, M.; Lau, G.K.; Abbas, Z.; Chan, H.L.; Chen, C.J.; Chen, D.S.; Chen, H.L.; Chen, P.J.; Chien, R.N.; et al. Asian-Pacific clinical practice guidelines on the management of hepatitis B: A 2015 update. Hepatol. Int. 2016, 10, 1–98. [Google Scholar] [CrossRef] [PubMed]
- European Association for the Study of the Liver. EASL 2017 Clinical Practice Guidelines on the management of hepatitis B virus infection. J. Hepatol. 2017, 67, 370–398. [Google Scholar] [CrossRef]
- Terrault, N.A.; Bzowej, N.H.; Chang, K.M.; Hwang, J.P.; Jonas, M.M.; Murad, M.H. AASLD guidelines for treatment of chronic hepatitis B. Hepatology 2016, 63, 261–283. [Google Scholar] [CrossRef] [PubMed]
- Lok, A.S.; McMahon, B.J.; Brown, R.S.; Wong, J.B.; Ahmed, A.T.; Farah, W.; Almasri, J.; Alahdab, F.; Benkhadra, K.; Mouchli, M.A.; et al. Antiviral therapy for chronic hepatitis B viral infection in adults: A systematic review and meta-analysis. Hepatology 2016, 63, 284–306. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.Y.; Chien, R.N.; Liaw, Y.F. Hepatitis B virus-related decompensated liver cirrhosis: Benefits of antiviral therapy. J. Hepatol. 2012, 57, 442–450. [Google Scholar] [CrossRef] [PubMed]
- Singal, A.K.; Fontana, R.J. Meta-analysis: Oral anti-viral agents in adults with decompensated hepatitis B virus cirrhosis. Aliment. Pharmacol. Ther. 2012, 35, 674–689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.L.; Morgan, T.R. The Natural History of Hepatitis C Virus (HCV) Infection. Int. J. Med. Sci. 2006, 3, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Petruzziello, A.; Marigliano, S.; Loquercio, G.; Cozzolino, A.; Cacciapuoti, C. Global epidemiology of hepatitis C virus infection: An up-date of the distribution and circulation of hepatitis C virus genotypes. World J. Gastroenterol. 2016, 22, 7824–7840. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.S.; D’Souza, L.S.; Jacobson, I.M. Hepatitis C—A Clinical Review. J. Med. Virol. 2016, 88, 1844–1855. [Google Scholar] [CrossRef] [PubMed]
- Goossens, N.; Hoshida, Y. Hepatitis C virus-induced hepatocellular carcinoma. Clin. Mol. Hepatol. 2015, 21, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Westbrook, R.H.; Dusheiko, G. Natural history of hepatitis C. J. Hepatol. 2014, 61, S58–S68. [Google Scholar] [CrossRef] [PubMed]
- Bruno, S.; Crosignani, A.; Roffi, L.; De Lisi, S.; Rossi, S.; Boccaccio, V.; Zermiani, P.; Mondelli, M.; Maisonneuve, P. P465 SVR is associated with no risk reduction of HCC development in patients with HCV-related cirrhosis. A prospective, up to 23-years, cohort follow-up study. J. Hepatol. 2014, 60, S224. [Google Scholar] [CrossRef]
- Fiorino, S.; Bacchi-Reggiani, L.; de Biase, D.; Fornelli, A.; Masetti, M.; Tura, A.; Grizzi, F.; Zanello, M.; Mastrangelo, L.; Lombardi, R.; et al. Possible association between hepatitis C virus and malignancies different from hepatocellular carcinoma: A systematic review. World J. Gastroenterol. 2015, 21, 12896–12953. [Google Scholar] [CrossRef] [PubMed]
- Arcaini, L.; Merli, M.; Volpetti, S.; Rattotti, S.; Gotti, M.; Zaja, F. Indolent B-Cell Lymphomas Associated with HCV Infection: Clinical and Virological Features and Role of Antiviral Therapy. Clin. Dev. Immunol. 2012, 2012, 638185. [Google Scholar] [CrossRef] [PubMed]
- Tilly, H.; Gomes da Silva, M.; Vitolo, U.; Jack, A.; Meignan, M.; Lopez-Guillermo, A.; Walewski, J.; André, M.; Johnson, P.W.; Pfreundschuh, M.; et al. Diffuse large B-cell lymphoma (DLBCL): ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2015, 26, v116–v125. [Google Scholar] [CrossRef] [PubMed]
- El-Maadawy, E.A.; Talaat, R.M.; Sadek, R.F.; El-Sherbini, S.M.; Abdel-Bary, N.; Abdel-Aziz, A.A. Hepatitis C Virus Associations with Non Hodgkin’s Lymphoma: Insights on Inflammation/Angiogenesis and CD Markers. Asian Pac. J. Cancer Prev. 2016, 17, 4415–4420. [Google Scholar] [PubMed]
- De Re, V.; Caggiari, L.; Garziera, M.; De Zorzi, M.; Repetto, O. Molecular signature in HCV-positive lymphomas. Clin. Dev. Immunol. 2012, 2012, 623465. [Google Scholar] [CrossRef] [PubMed]
- Sukowati, C.H.; El-Khobar, K.E.; Ie, S.I.; Anfuso, B.; Muljono, D.H.; Tiribelli, C. Significance of hepatitis virus infection in the oncogenic initiation of hepatocellular carcinoma. World J. Gastroenterol. 2016, 22, 1497–1512. [Google Scholar] [CrossRef] [PubMed]
- Lemon, S.M.; McGivern, D.R. Is Hepatitis C Virus Carcinogenic? Gastroenterology 2012, 142, 1274–1278. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.H.; Xie, Y.T.; Cai, Y.P.; Ren, J.; Ma, T. Effects of hepatitis C virus core protein and nonstructural protein 4B on the Wnt/β-catenin pathway. BMC Microbiol. 2017, 17, 124. [Google Scholar] [CrossRef] [PubMed]
- McGivern, D.R.; Masaki, T.; Lovell, W.; Hamlett, C.; Saalau-Bethell, S.; Graham, B. Protease Inhibitors Block Multiple Functions of the NS3/4A Protease-Helicase during the Hepatitis C Virus Life Cycle. J. Virol. 2015, 89, 5362–5370. [Google Scholar] [CrossRef] [PubMed]
- Gouttenoire, J.; Montserret, R.; Paul, D.; Castillo, R.; Meister, S.; Bartenschlager, R.; Penin, F.; Moradpour, D. Aminoterminal Amphipathic α-Helix AH1 of Hepatitis C Virus Nonstructural Protein 4B Possesses a Dual Role in RNA Replication and Virus Production. PLoS Pathog. 2014, 10, e1004501. [Google Scholar] [CrossRef] [PubMed]
- Scheel, T.K.H.; Rice, C.M. Understanding the hepatitis C virus life cycle paves the way for highly effective therapies. Nat. Med. 2013, 19, 837–849. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, Z.; Tang, J.; Tang, R.; Shan, X.; Zhang, W.; Chen, Q.; Zhou, F.; Chen, K.; Huang, A.; et al. Hepatitis C virus core protein activates Wnt/β-catenin signaling through multiple regulation of upstream molecules in the SMMC-7721 cell line. Arch. Virol. 2011, 156, 1013–1023. [Google Scholar] [CrossRef] [PubMed]
- Park, C.Y.; Choi, S.H.; Kang, S.M.; Kang, J.I.; Ahn, B.Y.; Kim, H.; Jung, G.; Choi, K.Y.; Hwang, S.B. Nonstructural 5A protein activates beta-catenin signaling cascades: Implication of hepatitis C virus-induced liver pathogenesis. J. Hepatol. 2009, 51, 853–864. [Google Scholar] [CrossRef] [PubMed]
- Vilchez, V.; Turcios, L.; Marti, F.; Gedaly, R. Targeting Wnt/β-catenin pathway in hepatocellular carcinoma treatment. World J. Gastroenterol. 2016, 22, 823–832. [Google Scholar] [CrossRef] [PubMed]
- Vescovo, T.; Refolo, G.; Vitagliano, G.; Fimia, G.M.; Piacentini, M. Molecular mechanisms of hepatitis C virus–induced hepatocellular carcinoma. Clin. Microbiol. Infect. 2016, 22, 853–861. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.K.; Jeng, K.S.; Machida, K.; Cheng, Y.S.; Lai, M.M. Hepatitis C virus NS3/4A protein interacts with ATM, impairs DNA repair and enhances sensitivity to ionizing radiation. Virology 2008, 370, 295–309. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Xiao, Z.; Wang, F. Hepatitis C Virus Nonstructural 5A Protein (HCV-NS5A) Inhibits Hepatocyte Apoptosis through the NF-κb/miR-503/bcl-2 Pathway. Mol. Cells 2017, 40, 202–210. [Google Scholar] [CrossRef] [PubMed]
- Higgs, M.R.; Chouteau, P.; Lerat, H. ‘Liver let die’: Oxidative DNA damage and hepatotropic viruses. J. Gen. Virol. 2014, 95, 991–1004. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L. Mechanisms of hepatic fibrogenesis. Gastroenterology 2008, 134, 1655–1669. [Google Scholar] [CrossRef] [PubMed]
- Abe, M.; Koga, H.; Yoshida, T.; Masuda, H.; Iwamoto, H.; Sakata, M.; Hanada, S.; Nakamura, T.; Taniguchi, E.; Kawaguchi, T.; et al. Hepatitis C virus core protein upregulates the expression of vascular endothelial growth factor via the nuclear factor-κB/hypoxia-inducible factor-1α axis under hypoxic conditions. Hepatol. Res. 2012, 42, 591–600. [Google Scholar] [CrossRef] [PubMed]
- Vrancken, K.; Paeshuyse, J.; Liekens, S. Angiogenic activity of hepatitis B and C viruses. Antivir. Chem. Chemother. 2012, 22, 159–170. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, K.; Murata, M.; Yoshida, K.; Sekimoto, G.; Uemura, Y.; Sakaida, N.; Kaibori, M.; Kamiyama, Y.; Nishizawa, M.; Fujisawa, J.; et al. Chronic inflammation associated with hepatitis C virus infection perturbs hepatic transforming growth factor β signaling, promoting cirrhosis and hepatocellular carcinoma. Hepatology 2007, 46, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Elia, G.; Fallahi, P. Hepatocellular carcinoma and CXCR3 chemokines: A narrative review. Clin. Ther. 2017, 168, e37–e41. [Google Scholar] [CrossRef]
- Haybaeck, J.; Zeller, N.; Wolf, M.J.; Weber, A.; Wagner, U.; Kurrer, M.O.; Bremer, J.; Iezzi, G.; Graf, R.; Clavien, P.A.; et al. A lymphotoxin-driven pathway to hepatocellular carcinoma. Cancer Cell 2009, 16, 295–308. [Google Scholar] [CrossRef] [PubMed]
- Park, E.J.; Lee, J.H.; Yu, G.Y.; He, G.; Ali, S.R.; Holzer, R.G.; Osterreicher, C.H.; Takahashi, H.; Karin, M. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell 2010, 140, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Wolf, M.J.; Adili, A.; Piotrowitz, K.; Abdullah, Z.; Boege, Y.; Stemmer, K.; Ringelhan, M.; Simonavicius, N.; Egger, M.; Wohlleber, D.; et al. Metabolic activation of intrahepatic CD8+ T cells and NKT cells causes nonalcoholic steatohepatitis and liver cancer via cross-talk with hepatocytes. Cancer Cell 2014, 26, 549–564. [Google Scholar] [CrossRef] [PubMed]
- European Association for the Study of the Liver. EASL Recommendations on Treatment of Hepatitis C 2016. J. Hepatol. 2016, 66, 153–194. [Google Scholar] [CrossRef]
- Khan, F.; Peltekian, K.M.; Peterson, T.C. Effect of interferon-alpha, ribavirin, pentoxifylline, and interleukin-18 antibody on hepatitis C sera-stimulated hepatic stellate cell proliferation. J. Interferon Cytokine Res. 2008, 28, 643–651. [Google Scholar] [CrossRef] [PubMed]
- Zamor, P.J.; deLemos, A.S.; Russo, M.W. Viral hepatitis and hepatocellular carcinoma: Etiology and management. J. Gastrointest. Oncol. 2017, 8, 229–242. [Google Scholar] [CrossRef] [PubMed]
- Zeuzem, S.; Berg, T.; Moeller, B.; Hinrichsen, H.; Mauss, S.; Wedemeyer, H.; Sarrazin, C.; Hueppe, D.; Zehnter, E.; Manns, M.P. Expert opinion on the treatment of patients with chronic hepatitis C. J. Viral Hepat. 2009, 16, 75–90. [Google Scholar] [CrossRef] [PubMed]
- Serti, E.; Park, H.; Keane, M.; O’Keefe, A.C.; Rivera, E.; Liang, T.J.; Ghany, M.; Rehermann, B. Rapid decrease in hepatitis C viremia by direct acting antivirals improves the natural killer cell response to IFNα. Gut 2017, 66, 724–735. [Google Scholar] [CrossRef] [PubMed]
- The American Association for the Study of Liver Diseases and the Infectious Diseases Society of America (AASLD/IDSA). HCV Guidance: Recommendations for Testing, Managing, and Treating Hepatitis C. 2017. Available online: http://www.hcvguidelines.org/ (accessed on 10 August 2017).
- Gallo, R.C. The discovery of the first human retrovirus: HTLV-1 and HTLV-2. Retrovirology 2005, 2, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panfil, A.R.; Martinez, M.P.; Ratner, L.; Green, P.L. Human T-cell leukemia virus-associated malignancy. Curr. Opin. Virol. 2016, 20, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Osame, M.; Usuku, K.; Izumo, S.; Ijichi, N.; Amitani, H.; Igata, A.; Matsumoto, M.; Tara, M. HTLV-I associated myelopathy, a new clinical entity. Lancet 1986, 328, 1031–1032. [Google Scholar] [CrossRef]
- LaGrenade, L.; Hanchard, B.; Fletcher, V.; Cranston, B.; Blattner, W. Infective dermatitis of Jamaican children: A marker for HTLV-I infection. Lancet 1990, 336, 1345–1347. [Google Scholar] [CrossRef] [PubMed]
- Manns, A.; Hisada, M.; La Grenade, L. Human T-lymphotropic virus type I infection. Lancet 1999, 353, 1951–1958. [Google Scholar] [CrossRef]
- Gessain, A.; Cassar, O. Epidemiological Aspects and World Distribution of HTLV-1 Infection. Front. Microbiol. 2012, 3, 388. [Google Scholar] [CrossRef] [PubMed]
- Iwanaga, M.; Watanabe, T.; Yamaguchi, K. Adult T-cell leukemia: A review of epidemiological evidence. Front. Microbiol. 2012, 3, 322. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, J.E.; Osame, M.; Kubota, H.; Igata, A.; Nishitani, H.; Maeda, Y.; Khabbaz, R.F.; Janssen, R.S. The risk of development of HTLV-I-associated myelopathy/tropical spastic paraparesis among persons infected with HTLV-I. J. Acquir. Immune Defic. Syndr. 1990, 3, 1096–1101. [Google Scholar] [PubMed]
- Proietti, F.A.; Carneiro-Proietti, A.B.; Catalan-Soares, B.C.; Murphy, E.L. Global epidemiology of HTLV-I infection and associated diseases. Oncogene 2005, 24, 6058–6068. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, D.U.; Proietti, F.A.; Ribas, J.G.; Araújo, M.G.; Pinheiro, S.R.; Guedes, A.C.; Carneiro-Proietti, A.B. Epidemiology, treatment, and prevention of human T-cell leukemia virus type 1-associated diseases. Clin. Microbiol. Rev. 2010, 23, 577–589. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control (CDC). Human T-lymphotropic virus type I screening in volunteer blood donors—United States, 1989. MMWR Morb. Mortal. Wkly. Rep. 1990, 39, 915–921. [Google Scholar]
- Okochi, K.; Sato, H.; Hinuma, Y. A retrospective study on transmission of adult T cell leukemia virus by blood transfusion: Seroconversion in recipients. Vox Sang. 1984, 46, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Hino, S. Establishment of the milk-borne transmission as a key factor for the peculiar endemicity of human T-lymphotropic virus type 1 (HTLV-1): The ATL Prevention Program Nagasaki. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2011, 87, 152–166. [Google Scholar] [CrossRef] [PubMed]
- Roucoux, D.F.; Wang, B.; Smith, D.; Nass, C.C.; Smith, J.; Hutching, S.T.; Newman, B.; Lee, T.H.; Chafets, D.M.; Murphy, E.L.; et al. A prospective study of sexual transmission of human T lymphotropic virus (HTLV)-I and HTLV-II. J. Infect. Dis. 2005, 191, 1490–1497. [Google Scholar] [CrossRef] [PubMed]
- Kajiyama, W.; Kashiwagi, S.; Ikematsu, H.; Hayashi, J.; Nomura, H.; Okochi, K. Intrafamilial transmission of adult T cell leukemia virus. J. Infect. Dis. 1986, 154, 851–857. [Google Scholar] [CrossRef] [PubMed]
- Laperche, S.; Worms, B.; Pillonel, J. European Network of Transfusion Medecine Societies; Steering Committee. Blood safety strategies for human T-cell lymphotropic virus in Europe. Vox Sang. 2009, 96, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Bangham, C.R.; Ratner, L. How does HTLV-1 cause adult T-cell leukaemia/lymphoma (ATL)? Curr. Opin. Virol. 2015, 14, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Hinuma, Y.; Nagata, K.; Hanaoka, M.; Nakai, M.; Matsumoto, T.; Kinoshita, K.I.; Shirakawa, S.; Miyoshi, I. Adult T-cell leukemia: Antigen in an ATL cell line and detection of antibodies to the antigen in human sera. Proc. Natl. Acad. Sci. USA 1981, 78, 6476–6480. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Takezaki, T.; Oki, T.; Kawakami, K.; Yashiki, S.; Fujiyoshi, T.; Usuku, K.; Mueller, N.; Osame, M.; Miyata, K. Inhibitory effect of maternal antibody on mother-to-child transmission of human T-lymphotropic virus type I. The Mother-to-Child Transmission Study Group. Int. J. Cancer 1991, 49, 673–677. [Google Scholar] [CrossRef] [PubMed]
- Do Valle, A.C.; Galhardo, M.C.; Leite, A.C.; Araújo, A.Q.; Cuzzi-Maya, T.; Maceira, J.P.; de Ameida Dobbin, J. Adult T-cell leukemia/lymphoma associated with HTLV-1 infection in a Brazilian adolescent. Rev. Inst. Med. Trop. Sao Paulo 2001, 43, 283–286. [Google Scholar] [CrossRef] [PubMed]
- Shimoyama, M. Diagnostic criteria and classification of clinical subtypes of adult T-cell leukaemia-lymphoma. A report from the Lymphoma Study Group (1984–87). Br. J. Haematol. 1991, 79, 428–437. [Google Scholar] [CrossRef] [PubMed]
- Kato, K.; Akashi, K. Recent Advances in Therapeutic Approaches for Adult T-cell Leukemia/Lymphoma. Viruses 2015, 7, 6604–6612. [Google Scholar] [CrossRef] [PubMed]
- Katsuya, H.; Ishitsuka, K.; Utsunomiya, A.; Hanada, S.; Eto, T.; Moriuchi, Y.; Saburi, Y.; Miyahara, M.; Sueoka, E.; Uike, N.; et al. Treatment and survival among 1594 patients with ATL. Blood 2015, 126, 2570–2577. [Google Scholar] [CrossRef] [PubMed]
- Phillips, A.A.; Shapira, I.; Willim, R.D.; Sanmugarajah, J.; Solomon, W.B.; Horwitz, S.M.; Savage, D.G.; Bhagat, G.; Soff, G.; Zain, J.M.; et al. A critical analysis of prognostic factors in North American patients with human T-cell lymphotropic virus type-1-associated adult T-cell leukemia/lymphoma: A multicenter clinicopathologic experience and new prognostic score. Cancer 2010, 116, 3438–3446. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T. Adult T-cell leukemia: Molecular basis for clonal expansion and transformation of HTLV-1-infected T cells. Blood 2017, 129, 1071–1081. [Google Scholar] [CrossRef] [PubMed]
- Ishitsuka, K.; Tamura, K. Human T-cell leukaemia virus type I and adult T-cell leukaemia-lymphoma. Lancet Oncol. 2014, 15, e517–e526. [Google Scholar] [CrossRef]
- Sawada, Y.; Hino, R.; Hama, K.; Ohmori, S.; Fueki, H.; Yamada, S.; Fukamachi, S.; Tajiri, M.; Kubo, R.; Yoshioka, M.; et al. Type of skin eruption is an independent prognostic indicator for adult T-cell leukemia/lymphoma. Blood 2011, 117, 3961–3967. [Google Scholar] [CrossRef] [PubMed]
- Boxus, M.; Willems, L. Mechanisms of HTLV-1 persistence and transformation. Br. J. Cancer 2009, 101, 1497–1501. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.H.; Coligan, J.E.; Homma, T.; McLane, M.F.; Tachibana, N.; Essex, M. Human T-cell leukemia virus-associated membrane antigens: Identity of the major antigens recognized after virus infection. Proc. Natl. Acad. Sci. USA 1984, 81, 3856–3860. [Google Scholar] [CrossRef] [PubMed]
- Nam, S.H.; Kidokoro, M.; Shida, H.; Hatanaka, M. Processing of gag precursor polyprotein of human T-cell leukemia virus type I by virus-encoded protease. J. Virol. 1988, 62, 3718–3728. [Google Scholar] [PubMed]
- Frederiks, W.M.; James, J. Isolation and separation of rat liver nuclei. Acta Histochem. Suppl. 1979, 20, 147–158. [Google Scholar] [PubMed]
- Bartoe, J.T.; Albrecht, B.; Collins, N.D.; Robek, M.D.; Ratner, L.; Green, P.L.; Lairmore, M.D. Functional role of pX open reading frame II of human T-lymphotropic virus type 1 in maintenance of viral loads in vivo. J. Virol. 2000, 74, 1094–1100. [Google Scholar] [CrossRef] [PubMed]
- Collins, N.D.; Newbound, G.C.; Albrecht, B.; Beard, J.L.; Ratner, L.; Lairmore, M.D. Selective ablation of human T-cell lymphotropic virus type 1 p12I reduces viral infectivity in vivo. Blood 1998, 91, 4701–4707. [Google Scholar] [PubMed]
- Silverman, L.R.; Phipps, A.J.; Montgomery, A.; Ratner, L.; Lairmore, M.D. Human T-cell lymphotropic virus type 1 open reading frame II-encoded p30II is required for in vivo replication: Evidence of in vivo reversion. J. Virol. 2004, 78, 3837–3845. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T. The Role of HBZ in HTLV-1-Induced Oncogenesis. Viruses 2016, 8, 34. [Google Scholar] [CrossRef] [PubMed]
- Currer, R.; Van Duyne, R.; Jaworski, E.; Guendel, I.; Sampey, G.; Das, R.; Narayanan, A.; Kashanchi, F. HTLV tax: A fascinating multifunctional co-regulator of viral and cellular pathways. Front. Microbiol. 2012, 3, 406. [Google Scholar] [CrossRef] [PubMed]
- Pais-Correia, A.M.; Sachse, M.; Guadagnini, S.; Robbiati, V.; Lasserre, R.; Gessain, A.; Gout, O.; Alcover, A.; Thoulouze, M.I. Biofilm-like extracellular viral assemblies mediate HTLV-1 cell-to-cell transmission at virological synapses. Nat. Med. 2010, 16, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Meytes, D.; Schochat, B.; Lee, H.; Nadel, G.; Sidi, Y.; Cerney, M.; Swanson, P.; Shaklai, M.; Kilim, Y.; Elgat, M. Serological and molecular survey for HTLV-I infection in a high-risk Middle Eastern group. Lancet 1990, 336, 1533–1535. [Google Scholar] [CrossRef]
- Nogueira, C.M.; Cavalcanti, M.; Schechter, M.; Ferreira, O.C. Human T lymphotropic virus type I and II infections in healthy blood donors from Rio de Janeiro, Brazil. Vox Sang. 1996, 70, 47–48. [Google Scholar] [CrossRef] [PubMed]
- Nejmeddine, M.; Bangham, C.R. The HTLV-1 Virological Synapse. Viruses 2010, 2, 1427–1447. [Google Scholar] [CrossRef] [PubMed]
- Derse, D.; Heidecker, G. Virology. Forced entry–or does HTLV-I have the key? Science 2003, 299, 1670–1671. [Google Scholar] [CrossRef] [PubMed]
- Pique, C.; Jones, K.S. Pathways of cell-cell transmission of HTLV-1. Front. Microbiol. 2012, 3, 378. [Google Scholar] [CrossRef] [PubMed]
- Manel, N.; Kim, F.J.; Kinet, S.; Taylor, N.; Sitbon, M.; Battini, J.L. The ubiquitous glucose transporter GLUT-1 is a receptor for HTLV. Cell 2003, 115, 449–459. [Google Scholar] [CrossRef]
- Jones, K.S.; Petrow-Sadowski, C.; Bertolette, D.C.; Huang, Y.; Ruscetti, F.W. Heparan sulfate proteoglycans mediate attachment and entry of human T-cell leukemia virus type 1 virions into CD4+ T cells. J. Virol. 2005, 79, 12692–12702. [Google Scholar] [CrossRef] [PubMed]
- Lambert, S.; Bouttier, M.; Vassy, R.; Seigneuret, M.; Petrow-Sadowski, C.; Janvier, S.; Heveker, N.; Ruscetti, F.W.; Perret, G.; Jones, K.S.; et al. HTLV-1 uses HSPG and neuropilin-1 for entry by molecular mimicry of VEGF165. Blood 2009, 113, 5176–5185. [Google Scholar] [CrossRef] [PubMed]
- Jones, K.S.; Lambert, S.; Bouttier, M.; Bénit, L.; Ruscetti, F.W.; Hermine, O.; Pique, C. Molecular aspects of HTLV-1 entry: Functional domains of the HTLV-1 surface subunit (SU) and their relationships to the entry receptors. Viruses 2011, 3, 794–810. [Google Scholar] [CrossRef] [PubMed]
- Ghez, D.; Lepelletier, Y.; Jones, K.S.; Pique, C.; Hermine, O. Current concepts regarding the HTLV-1 receptor complex. Retrovirology 2010, 7, 99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliveira, P.D.; Farre, L.; Bittencourt, A.L. Adult T-cell leukemia/lymphoma. Rev. Assoc. Med. Bras. 2016, 62, 691–700. [Google Scholar] [CrossRef] [PubMed]
- Igakura, T.; Stinchcombe, J.C.; Goon, P.K.; Taylor, G.P.; Weber, J.N.; Griffiths, G.M.; Tanaka, Y.; Osame, M.; Bangham, C.R. Spread of HTLV-I between lymphocytes by virus-induced polarization of the cytoskeleton. Science 2003, 299, 1713–1716. [Google Scholar] [CrossRef] [PubMed]
- Fan, N.; Gavalchin, J.; Paul, B.; Wells, K.H.; Lane, M.J.; Poiesz, B.J. Infection of peripheral blood mononuclear cells and cell lines by cell-free human T-cell lymphoma/leukemia virus type I. J. Clin. Microbiol. 1992, 30, 905–910. [Google Scholar] [PubMed]
- Derse, D.; Hill, S.A.; Lloyd, P.A.; Chung, H.K.; Morse, B.A. Examining human T-lymphotropic virus type 1 infection and replication by cell-free infection with recombinant virus vectors. J. Virol. 2001, 75, 8461–8468. [Google Scholar] [CrossRef] [PubMed]
- Mazurov, D.; Ilinskaya, A.; Heidecker, G.; Lloyd, P.; Derse, D. Quantitative comparison of HTLV-1 and HIV-1 cell-to-cell infection with new replication dependent vectors. PLoS Pathog. 2010, 6, e1000788. [Google Scholar] [CrossRef] [PubMed]
- Nejmeddine, M.; Negi, V.S.; Mukherjee, S.; Tanaka, Y.; Orth, K.; Taylor, G.P.; Bangham, C.R. HTLV-1-Tax and ICAM-1 act on T-cell signal pathways to polarize the microtubule-organizing center at the virological synapse. Blood 2009, 114, 1016–1025. [Google Scholar] [CrossRef] [PubMed]
- Mesnard, J.M.; Barbeau, B.; Césaire, R.; Péloponèse, J.M. Roles of HTLV-1 basic Zip Factor (HBZ) in Viral Chronicity and Leukemic Transformation. Potential New Therapeutic Approaches to Prevent and Treat HTLV-1-Related Diseases. Viruses 2015, 7, 6490–6505. [Google Scholar] [CrossRef] [PubMed]
- Nicot, C. HTLV-I Tax-Mediated Inactivation of Cell Cycle Checkpoints and DNA Repair Pathways Contribute to Cellular Transformation: “A Random Mutagenesis Model”. J. Cancer Sci. 2015, 2. [Google Scholar] [CrossRef]
- Boxus, M.; Twizere, J.C.; Legros, S.; Dewulf, J.F.; Kettmann, R.; Willems, L. The HTLV-1 Tax interactome. Retrovirology 2008, 5, 76. [Google Scholar] [CrossRef] [PubMed]
- Kannian, P.; Green, P.L. Human T Lymphotropic Virus Type 1 (HTLV-1): Molecular Biology and Oncogenesis. Viruses 2010, 2, 2037–2077. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.C.; Yamaoka, S. Activation of NF-kappaB by HTLV-I and implications for cell transformation. Oncogene 2005, 24, 5952–5964. [Google Scholar] [CrossRef] [PubMed]
- Grassmann, R.; Aboud, M.; Jeang, K.T. Molecular mechanisms of cellular transformation by HTLV-1 Tax. Oncogene 2005, 24, 5976–5985. [Google Scholar] [CrossRef] [PubMed]
- Mulloy, J.C.; Kislyakova, T.; Cereseto, A.; Casareto, L.; LoMonico, A.; Fullen, J.; Lorenzi, M.V.; Cara, A.; Nicot, C.; et al. Human T-cell lymphotropic/leukemia virus type 1 Tax abrogates p53-induced cell cycle arrest and apoptosis through its CREB/ATF functional domain. J. Virol. 1998, 72, 8852–8860. [Google Scholar] [PubMed]
- Kibler, K.V.; Jeang, K.T. CREB/ATF-dependent repression of cyclin a by human T-cell leukemia virus type 1 Tax protein. J. Virol. 2001, 75, 2161–2173. [Google Scholar] [CrossRef] [PubMed]
- Nicot, C.; Opavsky, R.; Mahieux, R.; Johnson, J.M.; Brady, J.N.; Wolff, L.; Franchini, G. Tax oncoprotein trans-represses endogenous B-myb promoter activity in human T cells. AIDS Res. Hum. Retrovir. 2000, 16, 1629–1632. [Google Scholar] [CrossRef] [PubMed]
- Fujii, M.; Niki, T.; Mori, T.; Matsuda, T.; Matsui, M.; Nomura, N.; Seiki, M. HTLV-1 Tax induces expression of various immediate early serum responsive genes. Oncogene 1991, 6, 1023–1029. [Google Scholar] [PubMed]
- Takeda, S.; Maeda, M.; Morikawa, S.; Taniguchi, Y.; Yasunaga, J.; Nosaka, K.; Tanaka, Y.; Matsuoka, M. Genetic and epigenetic inactivation of tax gene in adult T-cell leukemia cells. Int. J. Cancer 2004, 109, 559–567. [Google Scholar] [CrossRef] [PubMed]
- Fujikawa, D.; Nakagawa, S.; Hori, M.; Kurokawa, N.; Soejima, A.; Nakano, K.; Yamochi, T.; Nakashima, M.; Kobayashi, S.; Tanaka, Y.; et al. Polycomb-dependent epigenetic landscape in adult T-cell leukemia. Blood 2016, 127, 1790–1802. [Google Scholar] [CrossRef] [PubMed]
- Bazarbachi, A.; Suarez, F.; Fields, P.; Hermine, O. How I treat adult T-cell leukemia/lymphoma. Blood 2011, 118, 1736–1745. [Google Scholar] [CrossRef] [PubMed]
- Satou, Y.; Yasunaga, J.; Yoshida, M.; Matsuoka, M. HTLV-I basic leucine zipper factor gene mRNA supports proliferation of adult T cell leukemia cells. Proc. Natl. Acad. Sci. USA 2006, 103, 720–725. [Google Scholar] [CrossRef] [PubMed]
- Mesnard, J.M.; Barbeau, B.; Devaux, C. HBZ, a new important player in the mystery of adult T-cell leukemia. Blood 2006, 108, 3979–3982. [Google Scholar] [CrossRef] [PubMed]
- Lavorgna, A.; Harhaj, E.W. Regulation of HTLV-1 tax stability, cellular trafficking and NF-κB activation by the ubiquitin-proteasome pathway. Viruses 2014, 6, 3925–3943. [Google Scholar] [CrossRef] [PubMed]
- Zhi, H.; Yang, L.; Kuo, Y.L.; Ho, Y.K.; Shih, H.M.; Giam, C.Z. NF-κB hyper-activation by HTLV-1 tax induces cellular senescence, but can be alleviated by the viral anti-sense protein HBZ. PLoS Pathog. 2011, 7, e1002025. [Google Scholar] [CrossRef] [PubMed]
- Saito, M.; Matsuzaki, T.; Satou, Y.; Yasunaga, J.; Saito, K.; Arimura, K.; Matsuoka, M.; Ohara, Y. In vivo expression of the HBZ gene of HTLV-1 correlates with proviral load, inflammatory markers and disease severity in HTLV-1 associated myelopathy/tropical spastic paraparesis (HAM/TSP). Retrovirology 2009, 6, 19. [Google Scholar] [CrossRef] [PubMed]
- Usui, T.; Yanagihara, K.; Tsukasaki, K.; Murata, K.; Hasegawa, H.; Yamada, Y.; Kamihira, S. Characteristic expression of HTLV-1 basic zipper factor (HBZ) transcripts in HTLV-1 provirus-positive cells. Retrovirology 2008, 5, 34. [Google Scholar] [CrossRef] [PubMed]
- Ohshima, K. Molecular Pathology of Adult T-Cell Leukemia/Lymphoma. Oncology 2015, 89, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Nasr, R.; Marçais, A.; Hermine, O.; Bazarbachi, A. Overview of Targeted Therapies for Adult T-Cell Leukemia/Lymphoma. Methods Mol. Biol. 2017, 1582, 197–216. [Google Scholar] [CrossRef] [PubMed]
- Iwanaga, M.; Watanabe, T.; Utsunomiya, A.; Okayama, A.; Uchimaru, K.; Koh, K.R.; Ogata, M.; Kikuchi, H.; Sagara, Y.; Uozumi, K.; et al. Human T-cell leukemia virus type I (HTLV-1) proviral load and disease progression in asymptomatic HTLV-1 carriers: A nationwide prospective study in Japan. Blood 2010, 116, 1211–1219. [Google Scholar] [CrossRef] [PubMed]
- Macnamara, A.; Rowan, A.; Hilburn, S.; Kadolsky, U.; Fujiwara, H.; Suemori, K.; Yasukawa, M.; Taylor, G.; Bangham, C.R.; Asquith, B. HLA class I binding of HBZ determines outcome in HTLV-1 infection. PLoS Pathog. 2010, 6, e1001117. [Google Scholar] [CrossRef] [PubMed]
- Cook, L.; Melamed, A.; Yaguchi, H.; Bangham, C.R. The impact of HTLV-1 on the cellular genome. Curr. Opin. Virol. 2017, 26, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Hatta, Y.; Koeffler, H.P. Role of tumor suppressor genes in the development of adult T cell leukemia/lymphoma (ATLL). Leukemia 2002, 16, 1069–1085. [Google Scholar] [CrossRef] [PubMed]
- Sakashita, A.; Hattori, T.; Miller, C.W.; Suzushima, H.; Asou, N.; Takatsuki, K.; Koeffler, H.P. Mutations of the p53 gene in adult T-cell leukemia. Blood 1992, 79, 477–480. [Google Scholar] [PubMed]
- Cesarman, E.; Chadburn, A.; Inghirami, G.; Gaidano, G.; Knowles, D.M. Structural and functional analysis of oncogenes and tumor suppressor genes in adult T-cell leukemia/lymphoma shows frequent p53 mutations. Blood 1992, 80, 3205–3216. [Google Scholar] [PubMed]
- Hatta, Y.; Yamada, Y.; Tomonaga, M.; Koeffler, H.P. Extensive analysis of the retinoblastoma gene in adult T cell leukemia/lymphoma (ATL). Leukemia 1997, 11, 984–989. [Google Scholar] [CrossRef] [PubMed]
- Hangaishi, A.; Ogawa, S.; Imamura, N.; Miyawaki, S.; Miura, Y.; Uike, N.; Shimazaki, C.; Emi, N.; Takeyama, K.; Hirosawa, S.; et al. Inactivation of multiple tumor-suppressor genes involved in negative regulation of the cell cycle, MTS1/p16INK4A/CDKN2, MTS2/p15INK4B, p53, and Rb genes in primary lymphoid malignancies. Blood 1996, 87, 4949–4958. [Google Scholar] [PubMed]
- Tsukasaki, K.; Hermine, O.; Bazarbachi, A.; Ratner, L.; Ramos, J.C.; Harrington, W.; O’Mahony, D.; Janik, J.E.; Bittencourt, A.L.; Taylor, G.P.; et al. Definition, prognostic factors, treatment, and response criteria of adult T-cell leukemia-lymphoma: A proposal from an international consensus meeting. J. Clin. Oncol. 2009, 27, 453–459. [Google Scholar] [CrossRef] [PubMed]
- Marçais, A.; Suarez, F.; Sibon, D.; Bazarbachi, A.; Hermine, O. Clinical trials of adult T-cell leukaemia/lymphoma treatment. Leuk. Res. Treat. 2012, 2012, 932175. [Google Scholar] [CrossRef] [PubMed]
- Bazarbachi, A.; Plumelle, Y.; Carlos Ramos, J.; Tortevoye, P.; Otrock, Z.; Taylor, G.; Gessain, A.; Harrington, W.; Panelatti, G.; Hermine, O. Meta-analysis on the use of zidovudine and interferon-alfa in adult T-cell leukemia/lymphoma showing improved survival in the leukemic subtypes. J. Clin. Oncol. 2010, 28, 4177–4183. [Google Scholar] [CrossRef] [PubMed]
- Bazarbachi, A.; El-Sabban, M.E.; Nasr, R.; Quignon, F.; Awaraji, C.; Kersual, J.; Dianoux, L.; Zermati, Y.; Haidar, J.H.; Hermine, O.; et al. Arsenic trioxide and interferon-alpha synergize to induce cell cycle arrest and apoptosis in human T-cell lymphotropic virus type I-transformed cells. Blood 1999, 93, 278–283. [Google Scholar] [PubMed]
- El-Sabban, M.E.; Nasr, R.; Dbaibo, G.; Hermine, O.; Abboushi, N.; Quignon, F.; Ameisen, J.C.; Bex, F.; de Thé, H.; et al. Arsenic-interferon-alpha-triggered apoptosis in HTLV-I transformed cells is associated with tax down-regulation and reversal of NF-kappa B activation. Blood 2000, 96, 2849–2855. [Google Scholar] [PubMed]
- Nasr, R.; Rosenwald, A.; El-Sabban, M.E.; Arnulf, B.; Zalloua, P.; Lepelletier, Y.; Bex, F.; Hermine, O.; Staudt, L.; de Thé, H.; et al. Arsenic/interferon specifically reverses 2 distinct gene networks critical for the survival of HTLV-1-infected leukemic cells. Blood 2003, 101, 4576–4582. [Google Scholar] [CrossRef] [PubMed]
- Kchour, G.; Rezaee, R.; Farid, R.; Ghantous, A.; Rafatpanah, H.; Tarhini, M.; Kooshyar, M.M.; El Hajj, H.; Berry, F.; Mortada, M.; et al. The combination of arsenic, interferon-alpha, and zidovudine restores an “immunocompetent-like” cytokine expression profile in patients with adult T-cell leukemia lymphoma. Retrovirology 2013, 10, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waldmann, T.A.; White, J.D.; Goldman, C.K.; Top, L.; Grant, A.; Bamford, R.; Roessler, E.; Horak, I.D.; Zaknoen, S.; Kasten-Sportes, C. The interleukin-2 receptor: A target for monoclonal antibody treatment of human T-cell lymphotrophic virus I-induced adult T-cell leukemia. Blood 1993, 82, 1701–1712. [Google Scholar] [PubMed]
- Waldmann, T.A.; White, J.D.; Carrasquillo, J.A.; Reynolds, J.C.; Paik, C.H.; Gansow, O.A.; Brechbiel, M.W.; Jaffe, E.S.; Fleisher, T.A.; Goldman, C.K.; et al. Radioimmunotherapy of interleukin-2R alpha-expressing adult T-cell leukemia with Yttrium-90-labeled anti-Tac. Blood 1995, 86, 4063–4075. [Google Scholar] [PubMed]
- Ishida, T.; Joh, T.; Uike, N.; Yamamoto, K.; Utsunomiya, A.; Yoshida, S.; Saburi, Y.; Miyamoto, T.; Takemoto, S.; Suzushima, H.; et al. Defucosylated anti-CCR4 monoclonal antibody (KW-0761) for relapsed adult T-cell leukemia-lymphoma: A multicenter phase II study. J. Clin. Oncol. 2012, 30, 837–842. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Utsunomiya, A.; Tobinai, K.; Tsukasaki, K.; Uike, N.; Uozumi, K.; Yamaguchi, K.; Yamada, Y.; Hanada, S.; Tamura, K.; et al. Phase I study of KW-0761, a defucosylated humanized anti-CCR4 antibody, in relapsed patients with adult T-cell leukemia-lymphoma and peripheral T-cell lymphoma. J. Clin. Oncol. 2010, 28, 1591–1598. [Google Scholar] [CrossRef] [PubMed]
- Moura, I.C.; Lepelletier, Y.; Arnulf, B.; England, P.; Baude, C.; Beaumont, C.; Bazarbachi, A.; Benhamou, M.; Monteiro, R.C.; Hermine, O. A neutralizing monoclonal antibody (mAb A24) directed against the transferrin receptor induces apoptosis of tumor T lymphocytes from ATL patients. Blood 2004, 103, 1838–1845. [Google Scholar] [CrossRef] [PubMed]
- Marçais, A.; Suarez, F.; Sibon, D.; Frenzel, L.; Hermine, O.; Bazarbachi, A. Therapeutic options for adult T-cell leukemia/lymphoma. Curr. Oncol. Rep. 2013, 15, 457–464. [Google Scholar] [CrossRef] [PubMed]
- Ando, S.; Hasegawa, A.; Murakami, Y.; Zeng, N.; Takatsuka, N.; Maeda, Y.; Masuda, T.; Suehiro, Y.; Kannagi, M. HTLV-1 Tax-Specific CTL Epitope-Pulsed Dendritic Cell Therapy Reduces Proviral Load in Infected Rats with Immune Tolerance against Tax. J. Immunol. 2017, 198, 1210–1219. [Google Scholar] [CrossRef] [PubMed]
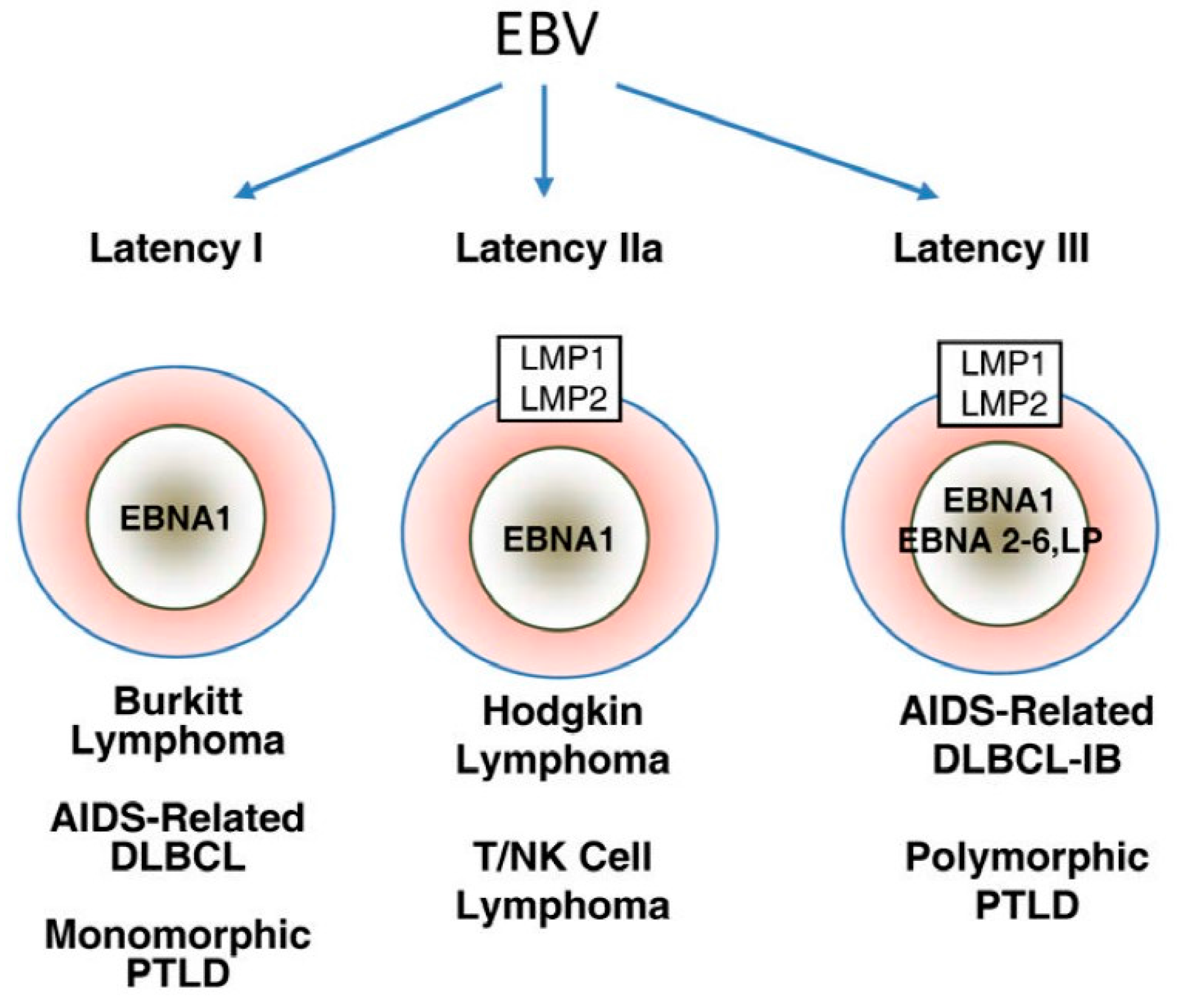
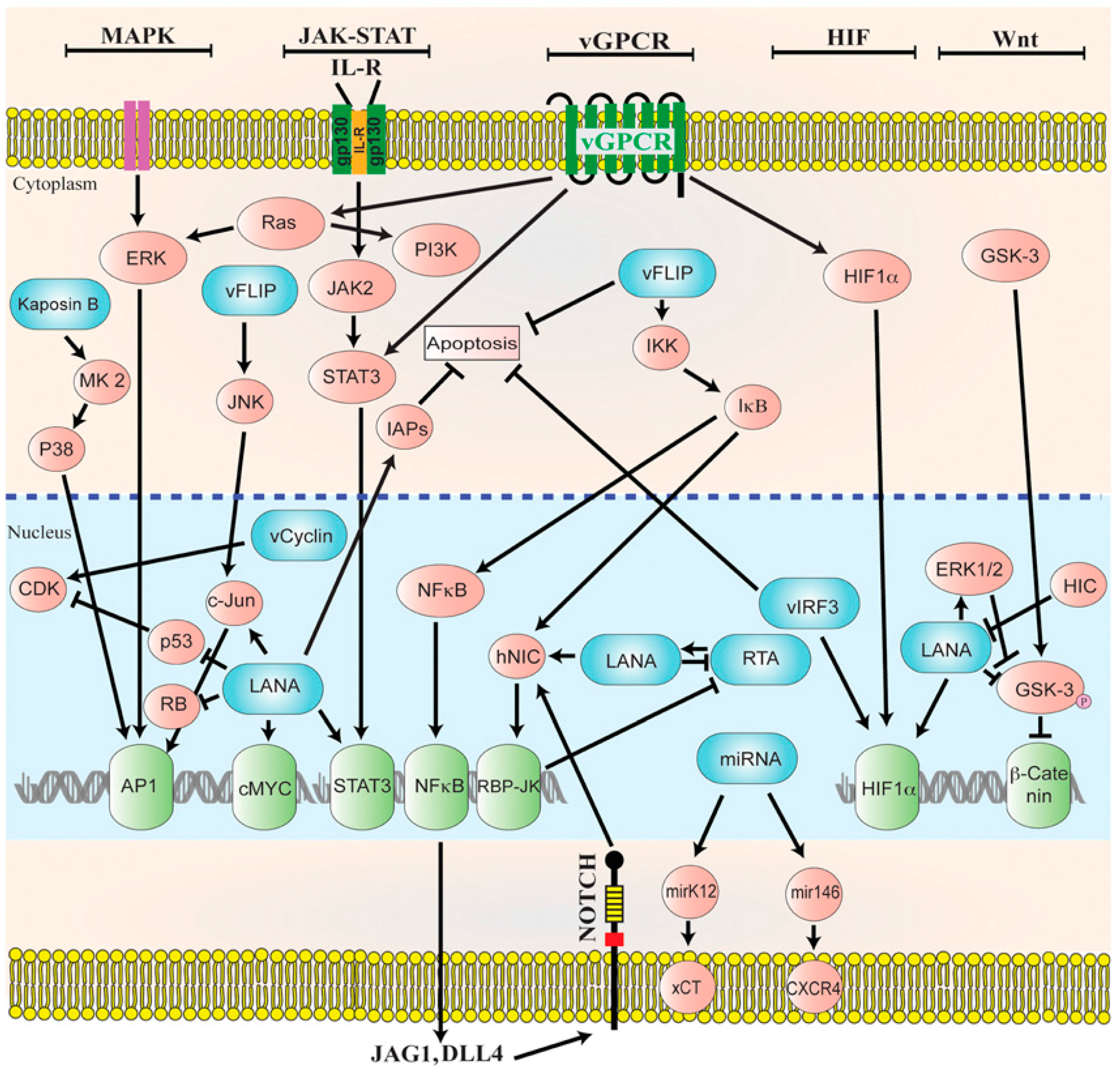
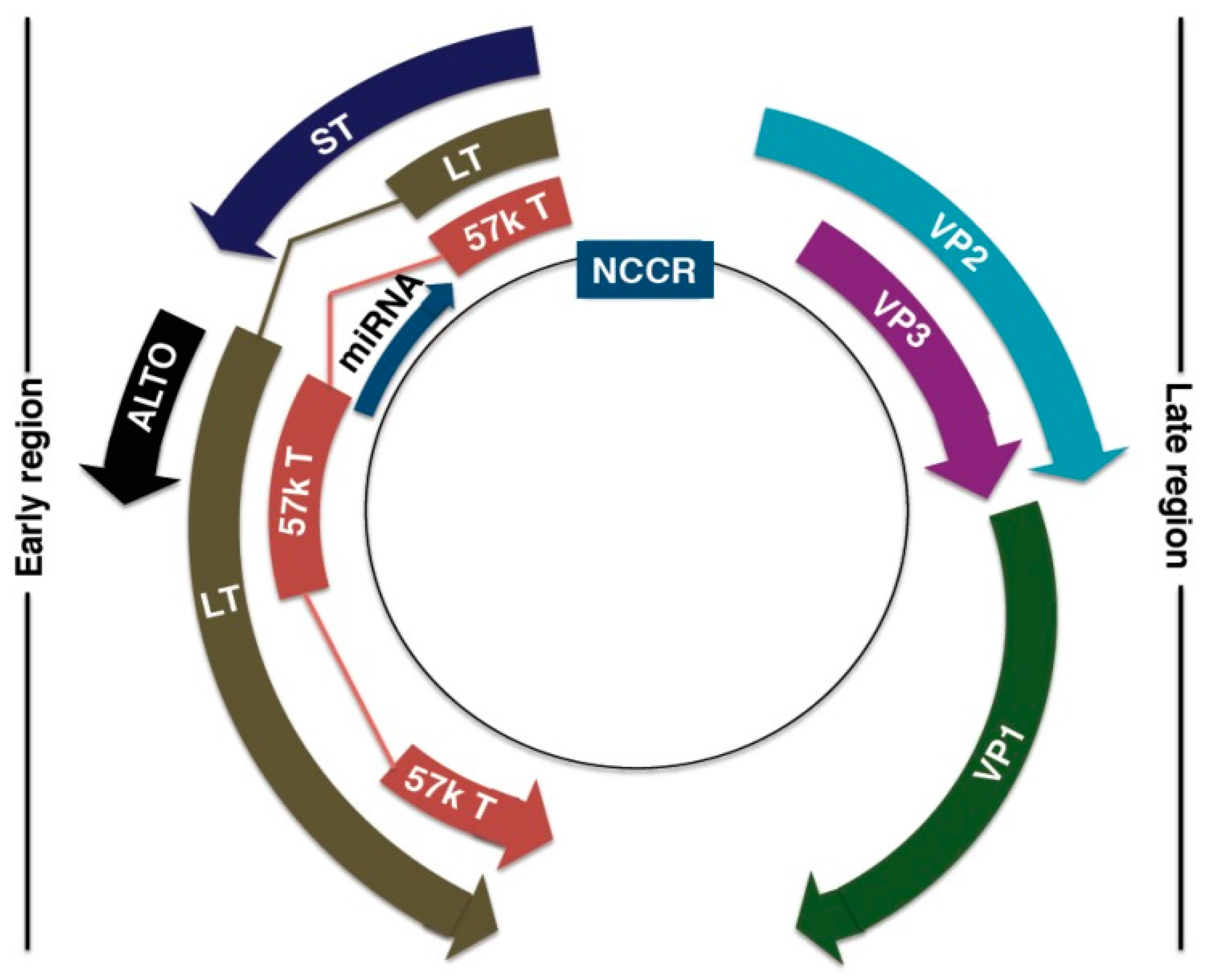
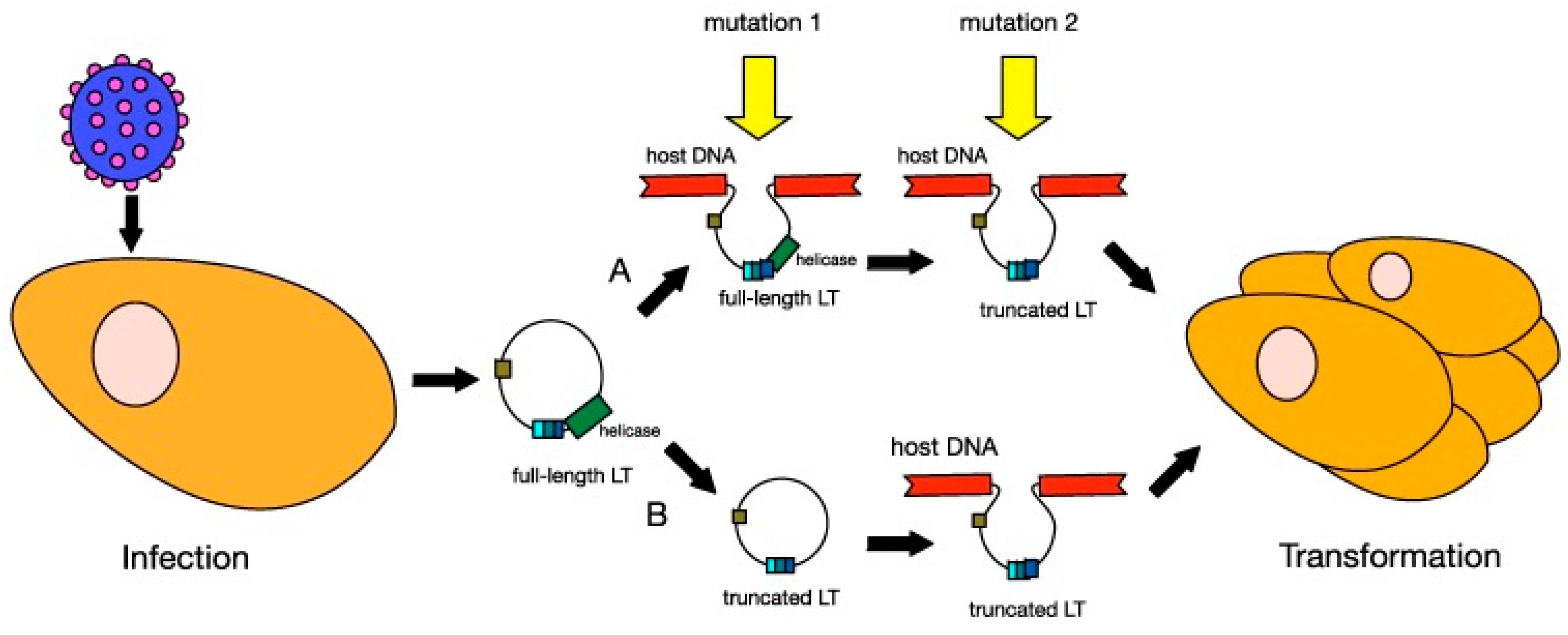

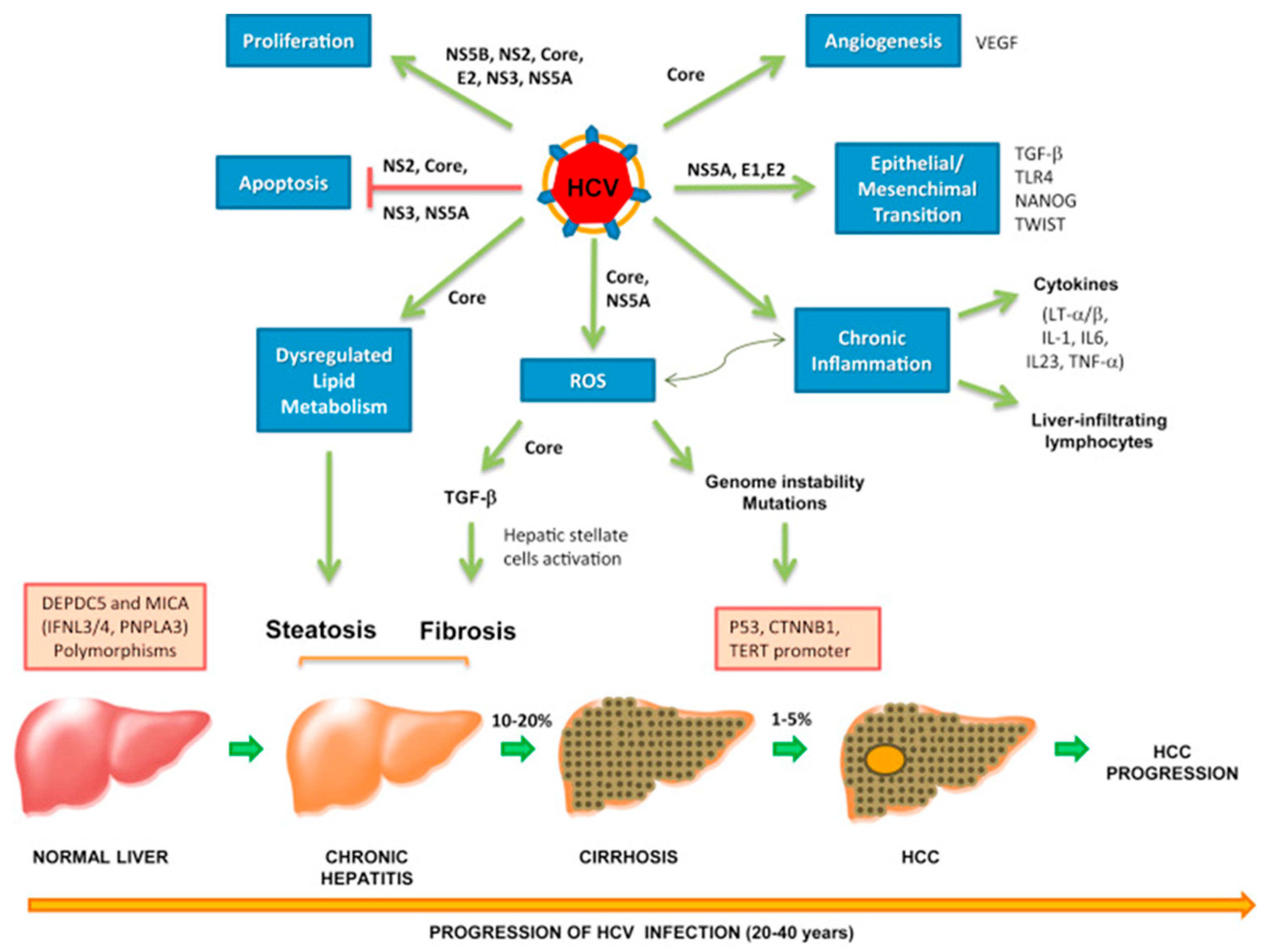
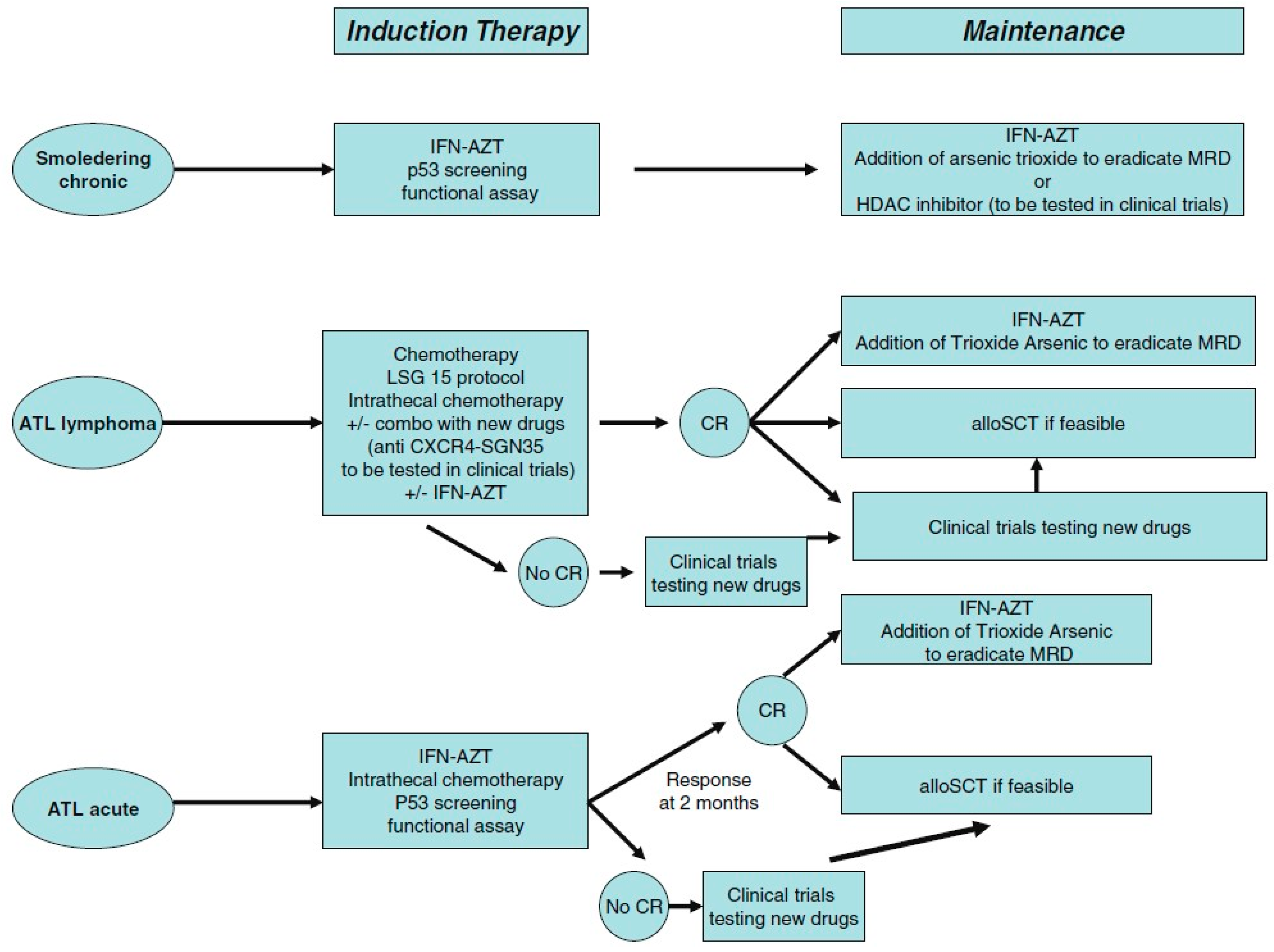

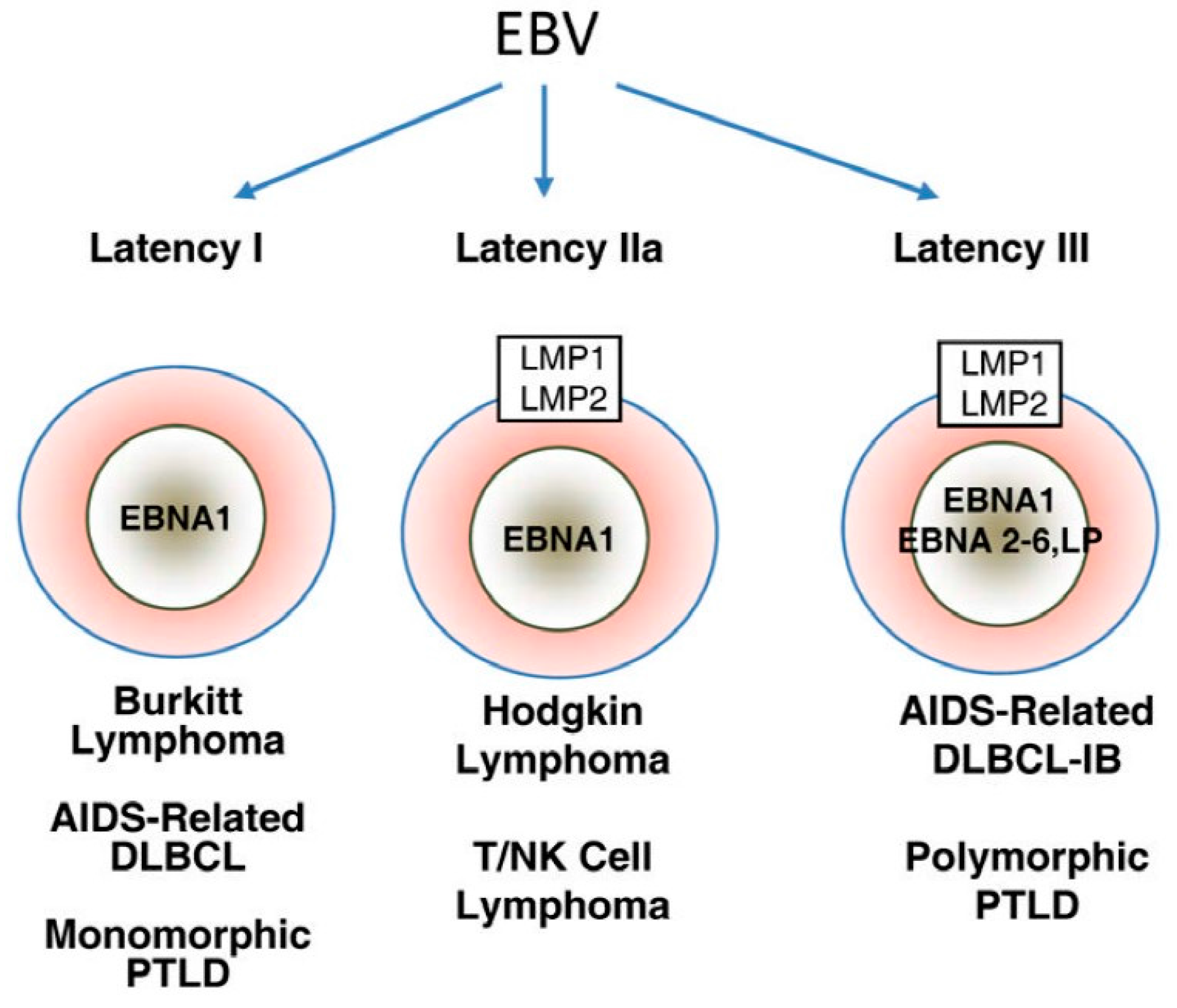{kind=link}
{kind=link}
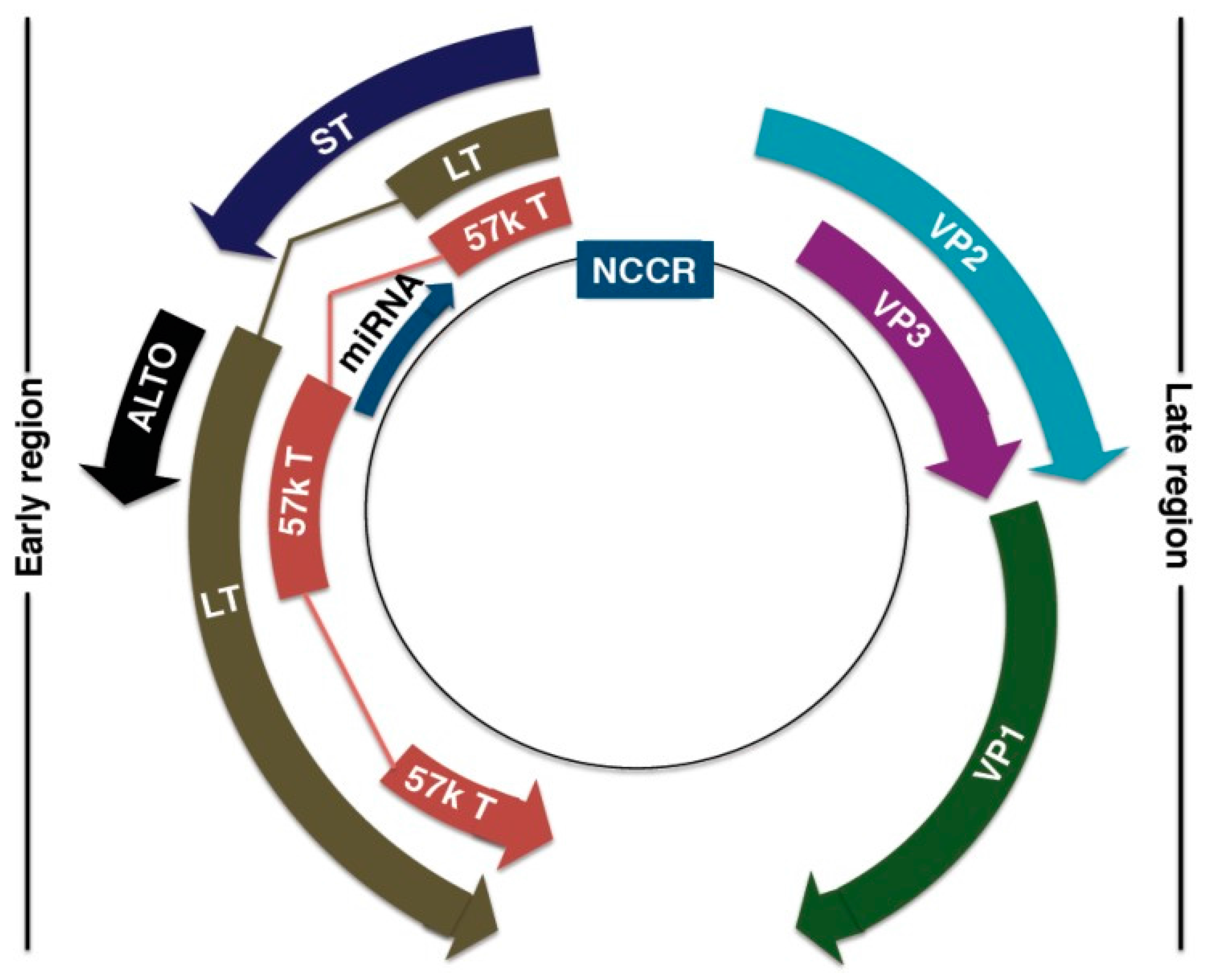{kind=link}
{kind=link}
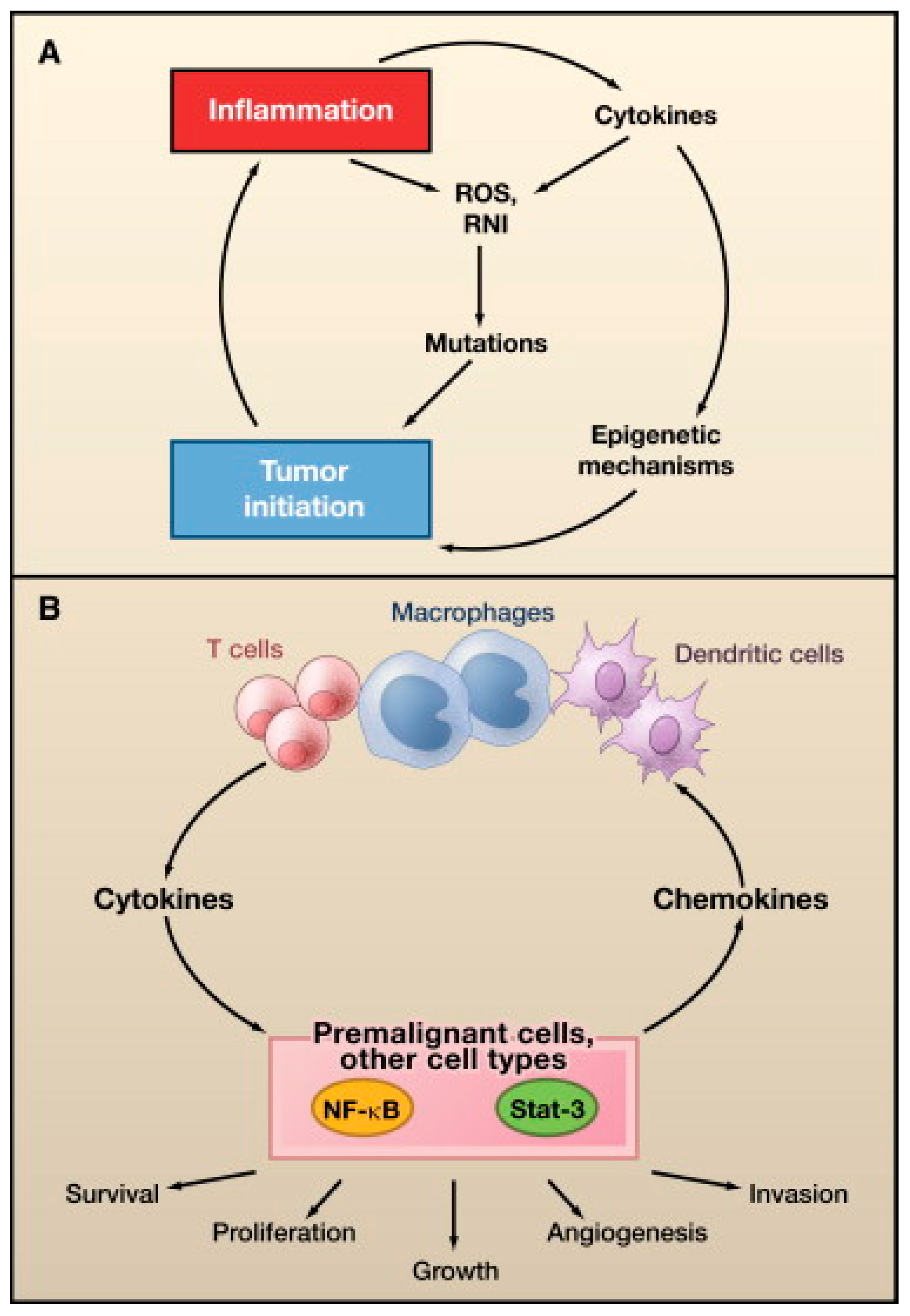{kind=link}
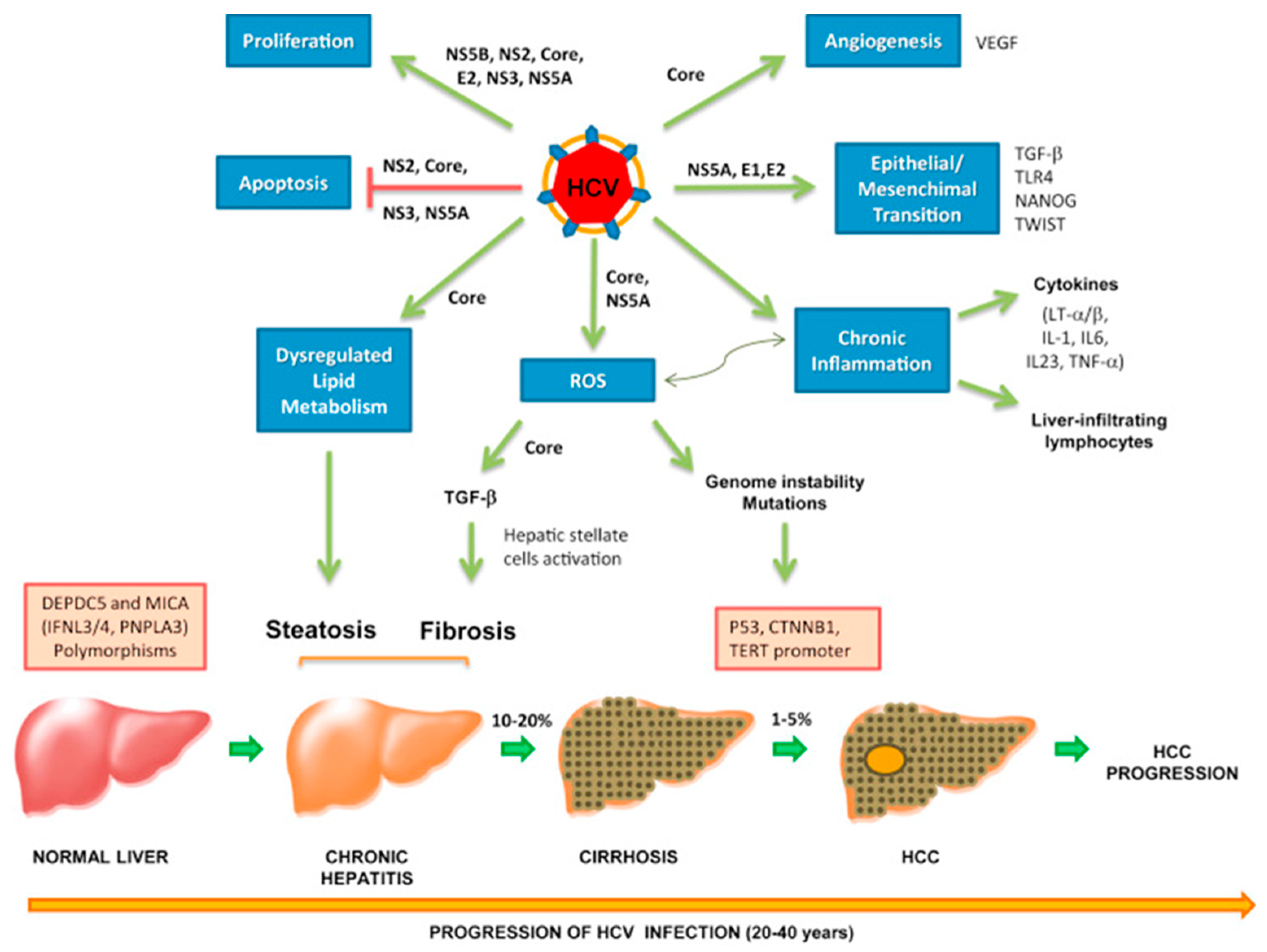{kind=link}
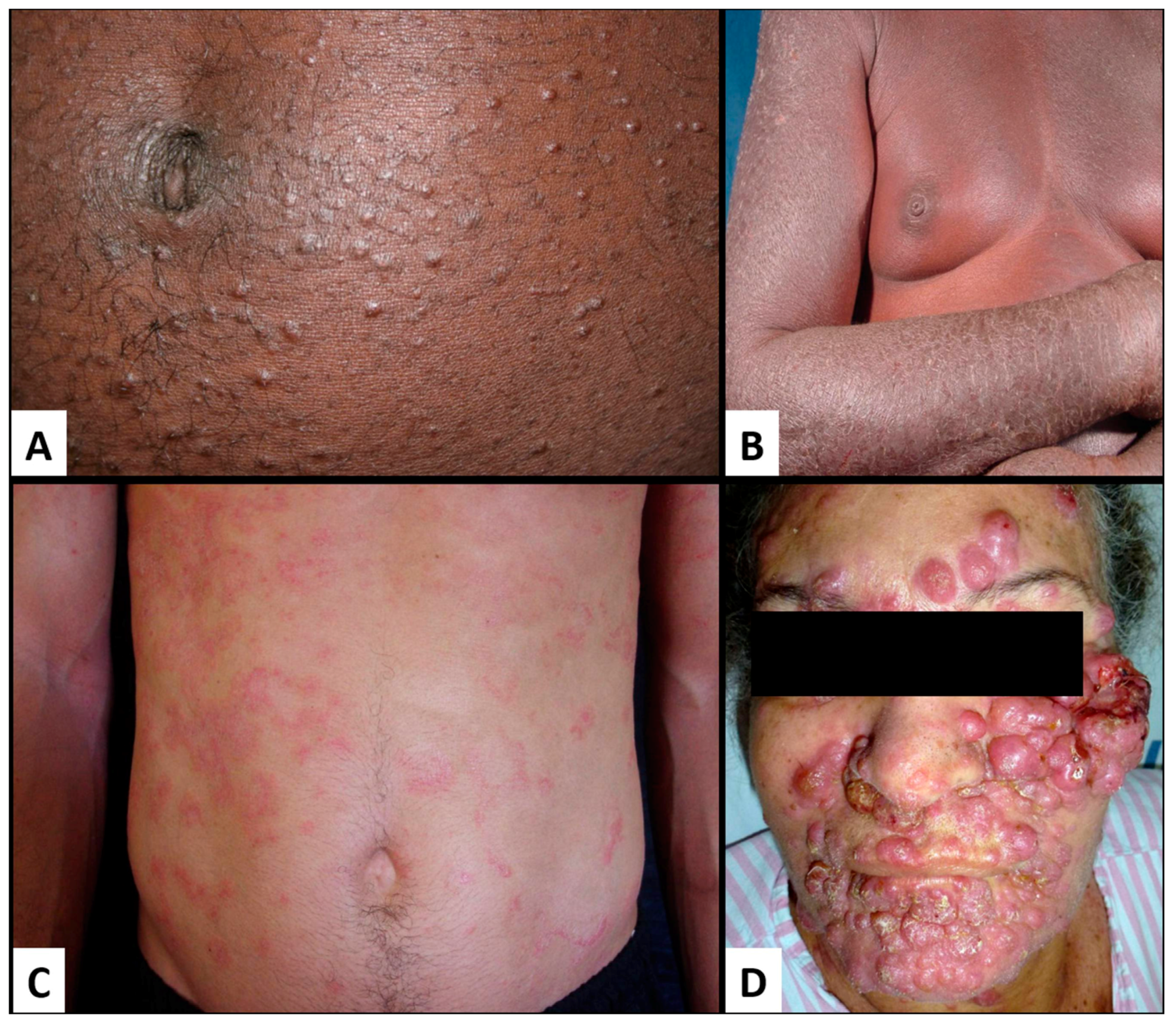{kind=link}
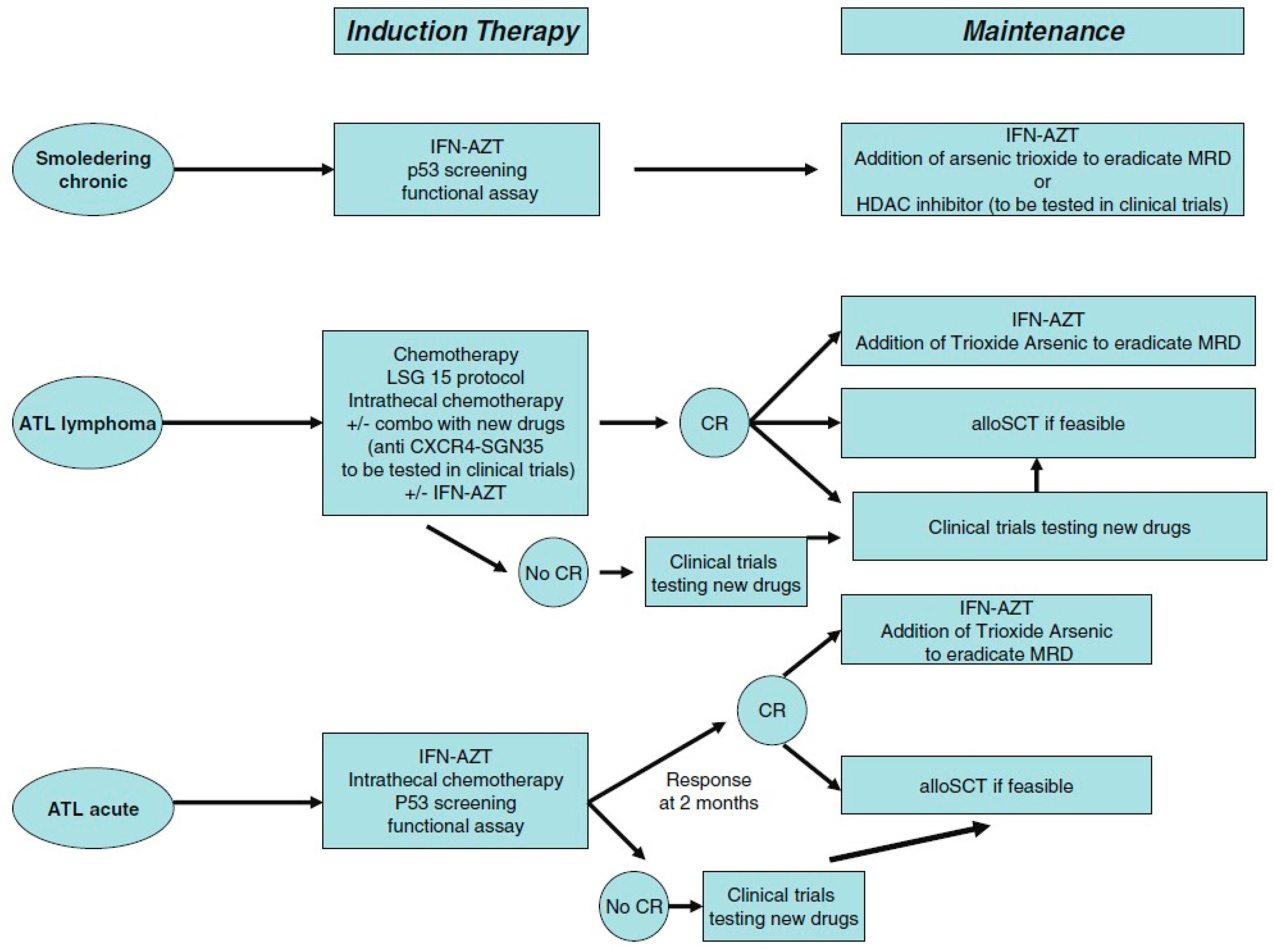{kind=link}
| Genotype | Treatment Options | Duration of Treatment | Strength of Evidence |
|---|---|---|---|
| Genotype 1a without/with compensated cirrhosis | Ledipasvir/sofosbuvir | 12 weeks | I, A |
| Velpatasvir/sofosbuvir | 12 weeks | I, A | |
| Grazoprevir/elbasvir | 12 weeks | I, A | |
| Paritaprevir/ritonavir/ombitasvir/dasabuvir ± ribavirin | 12 weeks/24 weeks | I, A | |
| Ledipasvir/sofosbuvir for patients who are non-black, HIV uninfected, without cirrhosis, and whose HCV RNA < 6 million IU/mL | 8 weeks | I, B | |
| Daclatasvir/sofosbuvir | 12 weeks/24 weeks | I, B/IIa, B | |
| Simeprevir/sofosbuvir ± ribavirin | 12 weeks/24 weeks | II, B | |
| Genotype 1b without/with compensated cirrhosis | Ledipasvir/sofosbuvir | 12 weeks | I, A |
| Velpatasvir/sofosbuvir | 12 weeks | I, A | |
| Grazoprevir/elbasvir | 12 weeks | I, A | |
| Paritaprevir/ritonavir/ombitasvir/dasabuvir | 12 weeks | I, A | |
| Ledipasvir/sofosbuvir for patients who are non-black, HIV uninfected, without cirrhosis, and whose HCV RNA < 6 million IU/mL | 8 weeks | I, B | |
| Daclatasvir/sofosbuvir | 12 weeks/24 weeks | I, B/IIa, B | |
| Simeprevir/sofosbuvir | 12 weeks/24 weeks | II, B | |
| Genotype 2 without/with compensated cirrhosis | Velpatasvir/sofosbuvir | 12 weeks | I, A |
| Sofosbuvir + weight-based RBV | 12 weeks/16 weeks | I, A/IIb, C | |
| Daclatasvir/sofosbuvir | 12 weeks/16–24 weeks | IIa, B | |
| Genotype 3 without/with compensated cirrhosis | Velpatasvir/sofosbuvir | 12 weeks | I, A |
| Daclatasvir/sofosbuvir ± ribavirin | 12 weeks/24 weeks | I, A/IIa, B | |
| Sofosbuvir + weight-based RBV + weekly peg-IFN | 12 weeks | I, A | |
| Sofosbuvir + weight-based RBV | 24 weeks | I, B | |
| Genotype 4 without/with compensated cirrhosis | Ledipasvir/sofosbuvir | 12 weeks | I, A/IIa, B |
| Velpatasvir/sofosbuvir | 12 weeks | I, A | |
| Grazoprevir/elbasvir | 12 weeks | I, A/IIa, B | |
| Paritaprevir/ritonavir/ombitasvir + weight-based RBV | 12 weeks | I, B | |
| Sofosbuvir + weight-based RBV | 24 weeks | IIa, B | |
| Sofosbuvir + weight-based RBV + weekly peg-IFN | 12 weeks | II, B | |
| Simeprevir/sofosbuvir | 12 weeks | II, B | |
| Daclatasvir/sofosbuvir | 12 weeks | II, B | |
| Genotype 5 and 6 with and without cirrhosis | Velpatasvir/sofosbuvir | 12 weeks | I, B |
| Daclatasvir/sofosbuvir | 12 weeks | I, B | |
| Ledipasvir/sofosbuvir | 12 weeks | IIa, B | |
| Sofosbuvir + weight-based RBV + weekly peg-IFN | 12 weeks | IIa, B |
| Smoldering | Chronic | Lymphoma | Acute | |
|---|---|---|---|---|
| Lymphocyte count (×103/L) | <4 | ≥4 | <4 | High |
| Flower cells (%) | <5 | ≥5 | ≤1 | High |
| LDH level | ≤1.5 times ULN | <2.5 times ULN | High | High |
| Ca2+ level | Normal | Normal | High | High |
| Skin and/or lung involvement | +/− | +/− | +/− | +/− |
| Lymph node involvement | No | +/− | Yes | +/− |
| Spleen/liver involvement | No | +/− | +/− | +/− |
| Central nervous system/bone/pleural/ascites | No | No | +− | +/− |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mui, U.N.; Haley, C.T.; Tyring, S.K. Viral Oncology: Molecular Biology and Pathogenesis. J. Clin. Med. 2017, 6, 111. https://doi.org/10.3390/jcm6120111
Mui UN, Haley CT, Tyring SK. Viral Oncology: Molecular Biology and Pathogenesis. Journal of Clinical Medicine. 2017; 6(12):111. https://doi.org/10.3390/jcm6120111
Chicago/Turabian StyleMui, Uyen Ngoc, Christopher T. Haley, and Stephen K. Tyring. 2017. "Viral Oncology: Molecular Biology and Pathogenesis" Journal of Clinical Medicine 6, no. 12: 111. https://doi.org/10.3390/jcm6120111