Clinical and Histopathologic Features of Interstitial Lung Disease in Erdheim–Chester Disease

 , , , and
, , , and
Abstract
:1. Introduction
2. Experimental Section
2.1. Patients and Molecular Studies
2.2. Pulmonary Physiology Testing
2.3. Radiologic Imaging
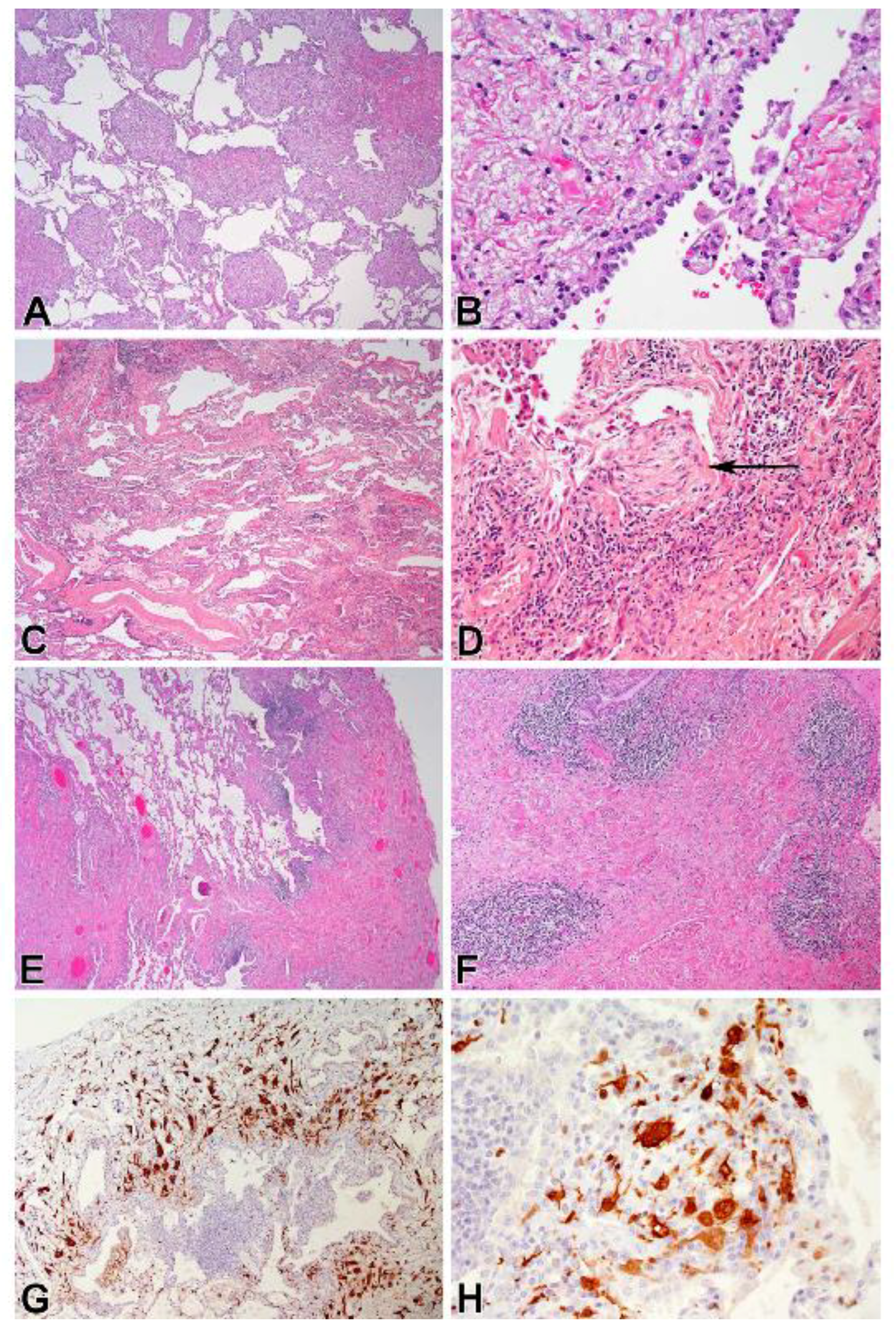
2.4. Histopathology and Immunohistochemistry
2.5. Statistical Analysis
3. Results
3.1. Clinical Features
3.2. Pulmonary Function Measurements
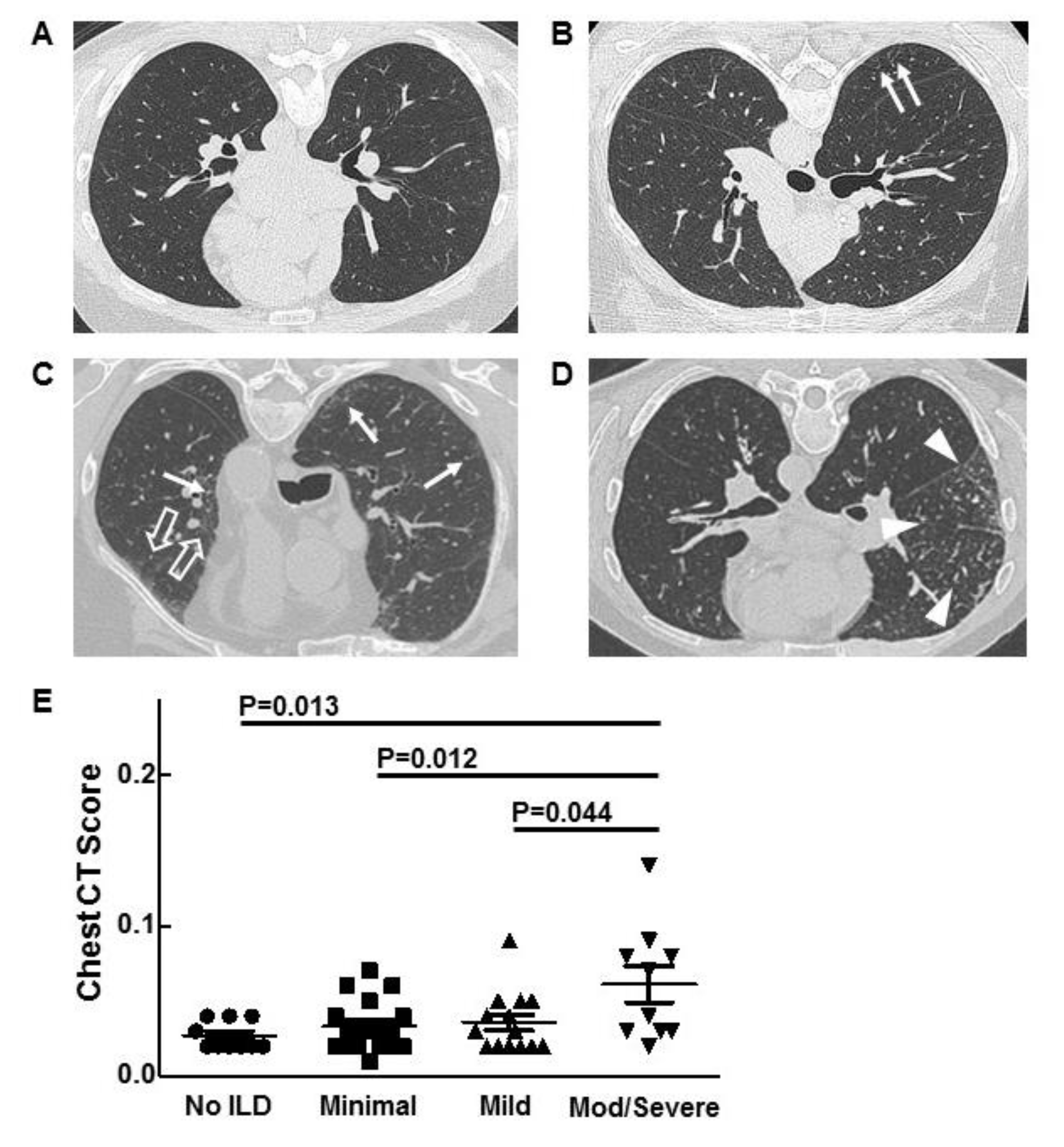
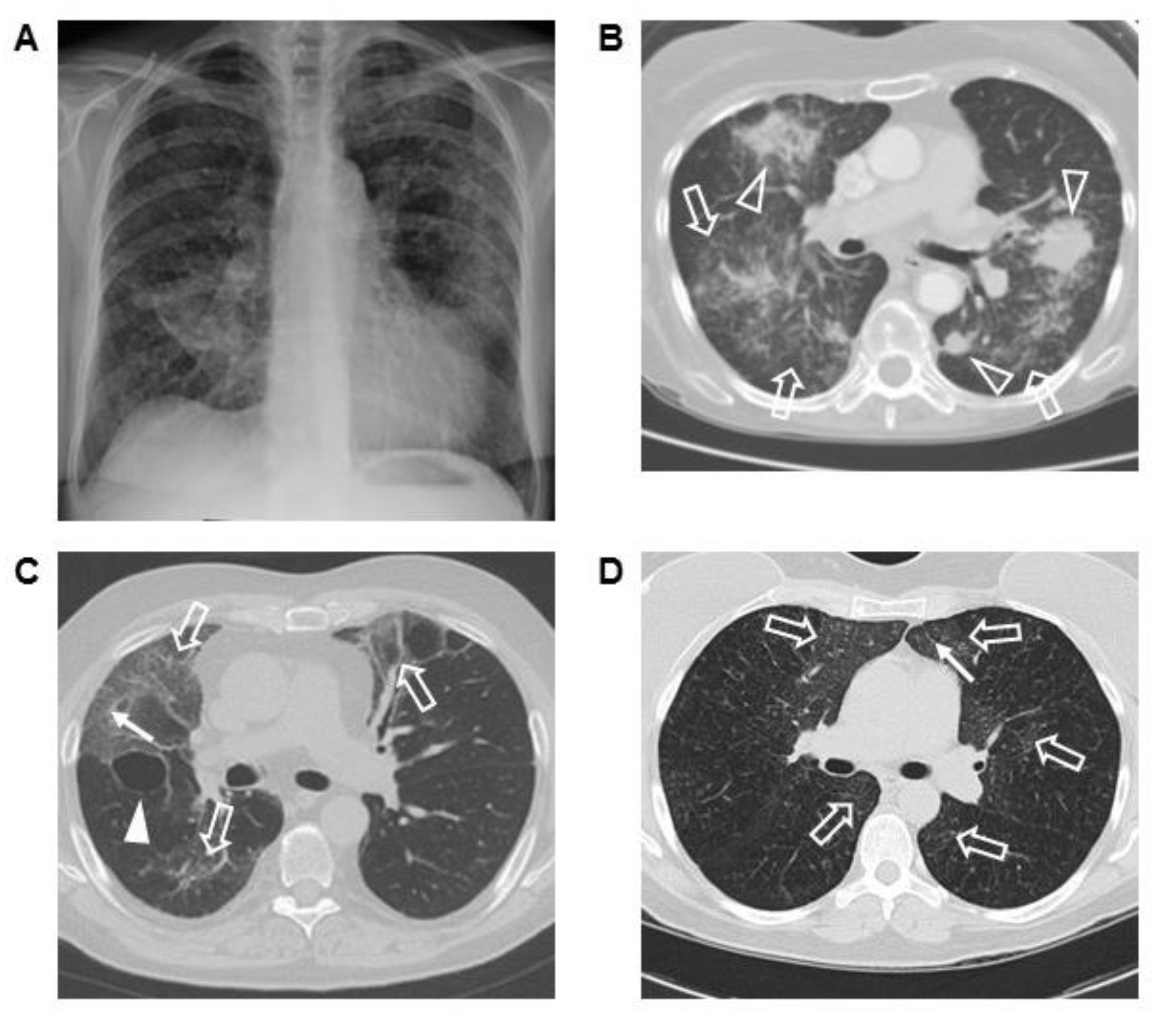
3.3. Chest Imaging and Scoring
3.4. Corresponding Radiographic and Histopathologic Findings
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chester, W. Uber lipoidgranulomatose. Virchows Arch. Pathol. Anat. 1930, 279, 561–602. [Google Scholar] [CrossRef]
- Haroche, J.; Arnaud, L.; Cohen-Aubart, F.; Hervier, B.; Charlotte, F.; Emile, J.F.; Amoura, Z. Erdheim-Chester disease. Curr. Rheumatol. Rep. 2014, 16, 412. [Google Scholar] [CrossRef] [PubMed]
- Courtillot, C.; Laugier Robiolle, S.; Cohen Aubart, F.; Leban, M.; Renard-Penna, R.; Drier, A.; Charlotte, F.; Amoura, Z.; Touraine, P.; Haroche, J. Endocrine manifestations in a monocentric cohort of 64 patients with Erdheim-Chester disease. J. Clin. Endocrinol. Metab. 2016, 101, 305–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Estrada-Veras, J.I.; O’Brien, K.J.; Boyd, L.C.; Dave, R.H.; Durham, B.; Xi, L.; Malayeri, A.A.; Chen, M.Y.; Gardner, P.J.; Alvarado-Enriquez, J.R.; et al. The clinical spectrum of Erdheim-Chester disease: An observational cohort study. Blood Adv. 2017, 1, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Ozkaya, N.; Rosenblum, M.K.; Durham, B.H.; Pichardo, J.D.; Abdel-Wahab, O.; Hameed, M.R.; Busam, K.J.; Travis, W.D.; Diamond, E.L.; Dogan, A. The histopathology of Erdheim-Chester disease: A comprehensive review of a molecularly characterized cohort. Mod. Pathol. 2018, 31, 581–597. [Google Scholar] [CrossRef] [PubMed]
- Emile, J.F.; Abla, O.; Fraitag, S.; Horne, A.; Haroche, J.; Donadieu, J.; Requena-Caballero, L.; Jordan, M.B.; Abdel-Wahab, O.; Allen, C.E.; et al. Revised classification of histiocytoses and neoplasms of the macrophage-dendritic cell lineages. Blood 2016, 127, 2672–2681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Badalian-Very, G.; Vergilio, J.A.; Degar, B.A.; MacConaill, L.E.; Brandner, B.; Calicchio, M.L.; Kuo, F.C.; Ligon, A.H.; Stevenson, K.E.; Kehoe, S.M.; et al. Recurrent BRAF mutations in Langerhans cell histiocytosis. Blood 2010, 116, 1919–1923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen-Aubart, F.; Emile, J.F.; Carrat, F.; Helias-Rodzewicz, Z.; Taly, V.; Charlotte, F.; Cluzel, P.; Donadieu, J.; Idbaih, A.; Barete, S.; et al. Phenotypes and survival in Erdheim-Chester disease: Results from a 165-patient cohort. Am. J. Hematol. 2018, 93, E114–E117. [Google Scholar] [CrossRef] [PubMed]
- Diamond, E.L.; Dagna, L.; Hyman, D.M.; Cavalli, G.; Janku, F.; Estrada-Veras, J.; Ferrarini, M.; Abdel-Wahab, O.; Heaney, M.L.; Scheel, P.J.; et al. Consensus guidelines for the diagnosis and clinical management of Erdheim-Chester disease. Blood 2014, 124, 483–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diamond, E.L.; Subbiah, V.; Lockhart, A.C.; Blay, J.Y.; Puzanov, I.; Chau, I.; Raje, N.S.; Wolf, J.; Erinjeri, J.P.; Torrisi, J.; et al. Vemurafenib for BRAF V600-Mutant Erdheim-Chester Disease and Langerhans Cell Histiocytosis: Analysis of Data From the Histology-Independent, Phase 2, Open-label VE-BASKET Study. JAMA Oncol. 2018, 4, 384–388. [Google Scholar] [CrossRef] [PubMed]
- Egan, A.J.M.; Boardman, L.A.; Tazelaar, H.D.; Swensen, S.J.; Jett, J.R.; Yousem, S.A.; Myers, J.L. Erdheim-Chester disease: Clinical, radiologic, and histopathologic findings in five patients with interstitial lung disease. Am. J. Surg. Pathol. 1999, 23, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Shamburek, R.D.; Brewer, H.B., Jr.; Gochuico, B.R. Erdheim-Chester disease: A rare multisystem histiocytic disorder associated with interstitial lung disease. Am. J. Med. Sci. 2001, 321, 66–75. [Google Scholar] [CrossRef] [PubMed]
- Arnaud, L.; Pierre, I.; Beigelman-Aubry, C.; Capron, F.; Brun, A.L.; Rigolet, A.; Girerd, X.; Weber, N.; Piette, J.C.; Grenier, P.A.; et al. Pulmonary involvement in Erdheim-Chester disease: A single-center study of thirty-four patients and a review of the literature. Arthritis Rheum. 2010, 62, 3504–3512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brun, A.L.; Touitou-Gottenberg, D.; Haroche, J.; Toledano, D.; Cluzel, P.; Beigelman-Aubry, C.; Piette, J.C.; Amoura, Z.; Grenier, P.A. Erdheim-Chester disease: CT findings of thoracic involvement. Eur. Radiol. 2010, 20, 2579–2587. [Google Scholar] [CrossRef] [PubMed]
- Mirmomen, S.M.; Sirajuddin, A.; Nikpanah, M.; Symons, R.; Paschall, A.K.; Papageorgiou, I.; Gahl, W.A.; O’Brien, K.; Estrada-Veras, J.I.; Malayeri, A.A. Thoracic involvement in Erdheim-Chester disease: Computed tomography imaging findings and their association with the BRAF(V600E) mutation. Eur. Radiol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.R.; Crapo, R.; Hankinson, J.; Brusasco, V.; Burgos, F.; Casaburi, R.; Coates, A.; Enright, P.; van der Grinten, C.P.; Gustafsson, P.; et al. General considerations for lung function testing. Eur. Respir. J. 2005, 26, 153–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosas, I.O.; Ren, P.; Avila, N.A.; Chow, C.K.; Franks, T.J.; Travis, W.D.; McCoy, J.P., Jr.; May, R.M.; Wu, H.P.; Nguyen, D.M.; et al. Early interstitial lung disease in familial pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2007, 176, 698–705. [Google Scholar] [CrossRef] [PubMed]
- Rosas, I.O.; Yao, J.; Avila, N.A.; Chow, C.K.; Gahl, W.A.; Gochuico, B.R. Automated quantification of high-resolution CT scan findings in individuals at risk for pulmonary fibrosis. Chest 2011, 140, 1590–1597. [Google Scholar] [CrossRef] [PubMed]
- Rouhani, F.N.; Brantly, M.L.; Markello, T.C.; Helip-Wooley, A.; O’Brien, K.; Hess, R.; Huizing, M.; Gahl, W.A.; Gochuico, B.R. Alveolar macrophage dysregulation in Hermansky-Pudlak syndrome type 1. Am. J. Respir. Crit. Care Med. 2009, 180, 1114–1121. [Google Scholar] [CrossRef] [PubMed]
- Gochuico, B.R.; Avila, N.A.; Chow, C.K.; Novero, L.J.; Wu, H.P.; Ren, P.; MacDonald, S.D.; Travis, W.D.; Stylianou, M.P.; Rosas, I.O. Progressive preclinical interstitial lung disease in rheumatoid arthritis. Arch. Intern. Med. 2008, 168, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Podolanczuk, A.J.; Oelsner, E.C.; Barr, R.G.; Hoffman, E.A.; Armstrong, H.F.; Austin, J.H.; Basner, R.C.; Bartels, M.N.; Christie, J.D.; Enright, P.L.; et al. High attenuation areas on chest computed tomography in community-dwelling adults: The MESA study. Eur. Respir. J. 2016, 48, 1442–1452. [Google Scholar] [CrossRef] [PubMed]
- Washko, G.R.; Hunninghake, G.M.; Fernandez, I.E.; Nishino, M.; Okajima, Y.; Yamashiro, T.; Ross, J.C.; Estepar, R.S.; Lynch, D.A.; Brehm, J.M.; et al. Lung volumes and emphysema in smokers with interstitial lung abnormalities. N. Engl. J. Med. 2011, 364, 897–906. [Google Scholar] [CrossRef] [PubMed]
- Huizing, M.; Malicdan, M.C.V.; Gochuico, B.R.; Gahl, W.A. Hermansky-Pudlak Syndrome. Available online: https://www.ncbi.nlm.nih.gov/books/NBK1287/ (accessed on 29 May 2018).[Green Version]
- Josan, E.S.; Green, J.W.; Zaidi, S.I.M.; Mehta, J.B. Isolated pulmonary involvement in Erdheim-Chester disease. Lung India 2017, 34, 555–558. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, K.; Miyoshi, K.; Mizutani, H.; Otani, S.; Sugimoto, S.; Yamane, M.; Oto, T. Successful lung transplantation for pulmonary disease associated with Erdheim-Chester disease. Ann. Thorac. Surg. 2017, 104, e13–e15. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| No ILD | Minimal ILD | Mild ILD | Mod/Severe ILD | |
|---|---|---|---|---|
| Gender (Male/Female) | 9/3 | 15/5 | 14/4 | 7/5 |
| Age (years) | 51.8 ± 2.5 | 49.2 ± 2.5 | 55.5 ± 3.1 | 55.8 ± 3.4 |
| Dyspnea | 25% | 25% | 50% | 58.3% |
| Cough | 8.3% | 15% | 16.7% | 33.3% |
| No Symptoms | 58.3% | 65% | 38.9% | 33% |
| Crackles | 0% | 0% | 16.7% | 16.7% |
| Wheezing | 0% | 0% | 5.6% | 16.7% |
| Previous Smoker | 8.3% | 10% | 22.2% | 16.7% |
| Current Smoker | 0% | 0% | 11.1% | 8.3% |
| Inhalational Exposures | 0% | 15% | 5.6% | 8.3% |
| Chemotherapy | 75% | 45% | 50% | 75% |
| BRAF Inhibitor | 16.7% | 10% | 11.1% | 16.7% |
| MEK Inhibitor | 0% | 5% | 5.6% | 0% |
| Rituximab | 0% | 5% | 5.6% | 8.3% |
| Interferon | 33.3% | 60% | 50% | 33.3% |
| BRAFV600E | 41.7% | 60% | 44.4% | 58.3% |
| ANA Positive | 33.3% | 25% | 22.2% | 18.2% |
| RF Positive | 8.3% | 0% | 11.1% | 18.2% |
| No ILD | Minimal ILD | Mild ILD | Mod/Severe ILD | p-Value | |
|---|---|---|---|---|---|
| FVC% | 98.6 ± 3.3 | 93.9 ± 3.2 | 85.1 ± 4.3 | 71.3 ± 4.7 | 0.345 * 0.031 † <0.001 ‡ 0.103 ** <0.001 †† 0.045 ‡‡ |
| TLC% | 94.8 ± 2.7 | 96.5 ± 3.1 | 85.5 ± 3.4 | 74.5 ± 4.5 | 0.717 * 0.056 † <0.001 ‡ 0.022 ** <0.001 †† 0.057 ‡‡ |
| DLCO% | 88.2 ± 2.9 | 77.2 ± 2.3 | 64.7 ± 2.0 | 58.2 ± 4.7 | 0.007 * <0.001 † <0.001 ‡ <0.001 ** <0.001 †† 0.171 ‡‡ |
| 6-MWT (m) | 504 ± 38 | 475 ± 19 | 432 ± 24 | 404 ± 37 | 0.461 * 0.106 † 0.077 ‡ 0.166 ** 0.070 †† 0.521 ‡‡ |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Haroutunian, S.G.; O’Brien, K.J.; Estrada-Veras, J.I.; Yao, J.; Boyd, L.C.; Mathur, K.; Gahl, W.A.; Mirmomen, S.M.; Malayeri, A.A.; Kleiner, D.E.; et al. Clinical and Histopathologic Features of Interstitial Lung Disease in Erdheim–Chester Disease. J. Clin. Med. 2018, 7, 243. https://doi.org/10.3390/jcm7090243
Haroutunian SG, O’Brien KJ, Estrada-Veras JI, Yao J, Boyd LC, Mathur K, Gahl WA, Mirmomen SM, Malayeri AA, Kleiner DE, et al. Clinical and Histopathologic Features of Interstitial Lung Disease in Erdheim–Chester Disease. Journal of Clinical Medicine. 2018; 7(9):243. https://doi.org/10.3390/jcm7090243
Chicago/Turabian StyleHaroutunian, Sara G., Kevin J. O’Brien, Juvianee I. Estrada-Veras, Jianhua Yao, Louisa C. Boyd, Kavya Mathur, William A. Gahl, S. Mojdeh Mirmomen, Ashkan A. Malayeri, David E. Kleiner, and et al. 2018. "Clinical and Histopathologic Features of Interstitial Lung Disease in Erdheim–Chester Disease" Journal of Clinical Medicine 7, no. 9: 243. https://doi.org/10.3390/jcm7090243